
Abstract
Adaptation of a cell behavior to the environment is possible due to the biochemical and physical information that is transmitted through molecular receptor present at the cell surface. Regulation of receptor distribution and trafficking is thus a key feature to allow cells to properly respond to extracellular signals. Many of the molecular mechanisms that support receptor trafficking occurs at a submicrometric scale and are highly dynamic. Because of its exceptional resolution and its piconewton sensitivity, atomic force microscope (AFM) is a powerful tool to study the trafficking of individual receptors in living cells under near-physiological conditions. In this review, we first describe the general principles of the AFM that allow the detection of single ligand–receptor interaction. We then turn to early studies that demonstrated the ability of AFM to detect individual receptors and map their distribution on the surface of living cell. Finally, we discuss how AFM in combination with optical imaging tools allow the simultaneous investigation of cellular biophysical properties and receptor-trafficking dynamics at the nanometer scale.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The binding of extracellular ligands to receptors allows living cells to constantly monitor and respond to changes in their environments. The control of receptor distribution and trafficking in a spatially and temporally ordered manner is required to modulate cell behaviors, which range from cell division to differentiation. In the last decades, large efforts have been made to characterize the molecular mechanisms governing receptor trafficking. This has led to the discovery that receptor trafficking depends on processes involving molecular complex formation, organization of the cell cytoskeleton, and exocytic insertion and endocytic removal of receptors as well as their lateral diffusion at the plasma membrane. Many of these processes happen over scales of tens of nanometers and milliseconds to minutes constituting a technical challenge for scientists. Few techniques can reach the spatial and temporal resolution necessary to gain insights into the molecular dynamics of receptors at the single-molecule level in living cells.
The atomic force microscope (AFM) represents one emerging technology that enables the simultaneous investigation of (1) the dynamics of receptor trafficking at the nanometer scale, (2) the biophysical properties of living cells, and (3) the cellular topology under near-physiological conditions.
In this review, we discuss the unique opportunity offered by the AFM to study receptor trafficking. We first describe the technical principles and the modes of operation of the AFM. We then discuss the advantages of the AFM applied to the study of ligand–receptor interaction in vitro and in vivo. Finally, we review recent advances in our understanding of receptor trafficking obtained using the AFM and how this technique can be combined with different forms of spectroscopy. The examples that we chose to discuss might help to illustrate the new perspectives provided by the use of the AFM into the study of the molecular mechanisms of receptor trafficking.
Principle of AFM
General principle
In 1986, Binnig et al. [6] invented a new scanning probe microscope, called atomic force microscope. The AFM probe sensitivity was based on short-range interaction forces, which enabled to investigate electric insulators as well as conductors, constituting a major advance compared to the scanning tunneling microscope, that requires conducting samples [7]. The AFM rapidly demonstrated its ability to image objects not only in air but also in liquids [15, 24], allowing to work on biological material under nearly physiological conditions. Moreover, the AFM provided a resolution exceeding the limits obtained with classical optical techniques. Nowadays, biological samples can be imaged by the AFM with a lateral resolution of 0.5 nm and a vertical resolution of 0.1 nm [17, 66].
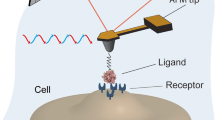
The AFM probe is a small tip (curvature radius of about 20 nm) mounted at the end of a flexible cantilever as depicted in Fig. 1. While probing the sample, the tip–sample interactions provoke deflections of the cantilever, which behaves as a micro-spring. These deflections are measured by a position-sensitive photodiode, collecting light from a laser beam reflected on the cantilever backside. The photodiode signal can either be used as a feedback to control the cantilever–sample distance (see “Imaging”) or as a direct measure of the tip–sample interactions (see “Force spectroscopy”).

Scheme of an AFM combined with an optical microscope for the study of biological sample such as living cells. The probe of the AFM is a sharp tip, placed at the end of a soft cantilever. A piezoelectric scanner allows precisely positioning the tip relative to the sample. A laser beam reflected on the cantilever backside and detected by photodiodes is used to measure the cantilever deflection. This signal is either used as a feedback to control the scanner (imaging mode) or measured and converted into a force (force spectroscopy mode). Since the cantilever is mounted under the scanner, the optical path is free and the AFM can be coupled to any inverted optical microscope. To increase clarity, the relative scale was not respected on the scheme
The sample is usually mounted on a piezoelectric scanner, allowing relative tip–sample displacements with sub-angstrom precision. However, in the new generation of AFM dedicated to life science, the scanner is situated above the cantilever and controls its movement, while the sample is steadily mounted on an inverted optical microscope, as depicted in Fig. 1. Therefore, it is now possible to probe living cells simultaneously with the AFM and with advanced optical microscopy techniques [35, 39, 54, 77].
