
Abstract
The distal convoluted tubule (DCT) is the shortest segment of the nephron and consists of an early (DCT1) and late part (DCT2). Here, several transport proteins, like the thiazide-sensitive NaCl cotransporter (NCC) and the epithelial magnesium (Mg2+) channel (TRPM6), are exclusively expressed. This makes the DCT the major site of active transcellular Mg2+ reabsorption determining the final excretion in the urine. Following the Mg2+ influx via the apically localized TRPM6, intracellular Mg2+ diffuses to the basolateral membrane where it is extruded to the blood compartment via still-unidentified Mg2+ transporters. Recent years have witnessed multiple breakthroughs in the field of transcellular Mg2+ reabsorption. Epidermal growth factor and estrogen were identified as magnesiotropic hormones by their effect on TRPM6 activity. Intracellularly, receptor of activated protein kinase C 1 and adenosine triphosphate were shown to inhibit TRPM6 activity through its α-kinase domain. Furthermore, dysregulation or malfunction of transcellular Mg2+ reabsorption in DCT has been associated with renal Mg2+ wasting. Mutations in TRPM6 are responsible for hypomagnesemia with secondary hypocalcemia. A defect in the γ-subunit of the Na+/K+-adenosine triphosphatase causes isolated dominant hypomagnesemia resulting from renal Mg2+ wasting. Moreover, in Gitelman’s syndrome, mutations in NCC also result in impaired transcellular Mg2+ reabsorption in DCT. This review highlights our recently obtained knowledge concerning the molecular regulation of transcellular Mg2+ reabsorption.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The distal convoluted tubule
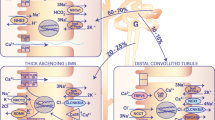
The renal distal tubule comprises anatomically discrete segments (Fig. 1), including the thick ascending limb of Henle (TAL) and the distal convoluted tubule (DCT), and ends in the connecting tubule (CNT) [65]. In rodents and humans, but not in rabbit, the segment of DCT can be further subdivided into an early (DCT1) and late (DCT2) portion [29–31]. The DCT plays an important role in fine-tuning renal excretion of sodium (Na+), calcium (Ca2+), and magnesium (Mg2+). Consequently, several transport proteins for these cations are specifically present in this nephron segment and are, therefore, frequently used as markers for DCT cells (Table 1). First of all, a thiazide-sensitive NaCl cotransporter (NCC) is a well-recognized marker for DCT [40]. NCC is the major apical Na+ transporter in this nephron segment. Its expression begins abruptly at the junction between TAL and DCT and ends at the junction between DCT and CNT [3]. Moreover, along the DCT, the abundance of NCC protein is high in the DCT1 cells and gradually decreases in the DCT2 cells [30, 31, 66]. Another apical transporter, the amiloride-sensitive epithelial Na+ channel (ENaC), is also present in DCT2 but to a much lesser extent. ENaC is predominantly expressed in CNT and cortical collecting duct (CCD) [31]. On the other hand, the Na+/K+-adenosine triphosphatase (ATPase) is consistently present at the basolateral membrane of the DCT. In addition, DCT1 is the major site of transcellular Mg2+ reabsorption. This notion is supported by the fact that the epithelial Mg2+ channel (TRPM6) is predominantly expressed in DCT1 with a lower abundance in DCT2 [66]. Furthermore, TRPM6 co-localizes with parvalbumin in DCT1 and with calbindin-D28K (CaBP28) in DCT2 [66]. Both parvalbumin and CaBP28 are cytoplasmic binding proteins for Ca2+ and Mg2+ with distinct distribution patterns. Parvalbumin is strongly expressed in DCT1 and only weakly in TAL and DCT2 [30, 31, 66]. Given that parvalbumin co-localizes with TRPM6 in DCT1, it could imply that parvalbumin may function as a Mg2+-binding protein. Furthermore, the abundance of CaBP28 is low in DCT1 but high in DCT2, suggesting a minor role for CaBP28 in Mg2+ reabsorption in DCT. Furthermore, Ca2+ transport proteins are confined to the DCT2 and CNT. Here, the epithelial Ca2+ channel TRPV5 is detected, and its expression pattern changes from prominent apical location in DCT2 to a progressively weaker apical to cytoplasmic location in CNT [30]. This suggests that DCT2 is the major site for transcellular Ca2+ reabsorption. CaBP28 primarily co-localizes with TRPV5 in DCT2, suggesting a critical role in transcellular Ca2+ reabsorption. On the basolateral side of DCT and CNT, the Na+/Ca2+ exchanger (NCX) and Ca2+-ATPase (PMCA1b) can be found. Both proteins have their highest abundance in DCT2 and CNT and a weaker expression in DCT1 [30, 31]. Thus, DCT contains all the ion transporters to facilitate the transcellular movement of cations, where DCT1 is critical in fine-tuning Mg2+ and DCT2 for Ca2+ handling. So far, the process of transcellular Ca2+ reabsorption has been investigated in great detail [19, 64]. In contrast, research on the molecular mechanism of transcellular Mg2+ reabsorption in DCT is still at an early stage. This could in part be due to the lack of specific blockers of Mg2+ reabsorption or an easy and reliable way to measure Mg2+ concentrations with fluorescent dyes or isotopes. Thus far, research on the molecular defect underlying inherited forms of hypomagnesemia has been instrumental in the elucidation of transport proteins important in transcellular Mg2+ reabsorption. This review highlights the recent breakthroughs in this field of research.

