
Abstract
The thiazide-sensitive NaCl cotransporter (NCC) plays key roles in renal electrolyte transport and blood pressure maintenance. Regulation of this cotransporter has received increased attention recently, prompted by the discovery that mutations in the with-no-lysine (WNK) kinases are the molecular explanation for pseudohypoaldosteronism type II (PHAII). Studies suggest that WNK4 regulates NCC via two distinct pathways, depending on its state of activation. Furthermore, an intact STE20-related proline–alanine-rich kinase (SPAK)/oxidative stress response 1 kinase (OSR1) pathway was found to be necessary for a WNK4 PHAII mutation to increase NCC phosphorylation and blood pressure in mice. The mouse protein 25α is a novel regulator of the SPAK/OSR1 kinase family, which greatly increases their activity. The phosphorylation status of NCC and the WNK is regulated by the serum- and glucocorticoid-inducible kinase 1, suggesting novel mechanisms whereby aldosterone modulates NCC activity. Dephosphorylation of NCC by protein phosphatase 4 strongly influences the activity of the cotransporter, confirming an important role for NCC phosphorylation. Finally, γ-adducin increases NCC activity. This stimulatory effect is dependent on the phosphorylation status of the cotransporter. γ-Adducin only binds the dephosphorylated cotransporter, suggesting that phosphorylation of NCC causes the dissociation of γ-adducin. Since γ-adducin is not a kinase, it is tempting to speculate that the protein exerts its function by acting as a scaffold between the dephosphorylated cotransporter and the regulatory kinase. As more molecular regulators of NCC are identified, the system-controlling NCC activity is becoming increasingly complex. This intricacy confers an ability to integrate a variety of stimuli, thereby regulating NCC transport activity and ultimately blood pressure.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Studies of electrolyte transport within the renal distal convoluted tubule (DCT) led to the identification of a thiazide-sensitive component that permits vectorial NaCl cotransport. The thiazide-sensitive NaCl cotransporter (NCC) was subsequently cloned from the urinary bladder of the winter flounder, Pseudopleuronectes americanus [16]. Gitelman’s syndrome is an autosomal recessive salt-losing disorder characterized by hypokalemic metabolic alkalosis, hypomagnesemia, and hypocalciuria [20, 43]. Genetic mapping studies in patients with Gitelman’s syndrome revealed that the defect was the result of mutations in the SLC12A3 gene, which encodes NCC [43]. Later studies led to the discovery of an intricate kinase network that regulates the function of NCC by modifying the phosphorylation level, trafficking, and lysosomal sorting of the protein. These discoveries were initially prompted by the genetic mapping of patients suffering from an autosomal dominant disorder associated with hypertension, namely pseudohypoaldosteronism type II (PHAII; also known as Gordon syndrome) [48].
Subsequent studies have shown that NCC plays a critical role in renal NaCl transport and blood pressure maintenance. Thiazides, the pharmacological inhibitor of NCC, remain one of the most effective and prescribed drugs in the treatment of hypertension. As such, knowledge regarding the regulation of NCC is a critical step in understanding the development and underlying pathogenesis of hypertension. This article aims to review the discovery of novel factors that regulate the cotransporter. These proteins appear to act as members of a cellular network that relay and integrate signals to alter NCC function and, consequently, blood pressure.
Mechanisms of NCC regulation
The majority of studies on NCC function have been conducted in the Xenopus laevis oocyte expression system. Injection of cRNA-encoding NCC into oocytes generates a stable and reproducible thiazide-sensitive uptake of 22Na+. The results are less consistent when mammalian cell systems are employed. Several groups have reported thiazide-sensitive 22Na+ uptake in mammalian cell lines expressing NCC, while others have failed to do so. For this reason, many still utilize the oocyte expression system to study NCC activity. The results obtained in the oocyte as well as mammalian cell systems are often in line with those obtained in vivo and thus suggest that these cellular models can be used to study NCC function. As will be evident in this review, the network of auxiliary proteins regulating NCC is expanding rapidly. Whether or not all members of this regulatory network are present in these cell models, it is uncertain and must be taken into account when analyzing the responses of the system.
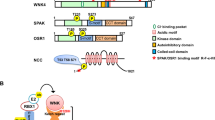
NCC can be regulated in a variety of ways including: changes in expression, trafficking, and phosphorylation. These processes all ultimately affect the net flow of NaCl across the DCT. Transcriptional regulation leading to changes in NCC abundance occurs after hormonal stimuli and is often thought of as a chronic adaptation to various alterations in the total body NaCl balance. More rapid regulatory processes include changes in trafficking and phosphorylation of the cotransporter. The amino N-terminal domain of NCC contains several phosphorylation sites (including Thr46, Thr55, Thr60, Ser73, and Ser91 in human NCC), quite a few of which are conserved among members of the SLC12 family [8, 18, 19, 34, 37] (Fig. 1). Although the understanding of the individual contributions of each phosphorylation site is incomplete, several observations have suggested that phosphorylation of these residues, especially that of Thr60, is critically important for the activation of NCC. When several of the N-terminal phosphorylation sites in NCC [9, 34] or Thr60 alone [21] is converted to constitutively inactive sites by substitutions to alanine, the transport activity of NCC is markedly decreased in the X. laevis expression system. Interestingly, the process seems to occur without altering membrane localization. This observation infers that phosphorylation at these sites is important for intrinsic activity of the cotransporter [21, 34]. Furthermore, while increasing the phosphorylation status of the cotransporter by incubating NCC-expressing oocytes in hypotonic, low Cl−-containing media increases transport activity without changing membrane abundance of the protein [34]. Studies in human embryonic kidney cells further demonstrate that mutation of Thr60 specifically prevents the phosphorylation of several of the other N-terminal residues and coincides with a reduction in thiazide-sensitive Na+ transport. This likely explains why individuals with a mutation in the Thr60 phosphorylation site (converted to methionine) present with Gitelman’s syndrome [29]. Moreover, it highlights the fact that intact NCC phosphorylation sites are necessary for renal NaCl reabsorption and the maintenance of blood pressure. Interestingly, while phosphorylation of the N-terminal residues in NCC seems dissociated from trafficking in the oocyte, studies suggest that the phosphorylated cotransporter is found only in the apical membrane of the DCT cell in rat [35]. These observations could indicate that phosphorylation of NCC at the N terminus only occurs when the cotransporter is anchored in the plasma membrane. Several novel phosphorylation sites including Ser124 (rat, equivalent to Ser126 in human NCC) and Ser811 in NCC have recently been identified; however at the present time, no function has been assigned to these residues [12, 23] (Fig. 1).