Imaging
Two imaging modes are mainly used to study biological samples by the AFM, the contact mode and the tapping mode. In contact mode imaging, the tip raster-scans the surface, while the cantilever is maintained in a constant deflected state (i.e., the tip exerts a small but constant force on the sample). The topology of the sample yields variations of the tip–sample interaction and therefore of the deflection of the cantilever. These variations in deflection are measured by the photodiode detector and transmitted through a feedback loop to the scanner, which adjusts its extension to maintain the constant cantilever deflection. The scanner movements are recorded and provide thus a topographic map of the sample. Contact mode was successfully used to image various biological samples including living cells [32, 56], proteins [53] and DNA molecules [25, 64]. In tapping mode imaging, the cantilever is oscillated at a constant frequency close to resonance. The tip–sample distance is precisely set such that the tip only lightly touches the sample at the lowest point of its oscillating cycle. In this setup, the topology of the sample provokes variations of the cantilever oscillation amplitude, which are detected by the photodiode and fed back to the scanner. The scanner adjusts its extension to maintain the constant amplitude of oscillation of the cantilever. Compared to contact mode, tapping mode reduces lateral and adhesive forces exerted on the sample allowing imaging of soft samples, like living biological specimens, weakly immobilized on a surface [26, 58]. Tapping mode was used to image delicate samples such as DNA in buffer [47, 73], and thus enabling the study of dynamic processes at a molecular level [17, 33, 74].
Force spectroscopy
In force spectroscopy, the scanner is not controlled by a feedback signal but describes linear extension–retraction cycles. Therefore, the tip and sample are alternately brought to and out of contact. The cantilever deflections are recorded during each extension–retraction cycle and displayed as two curves (approach and retraction curves, see Fig. 2). The cantilever deflection (d) can be converted into a force (F) using Hooke’s law (F = kd) where k is the cantilever spring constant. The curves monitoring the cantilever deflections are therefore generally named force–distance curves or simply force curves. As depicted in Fig. 2, a force curve usually has two parts, an in-contact region, where the tip pushes the sample and the cantilever is deflected upward, and an off-contact part, where the cantilever is not deflected at the same extent.

Force curves displaying the cantilever deflection as a function of the scanner extension for an approach–retraction cycle. When the tip approaches the surface (blue curve), the cantilever is first at rest and not deflected (1). When the tip touches the surface, the cantilever gets deflected upwards (2). During retraction (red curve), a ligand–receptor unbinding may be visible as a characteristic downwards nonlinear deflection of the cantilever (3), which reflects the stretching of the molecule crosslinkers. The unbinding itself is represented by an abrupt vertical segment following the downwards deflection (4). The cantilever then finally recovers its rest state (5). The ligand–receptor unbinding force F can be calculated by measuring the vertical segment d that characterize the unbinding and multiplying it by the cantilever spring constant k (Hooke’s law)
Force spectroscopy is widely used to measure intermolecular and intramolecular forces [12, 28, 75] and to probe the elastic properties of soft samples [59]. Measuring the forces between and inside molecules is achieved by attaching molecules on the tip on one side and on the substrate on the other side. The creation of a molecular interaction between the tip and the substrate then enables the exertion of a force on the molecules and the eventual unfolding of intramolecular structures or the unbinding of the two molecules. Force measurements between ligands and receptors form the basis of receptor-trafficking studies by atomic force microscopy and will be explained in more details below.
Biomechanical properties of living cells
Force curve measurements are also used to assess the elastic properties of biological samples. When the tip is pushing a soft sample, it deforms slightly and indents the sample. From the cantilever deflection and the scanner extension, it is possible to measure the indentation depth produced by a certain force, obtaining a force–indentation curve. Different models have been proposed to analyze indentation curves [1, 14, 34, 67]. The most widely used is the Hertzian model, which was adapted for AFM by Radmacher et al. in 1996 [60]. This model estimates Young’s elasticity modulus of the sample, which can be interpreted as a measure of stiffness. Elasticity measurements were performed on many cell types [30, 60, 62], and the contribution of cytoskeleton to cell elasticity was examined [34, 63, 76].
Atomic force microscope: an emerging tool to study receptor trafficking in living cells
Studying the density, spatial distribution, clustering and molecular changes of receptors at the level of single molecules in living biological specimens has been challenging. Beyond its initial imaging role, the AFM is capable of measuring molecular interactions. This property was applied to the study of ligand–receptor interactions using purified recombinant molecules as well as the trafficking of single receptors in their original environment such as the plasma membrane of living cells. In this section, we will describe the use of the AFM to characterize ligand–receptor interactions in vitro and discuss its ability to follow the trafficking of single receptors in living cell.