Model of transcellular Mg2+ reabsorption. The renal distal tubule in the nephron comprises anatomically discrete segments, including the thick ascending limb of the loop of Henle (TAL) and the distal convoluted tubule (DCT) that ends in the connecting tubule (CNT). DCT plays an important role in fine-tuning renal excretion of Mg2+. The epithelial Mg2+ channel (TRPM6) is predominantly expressed in DCT1 with a lower abundance in DCT2. TRPM6 co-localizes with NCC and parvalbumin in DCT1 and with calbindin-D28K in DCT2. Mg2+ influx via TRPM6 is controlled by the luminal and intracellular free Mg2+. Another intracellular TRPM6-associated protein is RACK1, which can inhibit TRPM6 activity by binding to its α-kinase domain. After Mg2+ influx via TRPM6, intracellular Mg2+ may be buffered and transported by a putative Mg2+-binding protein (MgBP). The basolateral membrane harbors epidermal growth factor receptor (EGFR), Na+/K+-ATPase, and possibly a Na+/Mg2+ exchanger and Mg2+-ATPase. The molecular identities of Na+/Mg2+ exchanger and Mg2+-ATPase are still elusive. EGFR activation by EGF can enhance Mg2+ influx by increasing TRPM6 membrane expression. FXYD2 may bind as γ-subunit with α- and β-subunit of Na+/K+-ATPase. A detailed mechanism by which FXYD2 regulates transcellular Mg2+ reabsorption is still under investigation
DCT is important in transcellular Mg2+ reabsorption in the kidney
In the nephron, the majority (∼85%) of filtered Mg2+ is reabsorbed in the proximal tubule and TAL via passive transport through the tight junctions. DCT reabsorbs ∼10% of the filtered Mg2+, which is 70–80% of that delivered from the loop of Henle (Fig. 1) [12]. Using micropuncture and microperfusion studies of the superficial nephron, it has been revealed that net Mg2+ reabsorption in DCT is essentially unidirectional since no secretion of Mg2+ into the lumen has been reported [12]. Accordingly, this segment is critical in determining the final urinary excretion. In general, blood Mg2+ levels are maintained between 0.7 and 1.1 mM [46], and intracellular Mg2+ concentration is ∼0.6 mM being buffered by adenosine triphosphate (ATP) [12, 62]. The luminal Mg2+ concentration in DCT, however, is around 0.2–0.7 mM [12], which is lower than the plasma and intracellular Mg2+ concentration. In addition, using immortalized mouse DCT cells, Dai and coworkers have demonstrated that the negative membrane potential directly determines the Mg2+ influx rate. Indeed, depolarization of the membrane diminished Mg2+ uptake in these cells [10]. This data would be in line with the operation of a voltage-driven Mg2+ permeable channel to support the entry of Mg2+ across the apical membrane [11]. Not long ago, this hypothesis was confirmed as being correct. Patients with hypomagnesemia with secondary hypocalcemia (HSH), a primary defect in intestinal and renal Mg2+ absorption, were found to carry mutations in TRPM6 [47, 50, 67]. TRPM6 is the first epithelial Mg2+-permeable channel predominantly present in the luminal membrane of epithelial cells in kidney and intestine [47, 50, 67]. In concert with TRPM6, the apical Na+ transporter NCC is indirectly involved in the transcellular movement of Mg2+ in DCT [38]. The evidence for this notion came from a NCC knockout mouse. Inactivation of the NCC gene resulted in a mouse model resembling Gitelman’s syndrome [31], a human disease characterized by mild renal Na+ wasting, hypocalciuria, hypomagnesemia, and hypokalemic alkalosis. Although it is still unclear how NCC dysfunction precisely leads to hypomagnesemia, their study provides further evidence to support the DCT as the major site for transcellular Mg2+ reabsorption. Following the Mg2+ influx in DCT cells, intracellular Mg2+ will be transported to the basolateral domain for extrusion to the blood compartment. A putative Na+/Mg2+ exchanger and Mg2+-ATPase have been proposed as candidates for the extrusion process [12]. So far, the molecular identities of these basolateral Mg2+ transporters are unknown. Furthermore, the basolateral membrane also harbors the receptors for magnesiotropic hormones, such as epidermal growth factor (EGF) and 17-β-estradiol [14, 15].
Mg2+ influx in DCT through TRPM6
TRPM6 as the gatekeeper of transcellular Mg2+ reabsorption
TRPM6 is the sixth member in the melastatin subfamily of transient receptor potential (TRP) ion channels. TRPM6 consists of 39 exons spanning 167 kb of genomic sequence and coding for a protein of 2,022 amino acids [47, 50, 67]. The TRPM6 protein has six transmembrane-spanning domains, in which the putative pore region is between the fifth and sixth domains. It also has a long amino (N)-terminus and a carboxyl (C)-terminus. TRPM6 has an α-kinase domain in the C-terminus. This kinase domain plays an important role in regulating TRPM6 activity. Further, TRPM6 also displays unique electrophysiological properties. When heterogeneously expressed in HEK293 cells, TRPM6 constitutes a channel that is characterized by extremely small inward but large outward currents [66]. External divalent cations such as Mg2+ and Ca2+ are permeable to TRPM6 and at the same time block monovalent cations permeating through the pore of the channel [66]. TRPM6 displays strong outward rectification, has a 5-fold higher affinity for Mg2+ than for Ca2+, and is blocked in a voltage-dependent manner by ruthenium red [66]. In other Ca2+-selective channels, there is a 10–1,000 lower affinity for Mg2+ than for Ca2+ [18], which further favors the TRPM6 identity as Mg2+-permeable channel. The permeation rank order determined from the inward current amplitude at −80 mV is Ba2+ ≥ Ni2+ > Mg2+ > Ca2+. Single-channel conductance of homomeric TRPM6 channel is 84 ± 2 pS [28]. Amino acid residues E1024, I1030, and D1031 seem important for channel function and account in the pore region for TRPM6 permeation properties [61]. In addition, Li et al. provided evidence that E1029 is also critical in regulating TRPM6 permeability [27]. It is remarkable that neutralization of the single amino acid residue, E1024Q, largely eliminates divalent permeation, converting the divalent selective TRPM6 to a virtually monovalent selective channel, and at the same time abolishes external pH sensitivity [27]. However, the Mg2+ selectivity filter of TRPM6 is still unclear. So far, two candidate regions in TRPM6 have been proposed as Mg2+ selectivity filters, including 1028GEIDVC1033 [61] and 1024EVYAGE1029 [27]. The missense mutations in TRPM6 identified in HSH lead to nonfunctional, mostly truncated, TRPM6 proteins [50, 67]. Collectively, these studies identified TRPM6 as a Mg2+-permeable ion channel.