Schematic diagram of human NCC, depicting the various phosphorylation sites within the cotransporter. Please note that Ser126 has not been described in human NCC, but has been identified as Ser124 in rat NCC. In addition, binding sites for the SPAK/OSR1 kinases as well as the γ-adducin-binding region are indicated in the figure
In addition to phosphorylation, trafficking of NCC from subapical vesicles to the plasma membrane may also be an important factor in regulating NaCl cotransport in the DCT. Although several of the N-terminal phosphorylation sites appear to regulate transport independently of changes in the subcellular localization of NCC, it remains unclear whether the trafficking process is dependent on phosphorylation of other sites within the cotransporter. Rapid alterations in membrane expression of NCC are seen after angiotensin II infusion in rats, which provokes movement of the cotransporter into the apical plasma membrane from subapical vesicles [41].
The WNK kinase family
The WNK family of serine–threonine kinases has a characteristic displacement of a catalytic lysine residue necessary for ATP binding, hence the name with-no-k (lysine) [46, 50]. The role of the WNK family in the maintenance of ambient blood pressure has been firmly established. PHAII patients have defects in either the WNK1 or WNK4 genes and suffer from increased renal reabsorption of NaCl and consequently elevated arterial pressure [24, 48]. Specifically, autosomal dominant mutations in the WNK4 gene or intronic deletions of the WNK1 gene (that massively increase its transcript) are responsible for PHAII [48].
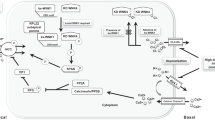
Thiazide diuretics are known to normalize blood pressure and correct electrolyte abnormalities when administered to PHAII patients [10], implicating a prominent role for NCC in this disease. WNK4 has subsequently been found to inhibit NCC transport by reducing plasma membrane abundance of the cotransporter in the oocyte expression system [49, 53]. Recent studies have shown that WNK4 inhibits NCC activity by diverting the cotransporter to the lysosomal compartment during forward trafficking. This likely occurs via a sortilin-dependent mechanism [44, 59] (Fig. 2a). The inhibitory effect of WNK4 occurs independently of NCC phosphorylation of Thr58 (equivalent to Thr60 in humans), which is important for NCC activity [21, 22]. Loss-of-function mutations in WNK4 have been the suggested molecular mechanism of PHAII. Here, mutations in WNK4 increase NCC abundance by reverting the inhibitory effect on lysosomal shuttling of NCC (Fig. 2b). One PHAII-causing mutation in WNK4 (Q562E) reduced the inhibitory effect of WNK4 on NCC activity and trafficking [49, 53]. However, in the same study, several other PHAII-causing mutations of WNK4 failed to inhibit the activity of the cotransporter [53].

The “inhibitory” pathway of WNK4. a WNK4 diverts NCC to the lysosomal compartment during forward trafficking of the cotransporter to the plasma membrane. WNK3 interacts with WNK4, potentially preventing the WNK4-induced shuttling of NCC to the lysosome. WNK1 also inhibits WNK4 by interacting with the protein. In addition, WNK1-KS acts by inhibiting WNK1, thereby releasing WNK1’s inhibition on WNK4, thus increasing the inhibitory effect of WNK4 upon NCC. b Potential mechanism by which loss-of-function mutations in WNK4 leads to PHAII. Loss-of-function mutations in WNK4 reduce the ability of the kinase to shuttle NCC to the lysosomes during forward trafficking, thereby increasing the abundance of the cotransporter in the apical plasma membrane. c An increase in the WNK1 protein occurs due to intronic deletion in its gene. Increased expression of WNK1 in this “inhibitory” pathway would lead to a marked inhibition of WNK4, thus increasing NCC membrane abundance and thereby causing PHAII
Contrasting these observations is the finding that WNK4 stimulates N-terminal phosphorylation and hence activity of NCC in the same cellular systems. This occurs via WNK4-dependent activation of the STE20 family of serine/threonine kinases, namely the STE20-related proline–alanine-rich kinase (SPAK) and oxidative stress response 1 kinase (OSR1) [47]. Additionally, angiotensin II increases NCC activity in the Xenopus oocyte system, but only in the presence of WNK4. This stimulatory effect of angiotensin II on NCC is dependent on SPAK activation and coincides with increased phosphorylation of the cotransporter [40] (Fig. 3a). Moreover, this study could not detect any additional stimulatory effect of angiotensin II on NCC activity when a PHAII mutant of WNK4 was expressed, suggesting that the mutated WNK4 was already in an activated state [40]. The positive effect of WNK4 on NCC activity may be due to an elevation of intracellular Ca2+, which is released after angiotensin II application. San-Cristobal et al., the authors of this study, highlighted the fact that PHAII mutations in WNK4 are situated in a negatively charged domain of the WNK4 protein. This part of WNK4 bears some similarity to a Ca2+-binding EF hand domain. Thus, it is suggested that PHAII mutations in this domain may switch WNK4 into an active state, thereby stimulating the SPAK and OSR1 kinases to phosphorylate NCC and subsequently causing the PHAII phenotype (Fig. 3b).