Ligand–receptor interactions
Measuring a ligand–receptor interaction at the level of single molecules is achieved by functionalizing an AFM tip with the ligand molecule probing either a surface coated with the receptor molecule [8, 65, 78] or a cell presenting the receptor at its surface [9, 57, 72, 79]. The first studies using the AFM to characterize ligand–receptor interactions were performed between biotin and streptavidin or avidin [18, 40, 46, 52]. Typically, biotin molecules covalently bound to bovine serum albumin, were adsorbed to the tip or to the substrate surface. Correspondingly, the substrate or the AFM tip was then coated with the conjugate molecules of streptavidin or avidin. Upon tip–substrate approach, a bond was formed between the biotin and streptavidin molecules. When the tip was retracted, the two molecules were stretched, inducing the deflection of the cantilever downwards, until the force exerted by the tip became strong enough to break the ligand–receptor interaction. Each breakage of one biotin–streptavidin/avidin interaction appeared as a small saw tooth event on the retraction curves as schematically described in Fig. 2. The cantilever deflection can be converted into an unbinding force using Hooke’s law (see previous section and Fig. 2). During the last 10 years, many different interactions of ligand–receptor complexes have been examined using the AFM [2, 3, 11, 28, 31, 78]. These studies demonstrated that it was possible to detect and quantify ligand–receptor binding forces, typically in the range of 50–250 pN, and also to study the structural parameters that characterize ligand–receptor interactions.
Detection of receptors at the cell surface
The ability of the AFM to detect small surface intermolecular forces at high lateral resolution makes it a powerful tool to study the distribution of individual receptors present at the surface of living cells. One of the early studies by Gad et al. [22] used AFM to map the position of polysaccharides on the surface of partially dehydrated yeast cells. To this purpose, they used a tip coated with concavalin A (conA), a lectin that recognizes mannose residues on the yeast cell wall. They estimated the binding force between conA and mannoses residues to be in the range of 75–200 pN. They demonstrated that the measured interaction was specific because it was suppressed when free mannose was added in the AFM chamber. However, while they detected specific events on the yeast cell wall, suggesting a nonuniform distribution of mannan, they were unable to resolve the spatial distribution of individual polysaccharides. Another study used a similar approach to define the integrin–ligand mechanics on bone cell cultures [43]. Using AFM tips coated with different ligands containing an Arg-Gly-Asp peptide (RGD) sequence, Lehenkari et al. showed that the RGD–integrin interaction is sequence specific and depends on the molecular context of the RGD sequence within a protein. They also demonstrated that the interaction force was cell specific, since it was significantly different among different bone cell types. However, in this study, bone-cell cultures were briefly chemically fixed before use. It is thus possible that fixation modified the mechanical properties of the cell or the biochemical environment of the receptor, suggesting the results may not fully reflect physiological conditions. Nevertheless, this study demonstrates that the AFM is useful for characterizing the properties of different extracellular matrix–ligand interactions with their receptors present at the surface of an intact cell. Moreover, these two examples show that the AFM enables the specific detection of various molecules present at the surface of intact whole cells.
Mapping single receptors at the cell surface
The first study that succeeded in mapping receptors at the surface of living cells in nearly physiological conditions investigated the distribution of calcitonin receptor on living osteoclasts [42]. The authors were first surprised to detect no calcitonin–calcitonin receptor interactions when they used a classical calcitonin-coated AFM tip because the cell was expected to present several millions of calcitonin receptors [42]. To increase the contact surface and therefore the probability of detecting ligand–receptor interactions, they attached microbeads to the AFM tip before coating with calcitonin. In this situation, they succeeded in measuring ligand–receptor interactions between calcitonin and its receptors. The resulting interaction map demonstrated a clear heterogeneous distribution of the receptors over the cell surface. Since some areas of the cell surface did not present any ligand–receptor interaction, they concluded that calcitonin binding was activated only on a small area of the cell.
Another study used similar methodology in combination with immunocytochemistry to visualize the distribution of one of the vitronectin (VN) receptor subunits, integrinβ5 at the surface of living osteoblastic cells [38]. The AFM measurements were performed with VN-coated beads attached to the tip, and the distribution of integrinβ5 at the surface of living osteoblastic cells was mapped over an area of 32 × 32 μm2. Comparing the distributions of integrinβ5 obtained by immunofluorescence and by the AFM showed similar but only partly overlapping localization. However, fluorescent images were acquired in the wide-field mode, which did not allow the identification of the population of integrinβ5 present only at the cell surface. More recently, the same authors used an AFM microscope coupled to a confocal microscope to study the distribution of overexpressed green fluorescent protein (GFP)-prostaglandin EP3 receptor at the surface of living Chinese Hamster Ovary (CHO) cell [37]. As before, they first attached microbeads to an AFM tip and then covalently linked anti-GFP antibodies to the beads. The distribution map of GFP-EP3 receptors obtained either by confocal microscopy or by the AFM over an area of 16 × 16 μm2 appeared to be heterogeneous at the surface of living CHO cells. In addition, the authors found a correlation between the fluorescent intensity of the GFP-EP3 receptors and the distribution of the interaction forces between the anti-GFP and the GFP-EP3 receptors. Finally, the authors quantified the energy corresponding to a single ligand–receptor unbinding event and then extrapolated that ~17,000 EP3 receptors were present on the cell surface, assuming the cell area to be ~5,000 μm2.