α-Kinase domain is critical in TRPM6 activity
TRPM6 is a channel kinase with an α-kinase domain fused to its C-terminus. These kinases display little sequence similarity with conventional protein kinases. Although α-kinases can predominantly phosphorylate residues present in α-helices [48], the α-kinase of TRPM6 is able to autophosphorylate the channel at several autophosphorylation sites [8]. Additionally, the autophosphorylation of the Ser/Thr-rich domain in the α-kinase is critical in the subsequent phosphorylation of protein substrates by providing access of the catalytic domain to the substrate [8]. The TRPM6 α-kinase domain is also a regulatory site for TRPM6 channel activity. For instance, deletion of the α-kinase domain reduces the TRPM6 current [59]. Furthermore, we have identified receptor of activated protein kinase C 1 (RACK1) as the first TRPM6-associated protein that inhibits TRPM6 channel activity depending on the phosphorylation of the threonine residue at position 1851 (T1851) in the α-kinase domain. RACK1 is a scaffold protein that binds phosphorylated protein kinase C (PKC) [45], by which RACK1 can bring the activated PKC into contact with its various substrates. Using the glutathione S-transferase (GST)-pull down technology, Cao and colleagues revealed that RACK1 is one of the associated proteins of TRPM6. RACK1 co-localizes with TRPM6 and NCC in DCT cells, suggesting a role for RACK1 in renal Mg2+ handling. Indeed, overexpressed RACK1 reduces TRPM6 currents in an α-kinase activity-dependent manner. Given that T1851 is crucial for the Mg2+-dependent autophosphorylation of TRPM6 [6] and channel activity, these data indicate that RACK1 confers, at least in part, the Mg2+ sensitivity of TRPM6 channel activity. Taken together, RACK1 is an intracellular TRPM6-associated protein that mediates TRMP6 channel activity and thus renal Mg2+ handling through the TRPM6 α-kinase domain.
Besides RACK1, intracellular ATP also regulates TRPM6 activity via its α-kinase domain [59]. In the TRPM6 α-kinase region, there is a conserved ATP-binding pocket [59]. By binding to this ATP-binding pocket, both Na+-ATP and Mg2+-ATP inhibited TRPM6 currents in a dose-dependent manner with a comparable IC50 of ∼1.3 mM [59]. Mutation of the conserved glycine residue in the ATP-binding pocket prevented the inhibitory effect of ATP on TRPM6 channel activity [59]. Interestingly, the α-kinase domain, but not its activity, is essential for ATP-induced inhibition on TRPM6 [59]. This result supports the critical role for the α-kinase domain in TRPM6 regulation. In contrast to ATP, Na+-CTP or Na+-GTP did not affect the TRPM6 current in transfected HEK293 cells [59], indicating the unique role of ATP in regulating the channel. However, it is still unknown to what extent and how ATP can modulate the transcellular Mg2+ reabsorption in DCT.
Regulation of TRPM6 by magnesiotropic hormones
Hormones primarily controlling the overall Mg2+ balance are not generally known (Table 2). Recently, EGF has been identified as a new magnesiotropic hormone regulating specifically the renal handling of Mg2+ [15, 35]. In a Dutch family with isolated recessive renal hypomagnesemia (IRH), Tiel Groenestege and colleagues employed a homozygosity-based mapping strategy and revealed a mutation in the pro-EGF gene where the highly conserved proline in the cytoplasmic 1067PKNP 1070 motif has been substituted by a leucine. This mutation diminishes basolateral release of pro-EGF and seriously hampers the EGF-dependent activation of the basolateral EGF receptor, leading to insufficient activation of TRPM6, less Mg2+ influx, and IRH [15]. Accordingly, it has been proposed that the pro-EGF gene mutation may result in improper basolateral sorting, leading to an inadequate secretion of the hormone into the circulation. Logically, the next question was how EGF activates TRPM6. To answer this question, it was demonstrated that EGF stimulates the mobility of subcellular TRPM6 to increase the plasma membrane expression of TRPM6 [58]. In detail, using the fluorescence recovery after photobleaching technology and cell surface biotinylation experiments, Thébault et al. visualized the EGF-enhanced TRPM6 trafficking from the cytosol to the plasma membrane [58]. This process is mediated via a sarcoma inducing gene (Src) kinase and Ras-related C3 botulinum toxin substrate 1 (Rac1) [58]. In line with this study, Ikari et al. reported that, in renal epithelial cells, EGF upregulates the TRPM6 expression by increasing the phosphorylation of extracellular signal-regulated kinases 1 and 2 (ERK1/2), leading to Mg2+ influx and an increase in cell proliferation [23]. In agreement with this study, the blockade of the EGF receptor (EGFR) induces hypomagnesemia. This is commonly seen in the colorectal cancer patients undertaking treatment with an EGFR-targeting antibody, cetuximab, and can now be explained by the inhibition of TRPM6 through the block of EGFR by cetuximab [15, 57]. Collectively, these data provide an insight into the mechanism of EGFR activation of TRPM6 and reiterate the importance of EGF in the overall Mg2+ homeostasis.
Another magnesiotropic hormone is 17-β-estradiol as evidenced by Tiel Groenestege et al., who showed that 17-β-estradiol upregulates mRNA level of TRPM6 in mouse kidney [14]. In line with this observation, surgical removal of ovary in rats caused a significantly reduction of TRPM6 mRNA level in the kidney, indicating the regulatory role of estrogen in the transcription process of TRPM6. In addition, subsequent treatment with 17-β-estradiol restored the TRPM6 mRNA levels [14], further supporting the candidature of 17-β-estradiol as a magnesiotropic hormone. In fact, this is also in agreement with a previous clinical study, showing that estrogen substitution therapy significantly decreases postmenopausal hypermagnesuria [32, 49]. Therefore, it is likely that 17-β-estradiol regulates transcellular Mg2+ reabsorption via an enhanced renal TRPM6 expression [14].