The “stimulatory” pathway of WNK4. a Activation of WNK4 by angiotensin II potentially occurring via elevations in intracellular Ca2+. In this state, WNK4 stimulates the SPAK/OSR1 pathway. Cl−-depletion and K+-depletion activates WNK1, thereby activating the SPAK and OSR1 kinases. In both cases, activation of the STE20 kinases leads to increased phosphorylation level of NCC. The augmented phosphorylation level of NCC increases cotransport activity. b Potential mechanism whereby gain-of-function mutations in WNK4 causes PHAII. Increased activity of WNK4 due to PHAII mutations increases its stimulatory effect on the SPAK/OSR1 pathway. This enhances phosphorylation of NCC and thereby increases NaCl transport, leading to PHAII. c An increase in the expression of WNK1 stimulates the activation of the SPAK/OSR1 kinases, which subsequently increases the phosphorylation level of NCC
Studies directed at understanding the effect of WNK4 on NCC function in these cellular systems seem contradictory. However, the studies can be reconciled if both the inhibitory and stimulatory pathways coexist within the cell. Thus, WNK4 may act as a potent stimulator or inhibitor of NCC, depending on its state of activation. Without adequate stimulation, WNK4 is likely to be inhibitory, shuttling NCC into lysosomes and reducing transport activity by doing so. However, in the presence of stimuli (such as angiotensin II), WNK4 will increase NCC transport via activation of the SPAK/OSR1 pathway. Although not yet described, there may also be factors that switch WNK4 in the opposite direction, thereby favoring the inhibitory pathway. A better understanding of the factors that are involved in changing WNK4’s state of activation will be important to our understanding of NCC biology. The biphasic effects of WNK4 may also help explain why in vivo experiments using mice with a genetically modified WNK4 pathway can yield conflicting results.
Hypomorphic mice with a targeted deletion of exon 7 in the Wnk4 gene have a partially functional WNK4. The hypomorphic strain presented with a lower blood pressure and increased NaCl excretion during dietary salt restriction, as well as reduced NCC phosphorylation [33]. These observations infer that WNK4 is predominantly stimulatory under basal conditions in vivo. This also raises the question whether PHAII occurs as the result of loss-of-function or gain-of-function mutations in WNK4. In transgenic knock-in mice with a PHAII-mutated Wnk4 gene (D561A), there is increased phosphorylation of NCC, suggesting that the SPAK/OSR1 pathway is activated [54]. Recent data from Chiga et al., utilizing triple knock-in mice strains with a PHAII-mutated WNK4 (D561A) and non-activatable SPAK and OSR1 (one allele) kinases, have shed light on this issue [5]. In contrast to the single knock-in WNK4 mutant mice strain, which have a PHAII-like phenotype and increased phosphorylation of NCC, triple knock-in mice fail to display a hypertensive phenotype and demonstrate reduced phosphorylation of NCC [5]. These data suggest that the WNK4-stimulated SPAK/OSR1 pathway is primarily responsible for the PHAII phenotype, at least for this particular WNK4 mutant. However, this does not exclude an important role for the inhibitory actions of WNK4 in the lysosomal sorting of NCC. Transgenic mice overexpressing another PHAII WNK4 mutant (Q562E) show a hypertensive phenotype similar to patients with PHAII. These mice display marked hyperplasia of the DCT and elevated expression of NCC. However, mice overexpressing wild-type WNK4 show the opposite phenotype, with lower blood pressure, atrophy of the DCT, and reduced NCC expression [27]. This suggests that a surplus of wild-type WNK4 in the intact animal can lead to an inhibition of NCC. The inhibitory pathway is likely to be operating during conditions when WNK4 is not in its activated form, as exemplified by mice engineered to have extra wild-type WNK4 alleles and in the study discussed below.
A recent study by Mu et al. showed that the WNK4 pathway plays an important role in salt-sensitive hypertension [31]. Activation of the adrenergic pathway in mice placed on a high-NaCl diet causes hypertension, increased expression of NCC, in addition to a reduced renal WNK4 content. These effects were not observed in β2-adrenergic receptor knockout mice. Interestingly, the authors found that the glucocorticoid receptor was necessary for the observed decrease in WNK4 abundance. This was elegantly demonstrated by utilizing mice with a targeted deletion of the glucocorticoid receptor in the distal nephron. This strain did not decrease WNK4 abundance or increase blood pressure when placed on high-NaCl diet and infused with β-adrenergic agonists [31]. The current study shows a clear role for WNK4 and NCC in salt-sensitive hypertension. Moreover, the data reinforce the notion that WNK4 can have an inhibitory effect on NCC in vivo during certain conditions. Although the amount of phosphorylated NCC is increased in this study, it is not clear whether this is a reflection of increased NCC expression or an actual increase in the phosphorylation level of the cotransporter. Therefore, it remains to be determined to what extent the SPAK/OSR1 pathway is activated in these experiments and what signaling cascade is involved in stimulating it. As discussed below, several members in the regulatory complex surrounding NCC act by modifying WNK4 function. Depending on WNK4’s state of activation, the role of the kinase may be dramatically different. Further research directed at dissecting out when each pathway predominates would aid tremendously in understanding how other regulatory proteins influence NCC function by modifying WNK4.
In the Xenopus oocyte system, WNK1 imposes an inhibitory effect on WNK4, abrogating its ability to shuttle NCC into lysosomes (Fig. 2a). This mechanism potentially explains why increased WNK1 protein expression results in the same PHAII phenotype caused by mutations in WNK4 [53] (Fig. 2c). The inhibitory effect of WNK1 on WNK4 might stem from interaction between the two kinases, leading to phosphorylation of WNK4 [39, 55]. However, WNK1 also activates the SPAK/OSR1 pathway during hypotonic Cl−-depleted or K+-depleted conditions [32, 37, 47] (Fig. 3a). Thus, overexpression of WNK1 in PHAII may increase the phosphorylation level and hence transport activity of NCC by activation of SPAK and OSR1 (Fig. 3c). The exact underlying molecular mechanism mediating PHAII via WNK1 overexpression is unclear. If WNK4 functions as a stimulator of NCC transport during basal conditions in the intact animal [33], it seems to reason that WNK1 overexpression in PHAII patients must result from activation of the SPAK/OSR1 pathway. Further data are necessary to confirm this, especially studies that dissect out all possible mechanisms by which WNK1 can inhibit WNK4. Regardless of the mechanism, it is clear from these data that WNK1 is a potent regulator of renal electrolyte transport and blood pressure, as is WNK4. This is consistent with the observation that heterozygous mice with genetic ablation of the Wnk1 gene have reduced blood pressure [58]. However, part of this response is likely caused by alterations in vascular tone, due to reduced phosphorylation of SPAK and NKCC1 in the arteries of this strain [1].
Although the role of WNK3 remains to be fully clarified, it is known that this kinase markedly stimulates the activity of NCC in the X. laevis expression system [38]. This stimulatory effect occurs by phosphorylating and consequently inhibiting WNK4, a mechanism that likely prevents shuttling of the cotransporter to the lysosomes [55]. The stimulatory effect of WNK3 on NCC coincides with increased plasma membrane expression of the cotransporter. This process occurs independently of Thr60 phosphorylation on NCC [21].