While these studies using a microbead attached to the AFM tip demonstrated clearly the ability of the AFM to map receptors at the surface of living cells, the increased contact surface between the microbead and the cell surface diminished dramatically the lateral resolution that could be obtained in the force-mapping approach by about one order of magnitude. In an attempt to study the distribution of receptor-associated protein (RAP) binding proteins at the surface of living fibroblasts with high resolution, Osada et al. [55] used an AFM tip coated with RAP. In this study, they used sulfur chemistry to introduce 2-pyridil disulfide groups into RAP to specifically orient the molecules on the coated AFM tip [29]. Recording force curves on different spots spread over a 4-μm2 area of living cells allowed them to simultaneously quantify the unbinding force between RAP and RAP binding protein and to map the location of RAP binding proteins at the cell surface. The specificity of the interaction was demonstrated by adding free RAP that abolished adhesion force between the functionalized tip and the cell surface. The authors also tried to give an estimate of the total number of receptors that might be present at the surface of a fibroblast. They calculated from the force mapping a total of about 36,000 receptors present at the surface of the cell. This study was the first to determine simultaneously the interaction force between a ligand and its receptor and to determine the distribution of single receptors at the surface of a living cell.
Measuring the interaction force between a ligand and its receptor and characterizing their distribution at the surface of a living cell can be useful for studying the mechanism of aggregation of receptors. One such example came from the study of the distribution of the nerve growth factor (NGF) receptor TrkA at the surface of living PC12 cells using a NGF-coated AFM tip [61]. The authors first quantified the force necessary to break apart the interaction between single NGF-TrkA dimer using a TrkA-coated substrate. Then, they recorded force maps of 1 × 1 μm2 at the surface of living PC12 cells and found that the unbinding forces measured were multiples of the value previously found for the single NGF-TrkA dimer. The authors interpreted this result as the consequence of multiple interactions between NGF molecules on the tip and TrkA at the cell surface. Moreover, they found that the force distribution was heterogeneous and varied depending on the location at the cell surface. They concluded, therefore, that TrkA aggregated at the surface in clusters of different sizes. Finally, the analysis of the distribution of TrkA revealed that the aggregates were randomly distributed and irrelevant to the cell topography.
More recently, a similar approach was used by Dupres et al. [16] to investigate the distribution of heparin-binding hemagglutinin adhesin (HBHA) on the surface of living bacteria. Using a heparin-coated tip, they found that the measured adhesion force between HBA and heparin across the cell surface of living mycobacteria had a bimodal distribution with mean values at 53 and 110 pN, suggesting that the HBHA–heparin interaction most likely occurs through multiple intermolecular bridges. In addition, spatially resolved adhesion maps of 300 × 300 nm revealed that the HBHA distribution was not random but concentrated into nanodomains.
These different studies showed that the AFM enables the detection of different ligand–receptor interactions at the surface of living cells in near physiological conditions. Its high lateral resolution allows the characterization of the distribution of individual receptors and the estimation of their total number present at the cell membranes. This has led to the discovery that receptors could be organized in nanodomains and distributed heterogeneously at the cell surface.
Receptor trafficking
As discussed previously, the AFM allows the detection of receptors at the surface of living cells and the study of their regional distribution at nanometer scale. However, studying receptor trafficking requires the ability to both follow changes in the distribution over time and to determine how external stimuli will affect their distribution. In a very recent study, Yersin et al. used the AFM to monitor the distribution and trafficking of single α-amino-3-hyroxy-5-methylisoxazole-4-proprionic acid (AMPA)-type glutamate receptors present at the surface of living hippocampal neurons [77]. The authors used an AFM tip coated with an anti-hemagglutinin (HA) antibody to detect exogenous AMPA receptors tagged with an extracellular HA-tag and map their distribution at the cell surface (Fig. 3). On average, they detected 55 unbinding events per 4 μm2 that corresponded to single HA-tagged AMPA receptors on the neuronal somas. The specificity of the interaction between the HA-coated tip and the HA-tagged AMPA receptor was validated in two different ways. First, they used a control tip coated with an anti-Myc antibody; in a second set of experiments, they used an anti HA-coated tip and measured force curve on living neurons that did not express HA-tagged AMPA receptors. In both cases, they found a dramatic decrease in the number of unbinding events detected compared to the experimental situation. Moreover, they also demonstrated that the average number of receptors at the surface of living neuron remained constant when they performed serial recordings during 90 min over the same area. They thus were able not only to characterize the distribution of single AMPA receptor present at the cell surface of hippocampal neurons but also to follow their distribution over time and correlate them with the topology and the elastic properties of the neurons.