Besides EGF and 17-β-estradiol, other hormones including calcitonin, glucagon, arginine vasopressin, mineralocorticoid hormones, prostaglandins, and insulin [12] have been indirectly implicated in the process of Mg2+ handling, but their effect on TRPM6 activity is unknown. It has, however, been suggested that parathyroid hormone (PTH) and 1,25-dihydroxyvitamin D3 (1,25(OH)2D3) do not regulate the transcription of TRPM6 in mouse [14]. This is interesting since both hormones have been suggested to increase the Mg2+ reabsorption in DCTs as evidenced from in vivo microperfusion experiments and immortalized mouse DCT cells from Mg2+ uptake experiments [12, 42]. Hence, there must be other pathways for PTH and 1,25(OH)2D3 to enhance transcellular Mg2+ reabsorption. In general, detailed molecular studies are needed to further substantiate the involvement of these latter hormones in transcellular Mg2+ reabsorption.
Other factors that regulate TRPM6
Other conditions can also regulate TRPM6 and thus renal Mg2+ handling, such as metabolic acidosis and alkalosis, immunosuppressive drugs, and thiazide diuretics (Table 2). Nijenhuis et al. reported that renal TRPM6 expression in kidney is decreased by chronic metabolic acidosis but increased by chronic metabolic alkalosis [37]. Furthermore, the immunosuppressive drugs tacrolimus and cyclosporin A both downregulate TRPM6 [22, 36]. Similarly, chronic application of thiazide diuretics, mimicking Gitelman’s syndrome, has been shown to reduce TRPM6 expression [31, 38]. Collectively, these data support the key role of TRPM6 in transcellular Mg2+ reabsorption and imply TRPM6 as a potential target for pharmacological treatment of various Mg2+-deficient disorders.
TRPM6 as distinct Mg2+ channel
TRPM6 and TRPM7 have an overall amino acid sequence identity of 52%, which increases to more then 80% in the region between the fifth and sixth transmembrane domains where the pore-forming loop is presumably located [66]. Moreover, both TRPM6 and TRPM7 have a C-terminal region with sequence similarities to the atypical α-kinase family. It is likely that TRPM6 and TRPM7 form distinct cation channels, but there is also experimental evidence that they can function as a heteromeric complex (TRPM6/7) [7, 28]. Importantly, there are several signature features of TRPM6 that distinguish this channel from homomeric TRPM7 and heteromeric TRPM6/7 channels. TRPM6 single-channel current is larger than TRPM7 or TRPM6/TRPM7 [28]. At micromolar concentrations, 2-aminoethoxydiphenyl borate (2-APB) enhanced TRPM6 but inhibited TRPM7 currents, whereas at millimolar concentrations, 2-APB increased TRPM7 and TRPM6/7 currents [28]. Hence, TRPM6 has different pharmacological properties from TRPM7 and TRPM6/7. Furthermore, the pH sensitivity of TRPM6 is distinctive from those of TRPM7 channels and TRPM6/7 complexes [28].
Mg2+-binding proteins
Several substances present in DCT cells have the potential to bind significant amounts of Mg2+, and thus could affect intracellular free Mg2+ levels. For instance, ATP, CaBP28, and parvalbumin are abundantly present in DCT [17, 66]. Furthermore, intracellular Mg2+ can also be modulated by the unidirectional transport of Mg2+ into subcellular organelles including nucleus, mitochondrion, endoplasmic reticulum, and ribosomes [17]. At present, evidence for a Mg2+-binding protein specifically controlling the process of transcellular Mg2+ reabsorption is lacking. This is in contrast to transcellular Ca2+ transport, where calbindins (CaBP28 in kidney and CaBP9 in intestine) specifically buffer Ca2+ to maintain low intracellular Ca2+ levels during the process of transcellular Ca2+ (re)absorption, which is a prerequisite for the signaling function of Ca2+. Additionally, the apical Ca2+ channel TRPV5 is strongly inhibited by intracellular Ca2+ in the submicromolar range [39]. Hence, without sufficient local Ca2+-buffering, the Ca2+ influx via TRPV5 would not be feasible. In contrast, extracellular and intracellular Mg2+ levels are both in the submillimolar range. Thus, there is virtually no chemical gradient of Mg2+ across the plasma membrane, and this questions the necessity of a specific Mg2+-binding protein. However, Voets and coworkers reported that the activity of TRPM6 is strongly controlled by the local Mg2+ concentrations with an IC50 of 0.5 mM, suggesting that adequate Mg2+ buffering could be important. Indeed, both parvalbumin and CaBP28 partly co-localize with TRPM6 in the DCT and could thus potentially function as local Mg2+ buffers. A recent study using parvalbumin knockout mouse failed to show a disturbance in the Mg2+ homeostasis, which seriously questions the candidateship of parvalbumin [4]. However, this may be due to a compensatory intestinal Mg2+ hyperabsorption, or increased high bone turnover could occur in these knockout mice. Similarly, this compensatory situation also happens in the CaBP28−/− mouse, which does not exhibit hypocalcemia [13]. Alternatively, ATP could, in addition to its direct effect on TRPM6 activity, also function as physiological and local Mg2+ buffer [59]. Taken together, further experimental studies are needed to fully understand the role of Mg2+-binding substances in transcellular Mg2+ reabsorption.
Mg2+ extrusion systems
A Na+/Mg2+ exchanger and a Mg2+ pump, in analogy with Ca2+ extrusion systems, have been postulated as candidates for Mg2+ extrusion system [12]. A putative Na+/Mg2+ exchanger has been studied functionally in many cells including epithelial cells, but its molecular identity remains unknown [16, 26, 52]. Schweigel and colleagues showed that sheep rumen epithelial cells take up Mg2+ when incubated in a Mg2+-containing medium and that this uptake is stimulated when the external medium contains no or small amounts (10 mM) of Na+ [52, 53]. Moreover, this Mg2+ influx was accompanied by a decrease in the intracellular Na+ concentration and reduced by quinidine and imipramine, which are both putative inhibitors of Na+/Mg2+ exchange [63]. Collectively, these data suggest the existence of a Na+/Mg2+ exchanger in epithelial cells, but its presence remains to be shown in the DCT. Alternatively, a Mg2+ pump, which molecular identity awaits experimental confirmation, has been proposed [51]. Taken together, further investigation is necessary to explore the molecular details of the Mg2+ extrusion across the basolateral membrane of DCT cells.