The aldosterone-regulated splice variant of WNK1, termed kidney specific WNK1 (WNK1-KS), inhibits the function of full-length WNK1, thereby reducing NCC activity [55]. This is in line with the observation that mice lacking WNK1-KS has increased mean arterial pressure as well as increased expression and phosphorylation of NCC [25].
The STE20 family of serine/threonine kinases
WNK-stimulated phosphorylation of NCC is largely mediated by the STE20 family of serine/threonine kinases, specifically the members SPAK and OSR1. Both kinases directly phosphorylate NCC on Thr46, Thr55, and Thr60 [37]. The kinases require a docking interaction between their conserved C-terminal domains and the SPAK/OSR1 binding motif (RFx[V/I]) in NCC to enable these phosphorylation events [37, 47] (Fig. 1). Moreover, phosphorylation of Ser71 is altered in SPAK-deficient mice (Ser73 in humans), suggesting that the kinase is required for phosphorylation of this site [56]. Recently, it was demonstrated that when the SPAK/OSR1 pathway is enhanced, phosphorylation of Ser73 is also increased in vitro [13]. Both WNK1 and WNK4 interact with SPAK and OSR1, and only the catalytically active WNKs are able to phosphorylate the SPAK/OSR1 kinases [47]. The WNKs contain SPAK/OSR1-binding motifs that allow them to interact with the STE20 kinases [15, 30, 47]. Further, phosphorylation of STE20 substrates by the WNKs increases both SPAK and OSR1 activity. Mutating one of these phosphorylation sites, namely the T-loop threonine residues in SPAK (Thr233) or OSR1 (Thr185), prevents activation of the kinases [5, 47].
Mutation of Thr185 in OSR1 to alanine causes embryonic lethality of the knock-in mice, while strains with Thr233 in SPAK are born at the expected Mendelian ratio [36]. These latter mice are hypotensive, a feature that is normalized by supplementing the animals with dietary NaCl. As expected, the animals have a marked reduction in NCC phosphorylation. In addition, a decrease in NCC abundance was also observed, without a concomitant reduction in mRNA, suggesting that the stability of the protein was somehow affected by inactive SPAK kinases [36]. Mice with a genetic deletion of the Spak gene show a similar phenotype as those with an inactivatable kinase, but several differences exist. For instance, these mice develop electrolyte abnormalities consistent with a Gitelman-like phenotype. Moreover, a reduction in endothelial NKCC1 phosphorylation is present, suggesting that impaired aortic contractility may contribute to the hypotensive phenotype [56].
Mouse protein-25Α (MO25α)
MO25α was recently discovered to be a master regulator of several members of the STE20 kinase family, including SPAK and OSR1 [13]. The protein is classically described as a scaffold that regulates the LKB1 tumor suppressor kinase complex. Conserved MO25α-binding sites were identified within both the SPAK and OSR1 kinases. Co-expression of MO25α with these kinases stimulated their activity as much as 70–100-fold. This remarkable increase in activation only occurs when the kinases are activated (i.e., by changing the T-loop threonine residues in SPAK (Thr233) and OSR1 (Thr185) to Glutamic acid residues). The WNK family phosphorylates these T-loops threonines and similar increases in the activity of wild-type OSR1 were observed when WNK1 was present. Knockdown of the MO25α protein did not affect the phosphorylation of SPAK/OSR1 by WNK1, suggesting that the binding of MO25α does not facilitate this process. As expected, phosphorylation of the N-terminal residues on NCC is markedly enhanced after this MO25α-dependent increase in activation of the SPAK/OSR1 kinases [13]. Many laboratories have reported difficulty establishing mammalian cell lines expressing NCC that display thiazide-sensitive transport. Perhaps co-expression of MO25α may help alleviate this problem by assuring maximal activation of NCC.
The serum- and glucocorticoid-inducible kinase 1
Serum- and glucocorticoid-inducible kinase 1 (SGK1) is involved in controlling several renal electrolyte transport processes. Part of the “early response” to aldosterone on renal electrolyte transport has been ascribed to translational upregulation of this kinase. The effect of SGK1 on epithelial transport has been well described for the epithelial Na+ channel (ENaC), where the kinase markedly stimulates channel activity [3]. This stimulatory effect on ENaC is achieved by phosphorylating the ubiquitin ligase Nedd4-2, preventing it from ubiquitinating ENaC and consequently marking it for degradation [28]. Genetic ablation of Sgk1 in mice results in salt wasting, which is only obvious when the mice are maintained on low dietary NaCl. In addition to a defect in ENaC cleavage, these mice present with a reduced expression of NCC [11]. When normal mice are placed on a low-NaCl diet, NCC abundance as well as phosphorylation of Thr53, Thr58, and Ser71 is increased (equivalent to Thr55, Thr60, and Ser73 in humans). Interestingly, this response is attenuated in mice with a targeted deletion of the Sgk1 gene, suggesting that SGK1 somehow affects NCC phosphorylation [45]. Whether the reduced expression of NCC is dependent upon a reduction in phosphorylation of the cotransporter, as reported by others, remains to be seen [5, 36, 56]. In the X. laevis system, WNK4 inhibits the activity of NCC under basal conditions by diverting the cotransporter to the lysosome. Here, SGK1 functions as an inhibitor of WNK4, reducing the negative effect of WNK4 upon NCC activity [39]. This inhibitory action on WNK4 occurs by binding of the SGK1 protein to WNK4, which results in SGK1-dependent phosphorylation at two sites within WNK4, namely Ser1169 and Ser1196. Converting these sites into aspartates, thereby mimicking active phosphorylation sites, completely reverts the inhibitory effect of WNK4 on NCC activity [39]. In an apparent feedback loop, WNK1 has been shown to activate SGK1. Interestingly, SGK1 activation is dependent on the N-terminal residues of WNK1 and not the catalytic activity of the kinase [51, 52]. This finding has subsequently been expanded to several of the WNKs, including WNK4 [26]. Conversely, SGK1 has also been shown to regulate WNK1 by phosphorylating it at Thr58 [4]. Whether these interactions results in altered NCC transport remains to be determined. Since SGK1 is an aldosterone-regulated protein, understanding its interaction with the WNK system will help delineate how aldosterone regulates NCC and distal nephron NaCl transport.