The authors also asked whether it was possible to detect changes in receptor distribution using pharmacological stimulation. It has recently been shown that stimulation of N-methyl-d-aspartate (NMDA)-type glutamate receptor with NMDA, leads to the internalization of AMPA receptors [41, 69]. In addition, it was also shown that blocking endogenous activity before NMDA stimulation still leads to their internalization, but the receptors are then recycled back to the plasma membrane. When the authors treated the neurons with NMDA, they measured a ~53% permanent decrease of the total number of HA-tagged AMPA receptors detected at the cell surface (Fig. 4a). This loss was attributed to the internalization of the receptors because blocking endocytosis prevented the decrease of surface AMPA receptors. In contrast to treatment with NMDA alone, when they blocked endogenous activity and consecutively applied NMDA, they measured first a decrease of HA-tagged AMPA receptors on the cell surface. However, the number of receptors increased again and reached control level after 30 min (Fig. 4b). This suggests that HA-tagged AMPA receptors were internalized and recycled back to the plasma membrane. Therefore, the AFM is not only suitable for mapping single receptors at the surface of living cell but also for revealing changes in their distribution due to mechanism associated with their trafficking such as internalization and/or insertion at the cells surface.

Online detection of AMPA receptors (AMPAR) at the surface of living hippocampal neurons by AFM, reproduced from Yersin et al. [77]. Hippocampal neurons were cotransfected with GFP and with the AMPA receptor subunit HA-GluR2. For confocal microscopy, the neuron was fixed and immunostained with a HA-antibody under nonpermeabilized conditions. The confocal sections here show the GFP signal (a) and the anti-HA-labeled surface GluR2 (b). Scale bar is 15 μm. c Experimental set-up used to detect AMPAR at the surface of living neurons. Indentation of the tip into the cell (bottom left) provides local elasticity measurements. Cantilever retraction (bottom right) is used to detect interactions between the anti-HA tip and surface HA-GluR2 (top right), which enables locating HA-tagged AMPAR at the cell surface. d Force volume measurements on a 2 × 2 μm area on the cell surface allowed simultaneous measurement of the cell topography (false three-dimensional), elasticity (color code), and the detection of AMPAR (asterisks). Figure from Yersin et al. [77], reproduced with permission. Copyright 2007 Biophysical Society

Online trafficking of AMPAR and elasticity measurements at the surface of living hippocampal neurons, reproduced from Yersin et al. [77]. AMPAR were detected at the cell surface with an AFM tip coated with antibodies, and the cell surface elasticity was simultaneously measured as explained in Fig. 3. a Number of detected binding–unbinding events (representing AMPARs) at the surface of GFP/HA-GluR2-cotransfected cells. During the first 30 min of measurements, the number of events detected remained stable. Cells were then stimulated for 2 min (arrow) with 50 μM NMDA (circles, n = 5 cells), or with vehicle alone (control, triangles, n = 6 cells). The number of events detected on cells receiving vehicle alone remained stable until the end of experiment, but on the cell stimulated with NMDA, the number of events rapidly decreased and remained on average 53% lower than the initial number measured before stimulation. b As in a except that neurons were preincubated with tetrodotoxin (TTX; 2 μM) during 60 min, before stimulation for 2 min with 50 μM NMDA (circles, n = 6 cells) or with vehicle alone (control, triangles, n = 6 cells). While the vehicle alone did not affect the number of events detected, NMDA/TTX produced a decrease in the number of events, which reached a minimum 16 min after stimulation. The number of events detected then increased again to recover the initial level 30 min after stimulation. This curve reflects the recycling of AMPAR after NMDA/TTX stimulation. d and e Elasticity modulus of AMPAR sites relative to their vicinity sites at 250 nm for the experiments described in a and b, respectively. In nonstimulated state, AMPAR were located in nanodomains on average 24% stiffer than the surrounding cell surface. Both NMDA and NMDA/TTX provoked the disappearance of these stiff nanodomains, without recovery for NMDA but with recovery for NMDA/TTX stimulation. It can be noticed that the variation of nanodomain elasticity follows a curve similar to the corresponding variation of event number with a slight temporal shift. Asterisks are the result of statistical t test; *P < 0.05, **P < 0.01, ***P < 0.001, t test. Figure from Yersin et al. [77], adapted with permission. Copyright 2007 Biophysical Society
Force curve measurements also provided estimates of local elasticity modulus, which reflects the stiffness of the cell surface. Yersin et al. were able to measure simultaneously the spatial distribution of single HA-tagged AMPA receptors and the relative elastic properties of the cell surface at any given time (Fig. 3c). Elasticity measurements show that AMPA receptors have on average an elasticity modulus 24% higher relative to their vicinity. This shows that AMPA receptors are located in stiff nanodomains of 250 nm diameter. In addition, they found that these nanodomains were influenced by NMDA stimulation, and changes in their mechanical properties preceded the internalization/reinsertion of AMPA receptors (Fig. 4c,d). It is likely that the variations in nanomechanical properties of the neuronal surface reflect changes of either the characteristics of the underlying cytoskeleton or the lipid or protein environments of the receptors. It was shown, for example, that lipid rafts (dynamic microdomains enriched in sphingolipids and cholesterol) are involved in local signaling at the membrane, protein trafficking, and regulation in cortical actin [68]. Moreover, a subpopulation of AMPA receptors were identified in lipid rafts [27]. It was shown that depletion of cholesterol/sphingolipid disturbs AMPA receptor internalization. It is thus possible that the nanodomains identified in this study correspond to membrane domains with specific chemical and physical properties such as lipid raft that are involved in the control of AMPA receptor distribution. Neuronal activity could thus change the specific biophysical properties of the microenvironment of AMPA receptor to promote their mobility and thus trafficking.