Diseases related to Mg2+ handling in DCT
Abnormal Mg2+ handling in the DCT, which is the major site of transcellular Mg2+ reabsorption, can cause sever hypomagnesemia [60], including HSH [50], isolated dominant hypomagnesemia (IDH) [5, 34], and Gitelman’s syndrome [31, 38].
TRPM6 mutation leads to HSH
The HSH is an autosomal recessive disease characterized by hypomagnesemia and hypocalcemia. The latter is possibly secondary to parathyroid failure resulting from Mg2+ deficiency due to Mg2+ mal(re)absorption [1]. Patients show neurologic symptoms of hypomagnesemia during infancy, including seizures and muscle spasms. Oral administration of high doses of Mg2+ can relieve clinical symptoms. Without treatment, it may be fatal or may result in neurological damage. In 2001, two different groups simultaneously discovered that HSH is caused by mutations in TRPM6 [50, 67]. This serendipitous discovery demonstrates that TRPM6 is the epithelial Mg2+ channel primarily controlling intestinal Mg2+ uptake and renal Mg2+ excretion.
FXYD2 mutant induces IDH
The FXYD2 gene encodes a single transmembrane protein that functions as the γ-subunit of Na+/K+-ATPase. A mutation of FXYD2 substituting glycine at position 41 to arginine (FXYD2-G41R) causes IDH, a dominant renal hypomagnesemia associated with hypocalciuria through a dominant negative mechanism [33, 34]. These disorders share the general symptoms of hypomagnesemia, tetany, and epileptiformic convulsions and often include secondary or associated disturbances in Ca2+ excretion. The γ-subunit is a small hydrophobic protein of 10 kDa, originally identified in purified preparations of the Na+/K+-ATPase. FXYD2 protein is highly expressed in the basolateral membrane of TAL and DCT [2, 41]. In kidneys of FXYD2 knockout mouse, Na+/K+-ATPase displayed a higher apparent affinity for Na+ without significant change in apparent affinity for K+ compared to wild-type animals [24]. The consequence of this alteration in affinity for the Mg2+ flux across the DCT is, despite intense research, not yet elucidated. Surprisingly, these knockout mice apparently do not exhibit a disturbance in overall Mg2+ balance as reflected by normal blood and urine Mg2+ values [24], indicating that the presence of wild-type FXYD2 per se is not required for the maintenance of the Mg2+ balance. Since a mutation (G41R) in, unlike a knockout of, FXYD2 causes renal Mg2+ wasting and consequently hypomagnesemia, the mutant protein possibly impairs another protein partner directly regulating the Mg2+ handling in DCT [34]. Now, the exact molecular consequence of FXYD2-G41R remains elusive. Indeed, it may cause malfunctioning of the Na+/K+-ATPase by destabilizing the association between FXYD2 and Na+/K+-ATPase [5] and thus directly depolarize the membrane potential [34]. Alternatively, both wild-type FXYD2 and FXYD2-G41R mutant form homomeric and heteromeric oligomers [5]. In Madin–Darby Canine Kidney (MDCK) cells, wild-type FXYD2 leads to an increase in transepithelial current, and this current is significantly reduced by co-transfection with the FXYD2-G41R mutant [54]. Hence, FXYD2-G41R mutant is dominant over wild-type FXYD2 [54]. In line with this observation, FXYD2-G41R has been shown to reduce the trafficking of wild-type FXYD2 to the plasma membrane [5]. Given that the dominant nature of the FXYD2-G41R mutation may be mediated through its association with wild-type FXYD2 [54], the oligomers of FXYD2-G41R mutant with FXYD2 wild-type seems essential for the occurrence of hypomagnesemia. Hypothetically, this small protein may directly regulate the activity of a relevant ion channel, including TRPM6, as FXYD proteins have in general been considered as regulators of ion channels [9].
NCC mutations cause Gitelman’s syndrome
Gitelman’s syndrome is one of the most frequently inherited renal tubular disorders [25]. It is an autosomal recessive Na+ wasting disease, characterized by hypomagnesemia, hypocalciuria, and secondary aldosteronism [25]. This leads to hypokalemia and metabolic alkalosis. Genetic analysis revealed that this syndrome is caused by mutations in NCC [55], which has been further confirmed by studies with NCC knockout mice and the chronic application of thiazide diuretics [31, 38]. It is still unclear how abolishment of NCC function induces hypomagnesemia. Nijenhuis et al. have shown that TRPM6 mRNA and protein abundance are reduced in a mouse model of Gitelman’s syndrome [38]. Furthermore, the hypomagnesemia in this mouse model is due to renal Mg2+ wasting [38]. Thus, reduced NCC activity seems to regulate transcellular Mg2+ reabsorption in DCT by controlling the expression of TRPM6. In addition, another study has revealed that aldosterone levels were significantly elevated in NCC knockout mice compared with wild-type mice [43]. Hypomagnesemia has been observed in primary aldosteronism [68]. Collectively, these data imply that aldosterone may be involved in hypomagnesemia in Gitelman’s syndrome. However, surprisingly, a recent report shows that aldosterone did not affect the TRPM6 expression in mouse kidney [56]. Thus, the molecular mechanism of TRPM6 downregulation and accompanied hypomagnesemia in Gitelman’s syndrome is still unclear.