Protein phosphatase 4
Regulation of NCC phosphorylation is critical in determining its activity. As such, protein phosphatases are important for regulating the activity of several other Slc12 family members. A recent report by Glover et al. found that protein phosphatase 4 (PP4) was able to strongly inhibit NCC activity in the Xenopus oocyte [22]. In line with previous studies, they observed that the inhibitory actions of PP4 on NCC occur independently of membrane trafficking. The effect of PP4 was clearly dependent on its phosphatase activity, as a phosphatase-dead mutant had no effect upon NCC activity. Moreover, the PP2B phosphatase had no effect on NCC activity, suggesting specificity of PP4. Consistent with this is the restricted expression of PP4 to the renal distal tubule. Finally, the inhibitory effect of PP4 was only found in NCC transporters with intact Thr58 phosphorylation sites (Thr60 in humans) [22]. Thus, these data indicate an important role for PP4 in the regulation of NCC activity, by modulating its phosphorylation level (Fig. 4, step 4).

Schematic model detailing the postulated mechanism whereby γ-adducin could stimulate NCC activity. This model is speculative at current. Since γ-adducin is not a kinase, it may act as a scaffold by anchoring a kinase (potentially SPAK or OSR1) to the dephosphorylated cotransporter (step 1). The anchored kinase phosphorylates NCC, which leads to an increase in transport activity (step 2). γ-Adducin dissociates from NCC after the kinase phosphorylates the cotrasporter (step 3). Dephosphorylation of NCC by the PP4 protein reduces NCC activity (step 4). The cycle can be repeated after the cotransporter has been dephosphorylated
γ-Adducin
The previously described studies highlight the fact that phosphorylation of the N-terminal domain of NCC is critically important for cotransporter activity. Dimke et al. utilized pull-down experiments with the N-terminal domain of NCC as bait to screen mouse kidney lysates for potential interactors of the cotransporter. Using this approach coupled to mass spectrometry, γ-adducin was identified as a novel auxiliary factor interacting with NCC [9]. The adducin gene family was originally characterized as cytoskeletal membrane proteins involved in spectrin–actin binding [17]. γ-Adducin is an interesting protein based on its proposed involvement in primary hypertension in humans and hypertensive rat models [2, 6, 57]. Moreover, γ-adducin co-localizes with NCC to the distal convoluted tubule of the kidney, making it an excellent candidate regulatory protein of NCC [9, 14]. When expressed in the oocyte system, γ-adducin markedly stimulated the activity of NCC. In addition, siRNA directed against the endogenous X. laevis γ-adducin reduced NCC-dependent uptake, further solidifying the role of γ-adducin in NCC regulation. To investigate whether binding of γ-adducin to the N terminus of NCC is required for its stimulatory effect, competition assays were performed. Injection of increasing amounts of the N-terminal domain into oocytes co-expressing γ-adducin and NCC completely reverted the stimulatory effect of γ-adducin, suggesting that binding to the N-terminal domain is necessary to increase NCC activity. Mapping of the γ-adducin-binding site in NCC revealed that it bound to a segment that encompasses several of the N-terminal phosphorylation sites. Another clue that γ-adducin could be involved in modulating the phosphorylation status of NCC was the observation that cotransporter trafficking was unaltered, despite increased activity. Mutating the phosphorylation sites in the N-terminal domain of NCC (Thr55, Thr60, and Ser73) into aspartates, thereby mimicking constitutively active sites, completely abolished the stimulatory effect of γ-adducin [9]. These data indicate that phosphorylation of NCC is integral for the stimulatory effect of γ-adducin. Moreover, γ-adducin was found to bind NCC only in conditions when the phosphorylation of the cotransporter was absent or low (i.e., in the GST-purified wild-type NCC N-terminal and the mutant N-terminal with the Thr55, Thr60, and Ser73 sites converted to alanines). However, when these phosphorylation sites were converted into aspartates, binding of γ-adducin to NCC was abolished [9]. This suggests that γ-adducin only binds NCC when the cotransporter is in its dephosphorylated state.
Acknowledging that γ-adducin is not a kinase, it may affect NCC activity by acting as a scaffold, bringing a kinase together with its substrate NCC. As phosphorylation of the N-terminal sites of NCC is mediated via SPAK and OSR1, these appear as likely candidates for the scaffolding functions of γ-adducin. At present, pull-down experiments with γ-adducin and the SPAK/OSR1 kinases have not been performed. Based on the initial data obtained, a potential mechanism of how γ-adducin binds NCC and stimulates its activity can be seen in Fig. 4. γ-Adducin may function by bringing the kinase together with the dephosphorylated N terminus of the cotransporter (Fig. 4, step 1). This inevitably leads to an increase in NCC phosphorylation and subsequently activity (Fig. 4, step 2). After the kinase has phosphorylated NCC (a feature that could lead to a conformational change in the binding domain of the cotransporter), γ-adducin dissociates from NCC and may even help the kinase dissociate from the cotransporter as well (Fig. 4, step 3). This theory is currently speculative and remains to be tested experimentally. Interaction studies directed at the potential binding of γ-adducin with the STE20 kinases will be crucial to increase our understanding of how γ-adducin affects NCC activity.
The adducin gene family has previously been implicated in arterial hypertension. Most remarkable is the role of α-adducin. However, the effect of this isoform is likely to occur by an alternative mechanism, as it is not expressed in the distal tubule [14] and α-adducin does not stimulate NCC activity in the oocyte system [9]. One single nucleotide polymorphism in the γ-adducin gene has been described, which is involved in systolic blood pressure regulation in certain individuals [7]. Moreover, systolic blood pressure and pulse rate remain stable in genetically modified mice lacking γ-adducin. However, this is expected since mice with a targeted deletion of NCC show no alteration in mean arterial pressure when maintained on normal amounts of dietary NaCl. However, when a reduction is imposed in dietary NaCl content, NCC-deficient mice develop hypotension [42]. Whether manipulations of NaCl in the diet are needed to provoke changes in blood pressure in γ-adducin-deficient mice remains to be determined.