Another recent study also identified changes in the mechanical properties of the cell surface associated with the clustering of the vascular endothelial growth factor (VEGF) receptors [4]. Using an AFM tip coated with an antibody that recognizes one of the VEGF receptor subtypes, the authors correlated the state of clustering of receptor and the local membrane elasticity. Interestingly, the clustering of VEGF receptors was accompanied by a decrease in membrane stiffness only in the region underlying the receptors and was reciprocally related to the receptor density. This suggested that a localized clustering of VEGF receptors could induce reorganization of the underlying cytoskeletal architecture necessary to induce the signal-transduction pathway linked to the VEGF receptors.
While the use of the AFM to study receptor trafficking is an emerging field, these two studies demonstrated its potential to detect individual receptors present at the cell surface of living cell and to characterize their behavior over time. In addition, the AFM offers a unique opportunity to study the link between receptor dynamics and the biophysical properties of the cell with nanometer resolution.
Perspectives and conclusions
The AFM has already proven its utility to acquire high-resolution images and force measurements of biological entities in near-physiological conditions [17, 20, 32, 33, 47; 51]. The AFM was shown to be particularly well suited to study the formation of macromolecular complexes in vitro [44, 45, 69, 78]. The recent demonstration that the AFM can be used to map single receptors at the surface of living cell and to correlate it with the mechanical properties of the cell membrane will open new avenues in our understanding of receptor trafficking and more generally of biological processes at the nanometer scale. Indeed, the recent discovery that populations of receptors are clustered in nanodomains biophysically isolated suggests an unsuspected level of organization whose molecular basis remains to be elucidated. Moreover, It was demonstrated that the AFM is able to distinguish mechanical changes due to different components of the cytoskeleton, suggesting a multilayer cell model [34]. Combining such an approach with the study of receptor trafficking would allow to investigate the effect of single ligand–receptor interactions on the local rearrangement of cellular architecture from the cell surface to deeper parts of the cell.
One of the limitations of the AFM is that it does not allow direct following of receptors trafficking inside the cell. However, development of the AFM combined with optical techniques should make possible simultaneous AFM measurements and imaging of intracellular processes linked to receptor trafficking. Indeed, while the AFM provides nanometer-scale resolution and the ability to study biophysical processes, fluorescence microscopy enables the characterization of biological mechanisms with a high temporal resolution and the possibility of functional imaging. For example, combining AFM with confocal microscopy allows performing simultaneous AFM measurements on living cells and at the same time visualizing labeled molecules [10, 19, 21, 23, 35, 48–50, 54]. Another example is the recent development of a spatially and temporally synchronized total internal fluorescence and AFM system (STIRF-AFM). STIRF-AFM allows collecting at each point in a sample both the fluorescence and topological or mechanical information of biological molecules [35]. This study demonstrated that element of the cytoskeleton stained with fluorescent marker and visualized by TIRF matched with surface topography elements imaged by AFM. However, TIRF microscopy is limited by the evanescent electromagnetic field that penetrates to a depth of only approximately 100 nm into the sample medium [5, 70]. STIRF-AFM will thus be useful to investigate very flat biological specimens or to examine the transmission of mechanical properties from the apical to the basal cell membrane [49].
Until recently, imaging and force spectroscopy were two distinct features of AFM that could not be done simultaneously. Although force mapping can also give topographical information in addition to receptor detection, its spatial resolution is rather poor (tens of nanometers scale) compared with the classical imaging mode. However, the new recognition imaging mode allows simultaneous high-resolution imaging and receptor detection [36, 71]. In this method, a tip is coated with a ligand as for force spectroscopy, but the cantilever is oscillated at high frequency as in tapping mode. Ligand–receptor binding and topography of the surface provoke distinct variations of cantilever amplitude that can be differentiated and used to create a topographical map and a recognition map. This technique allowed the mapping of receptor with nanometer scale on solid surface [71] and recently also at the cell surface [13].