Unresolved issues
The identification of TRPM6 as gatekeeper of transcellular Mg2+ reabsorption and of EGF and estrogen as magnesiotropic hormones acting on TRPM6 are recent breakthroughs in unraveling the molecular mechanism of transcellular Mg2+ reabsorption in DCT. Our picture of this process is, however, far from complete, and there are still many important questions unanswered. The activity of TRPM6 has been measured in heterogeneous expression systems like HEK293 cells. Here, the inward Mg2+ current is surprisingly small at physiological membrane potentials, and it is unknown how these small inward currents contribute to the overall transcellular Mg2+ reabsorption [66]. Furthermore, the contribution of organelles to the regulation of intracellular Mg2+ levels is unknown. Given that Mg2+ with an IC50 of 0.5 mM inhibits TRPM6 activity, it is important to delineate the role of subcellular organelles, like nucleus, mitochondria, and endoplasmic reticulum [44], in this process. Additionally, it is still unspecified how epithelial cells sense the extracellular Mg2+ concentration in order to trigger Mg2+ reabsorption. It has been established that the Ca2+-sensing receptor (CaSR) can also monitor extracellular Mg2+ concentration [20]. Additionally, CaSR activation may decrease protein kinase A activity, resulting in a decrease in phosphorylated claudin-16, the translocation of claudin-16 to lysosome, and a decrease in paracellular Mg2+ reabsorption [21]. Recently, our group discovered that CaSR is expressed in basolateral domain in the DCT cells (unpublished data) and regulates TRPV5 channel activity. Therefore, it would be interesting to test if this CaSR can sense extracellular Mg2+ concentration and modulate transcellular Mg2+ reabsorption. Finally, the relation between Ca2+ and Mg2+ reabsorption in the DCT needs further investigation. Hypomagnesemia is frequently accompanied by inappropriate renal Ca2+ wasting, but the molecular explanation of this phenomenon remains obscure. A multidisciplinary approach to explore disturbances in Mg2+ homeostasis will be necessary to further increase our understanding of transcellular Mg2+ reabsorption and ultimately develop new possibilities to treat Mg2+-related syndromes.
References
Anast CS, Mohs JM, Kaplan SL, Burns TW (1972) Evidence for parathyroid failure in magnesium deficiency. Science 177:606–608
Arystarkhova E, Wetzel RK, Asinovski NK, Sweadner KJ (1999) The gamma subunit modulates Na+ and K+ affinity of the renal Na,K-ATPase. J Biol Chem 274:33183–33185
Bachmann S, Velazquez H, Obermuller N, Reilly RF, Moser D, Ellison DH (1995) Expression of the thiazide-sensitive Na-Cl cotransporter by rabbit distal convoluted tubule cells. J Clin Invest 96:2510–2514
Belge H, Gailly P, Schwaller B, Loffing J, Debaix H, Riveira-Munoz E, Beauwens R, Devogelaer JP, Hoenderop JG, Bindels RJ, Devuyst O (2007) Renal expression of parvalbumin is critical for NaCl handling and response to diuretics. Proc Natl Acad Sci U S A 104:14849–14854
Cairo ER, Friedrich T, Swarts HG, Knoers NV, Bindels RJ, Monnens LA, Willems PH, De Pont JJ, Koenderink JB (2008) Impaired routing of wild type FXYD2 after oligomerisation with FXYD2-G41R might explain the dominant nature of renal hypomagnesemia. Biochim Biophys Acta 1778:398–404
Cao G, Thebault S, van der Wijst J, van der Kemp A, Lasonder E, Bindels RJ, Hoenderop JG (2008) RACK1 inhibits TRPM6 activity via phosphorylation of the fused α-kinase domain. Curr Biol 18:168–176
Chubanov V, Waldegger S, Mederos y Schnitzler M, Vitzthum H, Sassen MC, Seyberth HW, Konrad M, Gudermann T (2004) Disruption of TRPM6/TRPM7 complex formation by a mutation in the TRPM6 gene causes hypomagnesemia with secondary hypocalcemia. Proc Natl Acad Sci U S A 101:2894–2899
Clark K, Middelbeek J, Morrice NA, Figdor CG, Lasonder E, van Leeuwen FN (2008) Massive autophosphorylation of the Ser/Thr-rich domain controls protein kinase activity of TRPM6 and TRPM7. PLoS ONE 3:e1876
Crambert G, Geering K (2003) FXYD proteins: new tissue-specific regulators of the ubiquitous Na,K-ATPase. Sci STKE 2003:RE1
Dai LJ, Friedman PA, Quamme GA (1997) Cellular mechanisms of chlorothiazide and cellular potassium depletion on Mg2+ uptake in mouse distal convoluted tubule cells. Kidney Int 51:1008–1017
Dai LJ, Raymond L, Friedman PA, Quamme GA (1997) Mechanisms of amiloride stimulation of Mg2+ uptake in immortalized mouse distal convoluted tubule cells. Am J Physiol 272:F249–256
Dai LJ, Ritchie G, Kerstan D, Kang HS, Cole DE, Quamme GA (2001) Magnesium transport in the renal distal convoluted tubule. Physiol Rev 81:51–84
Gkika D, Hsu YJ, van der Kemp AW, Christakos S, Bindels RJ, Hoenderop JG (2006) Critical role of the epithelial Ca2+ channel TRPV5 in active Ca2+ reabsorption as revealed by TRPV5/calbindin-D28K knockout mice. J Am Soc Nephrol 17:3020–3027
Groenestege WM, Hoenderop JG, van den Heuvel L, Knoers N, Bindels RJ (2006) The epithelial Mg2+ channel transient receptor potential melastatin 6 is regulated by dietary Mg2+ content and estrogens. J Am Soc Nephrol 17:1035–1043
Groenestege WM, Thebault S, van der Wijst J, van den Berg D, Janssen R, Tejpar S, van den Heuvel LP, van Cutsem E, Hoenderop JG, Knoers NV, Bindels RJ (2007) Impaired basolateral sorting of pro-EGF causes isolated recessive renal hypomagnesemia. J Clin Invest 117:2260–2267