Conclusion
The kidney is a complex organ that responds to a variety of stimuli by altering urinary excretion of electrolytes. These adjustments depend on the signals the kidney receives from various sources throughout the body and from within the kidney itself. NCC plays a key role in renal NaCl transport thereby contributing to blood pressure maintenance. It is therefore not surprising that NCC is regulated at various levels. It is now clear that a larger network surrounds NCC. This network consists of kinases, scaffolds, and phosphatases that regulate different aspects of transporter function. Naturally, cross talk between components of this system must occur in order to relay the most important signal to the transporter. This integrated signaling complex regulates NCC activity by modifying phosphorylation status, trafficking, and lysosomal sorting. Future research into this area will help explain how this complex system regulates NCC. As such, it will be necessary to dissect out which proteins translate the diverse array of environmental signals into altered transporter activity. It will also be important to establish when WNK4 stimulates or inhibits NCC activity, in order to better understand this system.
References
Bergaya S, Faure S, Baudrie V, Rio M, Escoubet B, Bonnin P, Henrion D, Loirand G, Achard JM, Jeunemaitre X, Hadchouel J (2011) WNK1 regulates vasoconstriction and blood pressure response to {alpha}1-adrenergic stimulation in mice. Hypertension 58:439–445
Bianchi G, Tripodi G, Casari G, Salardi S, Barber BR, Garcia R, Leoni P, Torielli L, Cusi D, Ferrandi M et al (1994) Two point mutations within the adducin genes are involved in blood pressure variation. Proc Natl Acad Sci USA 91:3999–4003
Chen SY, Bhargava A, Mastroberardino L, Meijer OC, Wang J, Buse P, Firestone GL, Verrey F, Pearce D (1999) Epithelial sodium channel regulated by aldosterone-induced protein sgk. Proc Natl Acad Sci USA 96:2514–2519
Cheng CJ, Huang CL (2011) Activation of PI3-kinase stimulates endocytosis of ROMK via Akt1/SGK1-dependent phosphorylation of WNK1. J Am Soc Nephrol 22:460–471
Chiga M, Rafiqi FH, Alessi DR, Sohara E, Ohta A, Rai T, Sasaki S, Uchida S (2011) Phenotypes of pseudohypoaldosteronism type II caused by the WNK4 D561A missense mutation are dependent on the WNK-OSR1/SPAK kinase cascade. J Cell Sci 124:1391–1395
Cusi D, Barlassina C, Azzani T, Casari G, Citterio L, Devoto M, Glorioso N, Lanzani C, Manunta P, Righetti M, Rivera R, Stella P, Troffa C, Zagato L, Bianchi G (1997) Polymorphisms of alpha-adducin and salt sensitivity in patients with essential hypertension. Lancet 349:1353–1357
Cwynar M, Staessen JA, Ticha M, Nawrot T, Citterio L, Kuznetsova T, Wojciechowska W, Stolarz K, Filipovsky J, Kawecka-Jaszcz K, Grodzicki T, Struijker-Boudier HA, Thijs L, Van Bortel LM, Bianchi G (2005) Epistatic interaction between alpha- and gamma-adducin influences peripheral and central pulse pressures in white Europeans. J Hypertens 23:961–969
Darman RB, Forbush B (2002) A regulatory locus of phosphorylation in the N terminus of the Na-K-Cl cotransporter, NKCC1. J Biol Chem 277:37542–37550
Dimke H, San-Cristobal P, de Graaf M, Lenders JW, Deinum J, Hoenderop JG, Bindels RJ (2011) gamma-Adducin stimulates the thiazide-sensitive NaCl cotransporter. J Am Soc Nephrol 22:508–517
Farfel Z, Iaina A, Rosenthal T, Waks U, Shibolet S, Gafni J (1978) Familial hyperpotassemia and hypertension accompanied by normal plasma aldosterone levels: possible hereditary cell membrane defect. Arch Intern Med 138:1828–1832
Fejes-Toth G, Frindt G, Naray-Fejes-Toth A, Palmer LG (2008) Epithelial Na+ channel activation and processing in mice lacking SGK1. Am J Physiol Renal Physiol 294:F1298–F1305
Feric M, Zhao B, Hoffert JD, Pisitkun T, Knepper MA (2011) Large-scale phosphoproteomic analysis of membrane proteins in renal proximal and distal tubule. Am J Physiol Cell Physiol 300:C755–C770
Filippi BM, de Los HP, Mehellou Y, Navratilova I, Gourlay R, Deak M, Plater L, Toth R, Zeqiraj E, Alessi DR (2011) MO25 is a master regulator of SPAK/OSR1 and MST3/MST4/YSK1 protein kinases. EMBO J 30:1730–1741
Fowler L, Everitt J, Stevens JL, Jaken S (1998) Redistribution and enhanced protein kinase C-mediated phosphorylation of alpha- and gamma-adducin during renal tumor progression. Cell Growth Differ 9:405–413
Gagnon KB, England R, Delpire E (2006) Volume sensitivity of cation-Cl− cotransporters is modulated by the interaction of two kinases: Ste20-related proline–alanine-rich kinase and WNK4. Am J Physiol Cell Physiol 290:C134–C142
Gamba G, Saltzberg SN, Lombardi M, Miyanoshita A, Lytton J, Hediger MA, Brenner BM, Hebert SC (1993) Primary structure and functional expression of a cDNA encoding the thiazide-sensitive, electroneutral sodium-chloride cotransporter. Proc Natl Acad Sci U S A 90:2749–2753
Gardner K, Bennett V (1987) Modulation of spectrin-actin assembly by erythrocyte adducin. Nature 328:359–362
Gimenez I, Forbush B (2003) Short-term stimulation of the renal Na-K-Cl cotransporter (NKCC2) by vasopressin involves phosphorylation and membrane translocation of the protein. J Biol Chem 278:26946–26951