In this review, we have discussed the application of the AFM to follow the distribution and trafficking of receptors on living cell at the level of single molecules. While the AFM offers tremendous opportunities to investigate biophysical properties of the cell and the dynamics of receptor trafficking, its integration with optical techniques that allows the detection of single fluorescent molecules is particularly promising. Moreover, further development of the AFM to improve its spatial resolution and its scan rate will contribute to increase our understanding of biological processes at the molecular scale in vivo.
References
A-Hassan E, Heinz F, Antonik MD, D’Costa NP, Nageswaran S, Schoenenberger CA, Hoh JH (1998) Relative microelastic mapping of living cells by atomic force microscopy. Biophys J 74:1564–1578
Allen S, Rigby-Singleton SM, Harris H, Davies MC, O’Shea P (2003) Measuring and visualizing single molecular interactions in biology. Biochem Soc Trans 31:1052–1057
Allison DP, Hinterdorfer P, Han W (2002) Biomolecular force measurements and the atomic force microscope. Curr Opin Biotechnol 13:47–51
Almqvist N, Bhatia R, Primbs G, Desai N, Banerjee S, Lal R (2004) Elasticity and adhesion force mapping reveals real-time clustering of growth factor receptors and associated changes in local cellular rheological properties. Biophys J 86:1753–1762
Axelrod D (2001) Total internal reflection fluorescence microscopy in cell biology. Traffic 2:764–774
Binnig G, Quate CF, Gerber C (1986) Atomic force microscope. Phys Rev Lett 56:930–933
Binnig G, Rohrer H, Gerber C, Weibel E (1982) Tunneling through a controllable vacuum gap. Appl Phys Lett 40:178–180
Bonanni B, Kamruzzahan AS, Bizzarri AR, Rankl C, Gruber HJ, Hinterdorfer P, Cannistraro S (2005) Single molecule recognition between cytochrome C 551 and gold-immobilized azurin by force spectroscopy. Biophys J 89:2783–2791
Bustanji Y, Arciola CR, Conti M, Mandello E, Montanaro L, Samori B (2003) Dynamics of the interaction between a fibronectin molecule and a living bacterium under mechanical force. Proc Natl Acad Sci U S A 100:13292–13297
Charras GT, Horton MA (2002) Single cell mechanotransduction and its modulation analyzed by atomic force microscope indentation. Biophys J 82:2970–2981
Chen A, Moy VT (2000) Cross-linking of cell surface receptors enhances cooperativity of molecular adhesion. Biophys J 78:2814–2820
Chen A, Moy VT (2002) Single-molecule force measurements. Methods Cell Biol 68:301–309
Chtcheglova LA, Waschke J, Wildling L, Drenckhahn D, Hinterdorfer P (2007) Nano-scale dynamic recognition imaging on vascular endothelial cells. Biophys J 93:L11–13
Costa KD, Sim AJ, Yin FC (2006) Non-Hertzian approach to analyzing mechanical properties of endothelial cells probed by atomic force microscopy. J Biomech Eng 128:176–184
Drake B, Prater CB, Weisenhorn AL, Gould SA, Albrecht TR, Quate CF, Cannell DS, Hansma HG, Hansma PK (1989) Imaging crystals, polymers, and processes in water with the atomic force microscope. Science 243:1586–1589
Dupres V, Menozzi FD, Locht C, Clare BH, Abbott NL, Cuenot S, Bompard C, Raze D, Dufrene YF (2005) Nanoscale mapping and functional analysis of individual adhesins on living bacteria. Nat Methods 2:515–520
Engel A, Muller DJ (2000) Observing single biomolecules at work with the atomic force microscope. Nat Struct Biol 7:15–718
Florin EL, Moy VT, Gaub HE (1994) Adhesion forces between individual ligand–receptor pairs. Science 264:15–417
Formigli L, Meacci E, Sassoli C, Chellini F, Giannini R, Quercioli F, Tiribilli B, Squecco R, Bruni P, Francini F, Zecchi-Orlandini S (2005) Sphingosine 1-phosphate induces cytoskeletal reorganization in C2C12 myoblasts: physiological relevance for stress fibres in the modulation of ion current through stretch-activated channels. J Cell Sci 118:161–1171
Fotiadis D, Scheuring S, Muller SA, Engel A, Muller DJ (2002) Imaging and manipulation of biological structures with the AFM. Micron 33:85–397
Frankel DJ, Pfeiffer JR, Surviladze Z, Johnson AE, Oliver JM, Wilson BS, Burns AR (2006) Revealing the topography of cellular membrane domains by combined AFM/fluorescence imaging. Biophys J 90:404–2413