Gunther T (2007) Na+/Mg2+ antiport in non-erythrocyte vertebrate cells. Magnes Res 20:89–99
Gunther T (2007) Total and free Mg2+ contents in erythrocytes: a simple but still undisclosed cell model. Magnes Res 20:161–167
Hess P, Tsien RW (1984) Mechanism of ion permeation through calcium channels. Nature 309:453–456
Hoenderop JG, Nilius B, Bindels RJ (2005) Calcium absorption across epithelia. Physiol Rev 85:373–422
Ikari A, Nakajima K, Kawano K, Suketa Y (2001) Polyvalent cation-sensing mechanism increased Na+-independent Mg2+ transport in renal epithelial cells. Biochem Biophys Res Commun 287:671–674
Ikari A, Okude C, Sawada H, Sasaki Y, Yamazaki Y, Sugatani J, Degawa M, Miwa M (2008) Activation of a polyvalent cation-sensing receptor decreases magnesium transport via claudin-16. Biochim Biophys Acta 1778:283–290
Ikari A, Okude C, Sawada H, Takahashi T, Sugatani J, Miwa M (2007) Down-regulation of TRPM6-mediated magnesium influx by cyclosporin A. Naunyn Schmiedebergs Arch Pharmacol 377(4–6):333–343
Ikari A, Okude C, Sawada H, Yamazaki Y, Sugatani J, Miwa M (2008) TRPM6 expression and cell proliferation are up-regulated by phosphorylation of ERK1/2 in renal epithelial cells. Biochem Biophys Res Commun 369:1129–1133
Jones DH, Li TY, Arystarkhova E, Barr KJ, Wetzel RK, Peng J, Markham K, Sweadner KJ, Fong GH, Kidder GM (2005) Na,K-ATPase from mice lacking the gamma subunit (FXYD2) exhibits altered Na+ affinity and decreased thermal stability. J Biol Chem 280:19003–19011
Knoers NV, Levtchenko EN (2008) Gitelman syndrome. Orphanet J Rare Dis 3:22
LaBelle EF (1984) Reconstituted amiloride-inhibited sodium transporter from rabbit kidney medulla is responsible for Na+-H+ exchange. Biochim Biophys Acta 770:79–92
Li M, Du J, Jiang J, Ratzan W, Su LT, Runnels LW, Yue L (2007) Molecular determinants of Mg2+ and Ca2+ permeability and pH sensitivity in TRPM6 and TRPM7. J Biol Chem 282:25817–25830
Li M, Jiang J, Yue L (2006) Functional characterization of homo- and heteromeric channel kinases TRPM6 and TRPM7. J Gen physiol 127:525–537
Loffing J, Loffing-Cueni D, Macher A, Hebert SC, Olson B, Knepper MA, Rossier BC, Kaissling B (2000) Localization of epithelial sodium channel and aquaporin-2 in rabbit kidney cortex. Am J Physiol 278:F530–539
Loffing J, Loffing-Cueni D, Valderrabano V, Klausli L, Hebert SC, Rossier BC, Hoenderop JG, Bindels RJ, Kaissling B (2001) Distribution of transcellular calcium and sodium transport pathways along mouse distal nephron. Am J Physiol 281:F1021–1027
Loffing J, Vallon V, Loffing-Cueni D, Aregger F, Richter K, Pietri L, Bloch-Faure M, Hoenderop JG, Shull GE, Meneton P, Kaissling B (2004) Altered renal distal tubule structure and renal Na+ and Ca2+ handling in a mouse model for Gitelman’s syndrome. J Am Soc Nephrol 15:2276–2288
McNair P, Christiansen C, Transbol I (1984) Effect of menopause and estrogen substitutional therapy on magnesium metabolism. Miner Electrolyte Metab 10:84–87
Meij IC, Koenderink JB, De Jong JC, De Pont JJ, Monnens LA, Van Den Heuvel LP, Knoers NV (2003) Dominant isolated renal magnesium loss is caused by misrouting of the Na+,K+-ATPase gamma-subunit. Ann N Y Acad Sci 986:437–443
Meij IC, Koenderink JB, van Bokhoven H, Assink KF, Groenestege WT, de Pont JJ, Bindels RJ, Monnens LA, van den Heuvel LP, Knoers NV (2000) Dominant isolated renal magnesium loss is caused by misrouting of the Na+,K+-ATPase gamma-subunit. Nat Genet 26:265–266
Muallem S, Moe OW (2007) When EGF is offside, magnesium is wasted. J Clin Invest 117:2086–2089
Nijenhuis T, Hoenderop JG, Bindels RJ (2004) Downregulation of Ca2+ and Mg2+ transport proteins in the kidney explains tacrolimus (FK506)-induced hypercalciuria and hypomagnesemia. J Am Soc Nephrol 15:549–557
Nijenhuis T, Renkema KY, Hoenderop JG, Bindels RJ (2006) Acid-base status determines the renal expression of Ca2+ and Mg2+ transport proteins. J Am Soc Nephrol 17:617–626
Nijenhuis T, Vallon V, van der Kemp AW, Loffing J, Hoenderop JG, Bindels RJ (2005) Enhanced passive Ca2+ reabsorption and reduced Mg2+ channel abundance explains thiazide-induced hypocalciuria and hypomagnesemia. J Clin Invest 115:1651–1658
Nilius B, Prenen J, Vennekens R, Hoenderop JG, Bindels RJ, Droogmans G (2001) Modulation of the epithelial calcium channel, ECaC, by intracellular Ca2+. Cell Calcium 29:417–428
Obermuller N, Bernstein P, Velazquez H, Reilly R, Moser D, Ellison DH, Bachmann S (1995) Expression of the thiazide-sensitive Na-Cl cotransporter in rat and human kidney. Am J Physiol 269:F900–910
Pu HX, Cluzeaud F, Goldshleger R, Karlish SJ, Farman N, Blostein R (2001) Functional role and immunocytochemical localization of the gamma a and gamma b forms of the Na,K-ATPase gamma subunit. J Biol Chem 276:20370–20378
Quamme GA (1997) Renal magnesium handling: new insights in understanding old problems. Kidney Int 52:1180–1195
Riveira-Munoz E, Chang Q, Godefroid N, Hoenderop JG, Bindels RJ, Dahan K, Devuyst O (2007) Transcriptional and functional analyses of SLC12A3 mutations: new clues for the pathogenesis of Gitelman syndrome. J Am Soc Nephrol 18:1271–1283
Romani A (2007) Regulation of magnesium homeostasis and transport in mammalian cells. Arch Biochem Biophys 458:90–102