Gimenez I, Forbush B (2005) Regulatory phosphorylation sites in the NH2 terminus of the renal Na-K-Cl cotransporter (NKCC2). Am J Physiol Renal Physiol 289:F1341–F1345
Gitelman HJ, Graham JB, Welt LG (1966) A new familial disorder characterized by hypokalemia and hypomagnesemia. Trans Assoc Am Physicians 79:221–235
Glover M, Zuber AM, O'Shaughnessy KM (2009) Renal and brain isoforms of WNK3 have opposite effects on NCCT expression. J Am Soc Nephrol 20:1314–1322
Glover M, Mercier Zuber A, Figg N, O'Shaughnessy KM (2010) The activity of the thiazide-sensitive Na(+)-Cl(-) cotransporter is regulated by protein phosphatase PP4. Can J Physiol Pharmacol 88:986–995
Gonzales PA, Pisitkun T, Hoffert JD, Tchapyjnikov D, Star RA, Kleta R, Wang NS, Knepper MA (2009) Large-scale proteomics and phosphoproteomics of urinary exosomes. J Am Soc Nephrol 20:363–379
Gordon RD (1986) The syndrome of hypertension and hyperkalemia with normal glomerular filtration rate: Gordon’s syndrome. Aust N Z J Med 16:183–184
Hadchouel J, Soukaseum C, Busst C, Zhou XO, Baudrie V, Zurrer T, Cambillau M, Elghozi JL, Lifton RP, Loffing J, Jeunemaitre X (2010) Decreased ENaC expression compensates the increased NCC activity following inactivation of the kidney-specific isoform of WNK1 and prevents hypertension. Proc Natl Acad Sci U S A 107:18109–18114
Heise CJ, Xu BE, Deaton SL, Cha SK, Cheng CJ, Earnest S, Sengupta S, Juang YC, Stippec S, Xu Y, Zhao Y, Huang CL, Cobb MH (2010) Serum and glucocorticoid-induced kinase (SGK) 1 and the epithelial sodium channel are regulated by multiple with no lysine (WNK) family members. J Biol Chem 285:25161–25167
Lalioti MD, Zhang J, Volkman HM, Kahle KT, Hoffmann KE, Toka HR, Nelson-Williams C, Ellison DH, Flavell R, Booth CJ, Lu Y, Geller DS, Lifton RP (2006) Wnk4 controls blood pressure and potassium homeostasis via regulation of mass and activity of the distal convoluted tubule. Nat Genet 38:1124–1132
Lang F, Bohmer C, Palmada M, Seebohm G, Strutz-Seebohm N, Vallon V (2006) (Patho)physiological significance of the serum- and glucocorticoid-inducible kinase isoforms. Physiol Rev 86:1151–1178
Lin SH, Shiang JC, Huang CC, Yang SS, Hsu YJ, Cheng CJ (2005) Phenotype and genotype analysis in Chinese patients with Gitelman’s syndrome. J Clin Endocrinol Metab 90:2500–2507
Moriguchi T, Urushiyama S, Hisamoto N, Iemura S, Uchida S, Natsume T, Matsumoto K, Shibuya H (2005) WNK1 regulates phosphorylation of cation-chloride-coupled cotransporters via the STE20-related kinases, SPAK and OSR1. J Biol Chem 280:42685–42693
Mu S, Shimosawa T, Ogura S, Wang H, Uetake Y, Kawakami-Mori F, Marumo T, Yatomi Y, Geller DS, Tanaka H, Fujita T (2011) Epigenetic modulation of the renal beta-adrenergic-WNK4 pathway in salt-sensitive hypertension. Nat Med 17:573–580
Naito S, Ohta A, Sohara E, Ohta E, Rai T, Sasaki S, Uchida S (2010) Regulation of WNK1 kinase by extracellular potassium. Clin Exp Nephrol 15:195–202
Ohta A, Rai T, Yui N, Chiga M, Yang SS, Lin SH, Sohara E, Sasaki S, Uchida S (2009) Targeted disruption of the Wnk4 gene decreases phosphorylation of Na-Cl cotransporter, increases Na excretion and lowers blood pressure. Hum Mol Genet 18:3978–3986
Pacheco-Alvarez D, Cristobal PS, Meade P, Moreno E, Vazquez N, Munoz E, Diaz A, Juarez ME, Gimenez I, Gamba G (2006) The Na+:Cl− cotransporter is activated and phosphorylated at the amino-terminal domain upon intracellular chloride depletion. J Biol Chem 281:28755–28763
Pedersen NB, Hofmeister MV, Rosenbaek LL, Nielsen J, Fenton RA (2010) Vasopressin induces phosphorylation of the thiazide-sensitive sodium chloride cotransporter in the distal convoluted tubule. Kidney Int 78:160–169
Rafiqi FH, Zuber AM, Glover M, Richardson C, Fleming S, Jovanovic S, Jovanovic A, O'Shaughnessy KM, Alessi DR (2010) Role of the WNK-activated SPAK kinase in regulating blood pressure. EMBO Mol Med 2:63–75
Richardson C, Rafiqi FH, Karlsson HK, Moleleki N, Vandewalle A, Campbell DG, Morrice NA, Alessi DR (2008) Activation of the thiazide-sensitive Na+-Cl− cotransporter by the WNK-regulated kinases SPAK and OSR1. J Cell Sci 121:675–684
Rinehart J, Kahle KT, de Los HP, Vazquez N, Meade P, Wilson FH, Hebert SC, Gimenez I, Gamba G, Lifton RP (2005) WNK3 kinase is a positive regulator of NKCC2 and NCC, renal cation-Cl− cotransporters required for normal blood pressure homeostasis. Proc Natl Acad Sci U S A 102:16777–16782
Rozansky DJ, Cornwall T, Subramanya AR, Rogers S, Yang YF, David LL, Zhu X, Yang CL, Ellison DH (2009) Aldosterone mediates activation of the thiazide-sensitive Na-Cl cotransporter through an SGK1 and WNK4 signaling pathway. J Clin Invest 119:2601–2612
San-Cristobal P, Pacheco-Alvarez D, Richardson C, Ring AM, Vazquez N, Rafiqi FH, Chari D, Kahle KT, Leng Q, Bobadilla NA, Hebert SC, Alessi DR, Lifton RP, Gamba G (2009) Angiotensin II signaling increases activity of the renal Na-Cl cotransporter through a WNK4-SPAK-dependent pathway. Proc Natl Acad Sci U S A 106:4384–4389
Sandberg MB, Riquier AD, Pihakaski-Maunsbach K, McDonough AA, Maunsbach AB (2007) ANG II provokes acute trafficking of distal tubule Na+-Cl(−) cotransporter to apical membrane. Am J Physiol Renal Physiol 293:F662–F669
Schultheis PJ, Lorenz JN, Meneton P, Nieman ML, Riddle TM, Flagella M, Duffy JJ, Doetschman T, Miller ML, Shull GE (1998) Phenotype resembling Gitelman’s syndrome in mice lacking the apical Na+-Cl− cotransporter of the distal convoluted tubule. J Biol Chem 273:29150–29155
Simon DB, Nelson-Williams C, Bia MJ, Ellison D, Karet FE, Molina AM, Vaara I, Iwata F, Cushner HM, Koolen M, Gainza FJ, Gitleman HJ, Lifton RP (1996) Gitelman’s variant of Bartter’s syndrome, inherited hypokalaemic alkalosis, is caused by mutations in the thiazide-sensitive Na-Cl cotransporter. Nat Genet 12:24–30
Subramanya AR, Liu J, Ellison DH, Wade JB, Welling PA (2009) WNK4 diverts the thiazide-sensitive NaCl cotransporter to the lysosome and stimulates AP-3 interaction. J Biol Chem 284:18471–18480
Vallon V, Schroth J, Lang F, Kuhl D, Uchida S (2009) Expression and phosphorylation of the Na+-Cl− cotransporter NCC in vivo is regulated by dietary salt, potassium, and SGK1. Am J Physiol Renal Physiol 297:F704–F712
Verissimo F, Jordan P (2001) WNK kinases, a novel protein kinase subfamily in multi-cellular organisms. Oncogene 20:5562–5569
Vitari AC, Deak M, Morrice NA, Alessi DR (2005) The WNK1 and WNK4 protein kinases that are mutated in Gordon’s hypertension syndrome phosphorylate and activate SPAK and OSR1 protein kinases. Biochem J 391:17–24
Wilson FH, Disse-Nicodeme S, Choate KA, Ishikawa K, Nelson-Williams C, Desitter I, Gunel M, Milford DV, Lipkin GW, Achard JM, Feely MP, Dussol B, Berland Y, Unwin RJ, Mayan H, Simon DB, Farfel Z, Jeunemaitre X, Lifton RP (2001) Human hypertension caused by mutations in WNK kinases. Science 293:1107–1112
Wilson FH, Kahle KT, Sabath E, Lalioti MD, Rapson AK, Hoover RS, Hebert SC, Gamba G, Lifton RP (2003) Molecular pathogenesis of inherited hypertension with hyperkalemia: the Na-Cl cotransporter is inhibited by wild-type but not mutant WNK4. Proc Natl Acad Sci USA 100:680–684
Xu B, English JM, Wilsbacher JL, Stippec S, Goldsmith EJ, Cobb MH (2000) WNK1, a novel mammalian serine/threonine protein kinase lacking the catalytic lysine in subdomain II. J Biol Chem 275:16795–16801
Xu BE, Stippec S, Chu PY, Lazrak A, Li XJ, Lee BH, English JM, Ortega B, Huang CL, Cobb MH (2005) WNK1 activates SGK1 to regulate the epithelial sodium channel. Proc Natl Acad Sci U S A 102:10315–10320
Xu BE, Stippec S, Lazrak A, Huang CL, Cobb MH (2005) WNK1 activates SGK1 by a phosphatidylinositol 3-kinase-dependent and non-catalytic mechanism. J Biol Chem 280:34218–34223
Yang CL, Angell J, Mitchell R, Ellison DH (2003) WNK kinases regulate thiazide-sensitive Na-Cl cotransport. J Clin Invest 111:1039–1045
Yang SS, Morimoto T, Rai T, Chiga M, Sohara E, Ohno M, Uchida K, Lin SH, Moriguchi T, Shibuya H, Kondo Y, Sasaki S, Uchida S (2007) Molecular pathogenesis of pseudohypoaldosteronism type II: generation and analysis of a Wnk4(D561A/+) knockin mouse model. Cell Metab 5:331–344
Yang CL, Zhu X, Ellison DH (2007) The thiazide-sensitive Na-Cl cotransporter is regulated by a WNK kinase signaling complex. J Clin Invest 117:3403–3411
Yang SS, Lo YF, Wu CC, Lin SW, Yeh CJ, Chu P, Sytwu HK, Uchida S, Sasaki S, Lin SH (2010) SPAK-knockout mice manifest Gitelman syndrome and impaired vasoconstriction. J Am Soc Nephrol 21:1868–1877
Zagato L, Modica R, Florio M, Torielli L, Bihoreau MT, Bianchi G, Tripodi G (2000) Genetic mapping of blood pressure quantitative trait loci in Milan hypertensive rats. Hypertension 36:734–739
Zambrowicz BP, Abuin A, Ramirez-Solis R, Richter LJ, Piggott J, BeltrandelRio H, Buxton EC, Edwards J, Finch RA, Friddle CJ, Gupta A, Hansen G, Hu Y, Huang W, Jaing C, Key BW Jr, Kipp P, Kohlhauff B, Ma ZQ, Markesich D, Payne R, Potter DG, Qian N, Shaw J, Schrick J, Shi ZZ, Sparks MJ, Van Sligtenhorst I, Vogel P, Walke W, Xu N, Zhu Q, Person C, Sands AT (2003) Wnk1 kinase deficiency lowers blood pressure in mice: a gene-trap screen to identify potential targets for therapeutic intervention. Proc Natl Acad Sci U S A 100:14109–14114
Zhou B, Zhuang J, Gu D, Wang H, Cebotaru L, Guggino WB, Cai H (2009) WNK4 enhances the degradation of NCC through a sortilin-mediated lysosomal pathway. J Am Soc Nephrol 21:82–92
Acknowledgments
Dr. R. Todd Alexander and Dr. Catherine Morgan are thanked for critical reading of the manuscript. Professor René J. Bindels, Professor Joost G. Hoenderop, and Dr. Pedro San-Cristobal are thanked for the helpful discussions on the topic. This work was supported by the Danish Medical Research Council and an Alberta Innovates Health Solutions Award.
Conflicts of interest
The author has declared that no conflict of interest exists.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Dimke, H. Exploring the intricate regulatory network controlling the thiazide-sensitive NaCl cotransporter (NCC). Pflugers Arch - Eur J Physiol 462, 767–777 (2011). https://doi.org/10.1007/s00424-011-1027-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-011-1027-1