Ron D, Chen CH, Caldwell J, Jamieson L, Orr E, Mochly-Rosen D (1994) Cloning of an intracellular receptor for protein kinase C: a homolog of the beta subunit of G proteins. Proc Natl Acad Sci U S A 91:839–843
Rubin H (2007) The logic of the membrane, magnesium, mitosis (MMM) model for the regulation of animal cell proliferation. Arch Biochem Biophys 458:16–23
Ryazanov AG (2002) Elongation factor-2 kinase and its newly discovered relatives. FEBS Lett 514:26–29
Ryazanov AG, Pavur KS, Dorovkov MV (1999) Alpha-kinases: a new class of protein kinases with a novel catalytic domain. Curr Biol 9:R43–45
Schlemmer A, Podenphant J, Riis BJ, Christiansen C (1991) Urinary magnesium in early postmenopausal women. Influence of hormone therapy on calcium. Magnes Trace Elem 10:34–39
Schlingmann KP, Weber S, Peters M, Niemann Nejsum L, Vitzthum H, Klingel K, Kratz M, Haddad E, Ristoff E, Dinour D, Syrrou M, Nielsen S, Sassen M, Waldegger S, Seyberth HW, Konrad M (2002) Hypomagnesemia with secondary hypocalcemia is caused by mutations in TRPM6, a new member of the TRPM gene family. Nat Genet 31:166–170
Schweigel M, Martens H (2000) Magnesium transport in the gastrointestinal tract. Front Biosci 5:D666–677
Schweigel M, Park HS, Etschmann B, Martens H (2006) Characterization of the Na+-dependent Mg2+ transport in sheep ruminal epithelial cells. Am J Physiol 290:G56–65
Schweigel M, Vormann J, Martens H (2000) Mechanisms of Mg2+ transport in cultured ruminal epithelial cells. Am J Physiol 278:G400–408
Sha Q, Pearson W, Burcea LC, Wigfall DA, Schlesinger PH, Nichols CG, Mercer RW (2008) Human FXYD2 G41R mutation responsible for renal hypomagnesemia behaves as an inward-rectifying cation channel. Am J Physiol 295:F91–99
Simon DB, Nelson-Williams C, Bia MJ, Ellison D, Karet FE, Molina AM, Vaara I, Iwata F, Cushner HM, Koolen M, Gainza FJ, Gitleman HJ, Lifton RP (1996) Gitelman’s variant of Bartter’s syndrome, inherited hypokalaemic alkalosis, is caused by mutations in the thiazide-sensitive Na-Cl cotransporter. Nat Genet 12:24–30
Sontia B, Montezano AC, Paravicini T, Tabet F, Touyz RM (2008) Downregulation of renal TRPM7 and increased inflammation and fibrosis in aldosterone-infused mice: effects of magnesium. Hypertension 51:915–921
Tejpar S, Piessevaux H, Claes K, Piront P, Hoenderop JG, Verslype C, Van Cutsem E (2007) Magnesium wasting associated with epidermal-growth-factor receptor-targeting antibodies in colorectal cancer: a prospective study. Lancet Oncol 8:387–394
Thébault S, Alexander RT, Groenestege WM, Bindels RJ, Hoenderop JG (2008) EGF activates TRPM6 via a Src kinase and Rac1 mediated increase in plasma membrane expression. J Am Soc Nephrol 283:19999–20007
Thebault S, Cao G, Venselaar H, Xi Q, Bindels RJ, Hoenderop JG (2008) Role of the alpha-kinase domain in transient receptor potential melastatin 6 channel and regulation by intracellular ATP. J Biol Chem 283:19999–20007
Thebault S, Hoenderop JG, Bindels RJ (2006) Epithelial Ca2+ and Mg2+ channels in kidney disease. Adv Chronic Kidney Dis 13:110–117
Topala CN, Groenestege WT, Thebault S, van den Berg D, Nilius B, Hoenderop JG, Bindels RJ (2007) Molecular determinants of permeation through the cation channel TRPM6. Cell Calcium 41:513–523
Touyz RM (2008) Transient receptor potential melastatin 6 and 7 channels, magnesium transport, and vascular biology: implications in hypertension. Am J Physiol Heart Circ Physiol 294:H1103–1118
Touyz RM, Yao G (2003) Inhibitors of Na+/Mg2+ exchange activity attenuate the development of hypertension in angiotensin II-induced hypertensive rats. J Hypertens 21:337–344
van de Graaf SF, Bindels RJ, Hoenderop JG (2007) Physiology of epithelial Ca2+ and Mg2+ transport. Rev Physiol, Biochem Pharmacol 158:77–160
Velazquez H, Naray-Fejes-Toth A, Silva T, Andujar E, Reilly RF, Desir GV, Ellison DH (1998) Rabbit distal convoluted tubule coexpresses NaCl cotransporter and 11 β-hydroxysteroid dehydrogenase II mRNA. Kidney Int 54:464–472
Voets T, Nilius B, Hoefs S, van der Kemp AW, Droogmans G, Bindels RJ, Hoenderop JG (2004) TRPM6 forms the Mg2+ influx channel involved in intestinal and renal Mg2+ absorption. J Biol Chem 279:19–25
Walder RY, Landau D, Meyer P, Shalev H, Tsolia M, Borochowitz Z, Boettger MB, Beck GE, Englehardt RK, Carmi R, Sheffield VC (2002) Mutation of TRPM6 causes familial hypomagnesemia with secondary hypocalcemia. Nat Genet 31:171–174
Weber KT (2003) Aldosteronism revisited: perspectives on less well-recognized actions of aldosterone. J Lab Clin Med 142:71–82
Acknowledgment
This work was supported by the Netherlands Organization for Scientific Research, Zon-Mw 9120.6110, ALW 818.02.001), a EURYI award from the European Science Foundation, and the Dutch Kidney foundation (C03.6017, C05.2134, C08.2252). We would like to thank our colleagues at the Department of Physiology for valuable discussions and critical reading of this manuscript.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Xi, Q., Hoenderop, J.G.J. & Bindels, R.J.M. Regulation of magnesium reabsorption in DCT. Pflugers Arch - Eur J Physiol 458, 89–98 (2009). https://doi.org/10.1007/s00424-008-0601-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-008-0601-7