Gad M, Itoh A, Ikai A (1997) Mapping cell wall polysaccharides of living microbial cells using atomic force microscopy. Cell Biol Int 21:97–706
Gorelik J, Shevchuk A, Ramalho M, Elliott M, Lei C, Higgins CF, Lab MJ, Klenerman D, Krauzewicz N, Korchev Y (2002) Scanning surface confocal microscopy for simultaneous topographical and fluorescence imaging: application to single virus-like particle entry into a cell. Proc Natl Acad Sci U S A 99:6018–16023
Haberle W, Horber JKH, Binnig G (1991) Force microscopy on living cells. J Vac Sci Technol B 9:210–1213
Hansma HG, Vesenka J, Siegerist C, Kelderman G, Morrett H, Sinsheimer RL, Elings V, Bustamante C, Hansma PK (1992) Reproducible imaging and dissection of plasmid DNA under liquid with the atomic force microscope. Science 256:180–1184
Hansma PK, Cleveland JP, Radmacher M, Walters DA, Hillner PE, Bezanilla M, Fritz M, Vie D, Hansma HG, Prater CB et al (1994) Tapping mode atomic-force microscopy in liquids. Appl Phys Lett 64:738–1740
Hering H, Lin CC, Sheng M (2003) Lipid rafts in the maintenance of synapses, dendritic spines, and surface AMPA receptor stability. J Neurosci 23:3262–3271
Hinterdorfer P (2002) Atomic force microscopy in cell biology. In: Jena BP, Hörber JKH (eds) Methods cell biol. Academic, San Diego, pp 115–139
Hinterdorfer P, Dufrene YF (2006) Detection and localization of single molecular recognition events using atomic force microscopy. Nat Methods 3:347–355
Hofmann UG, Rotsch C, Parak WJ, Radmacher M (1997) Investigating the cytoskeleton of chicken cardiocytes with the atomic force microscope. J Struct Biol 119:84–91
Horton M, Charras G, Lehenkari P (2002) Analysis of ligand–receptor interactions in cells by atomic force microscopy. J Recept Signal Transduct Res 22:169–190
Kasas S, Gotzos V, Celio MR (1993) Observation of living cells using the atomic force microscope. Biophys J 64:539–544
Kasas S, Thomson NH, Smith BL, Hansma HG, Zhu X, Guthold M, Bustamante C, Kool ET, Kashlev M, Hansma PK (1997) Escherichia coli RNA polymerase activity observed using atomic force microscopy. Biochemistry 36:461–468
Kasas S, Wang X, Hirling H, Marsault R, Huni B, Yersin A, Regazzi R, Grenningloh G, Riederer B, Forro L et al (2005) Superficial and deep changes of cellular mechanical properties following cytoskeleton disassembly. Cell Motil Cytoskelet 62:124–132
Kellermayer MSZ, Karsai A, Kengyel A, Nagy A, Bianco P, Huber T, Kulcsar A, Niedetzky C, Proksch R, Grama L (2006) Spatially and temporally synchronized atomic force and total internal reflection fluorescence microscopy for imaging and manipulating cells and biomolecules. Biophys J 91(7):2665–2677
Kienberger F, Ebner A, Gruber HJ, Hinterdorfer P (2006) Molecular recognition imaging and force spectroscopy of single biomolecules. Acc Chem Res 39:29–36
Kim H, Arakawa H, Hatae N, Sugimoto Y, Matsumoto O, Osada T, Ichikawa A, Ikai A (2006) Quantification of the number of EP3 receptors on a living CHO cell surface by the AFM. Ultramicroscopy 106:652–662
Kim H, Arakawa H, Osada T, Ikai A (2003) Quantification of cell adhesion force with AFM: distribution of vitronectin receptors on a living MC3T3-E1 cell. Ultramicroscopy 97:359–363
Kodama T, Ohtani H, Arakawa H, Ikai A (2004) Development of confocal laser scanning microscope/atomic force microscope system for force curve measurement. Jpn J Appl Phys 1 43:4580–4583
Lee GU, Kidwell DA, Colton RJ (1994) Sensing discrete streptavidin biotin interactions with atomic-force microscopy. Langmuir 10:354–357
Lee SH, Simonetta A, Sheng M (2004) Subunit rules governing the sorting of internalized AMPA receptors in hippocampal neurons. Neuron 43:221–236
Lehenkari PP, Charras GT, Nykanen A, Horton MA (2000) Adapting atomic force microscopy for cell biology. Ultramicroscopy 82:289–295
Lehenkari PP, Horton MA (1999) Single integrin molecule adhesion forces in intact cells measured by atomic force microscopy. Biochem Biophys Res Commun 259:645–650
Litvinov RI, Bennett JS, Weisel JW, Shuman H (2005) Multi-step fibrinogen binding to the integrin {alpha}IIb{beta}3 detected using force spectroscopy. Biophys J 89:2824–2834
Liu W, Montana V, Bai J, Chapman ER, Mohideen U, Parpura V (2006) Single molecule mechanical probing of the SNARE protein interactions. Biophys J 91:744–758
Ludwig M, Moy VT, Rief M, Florin EL, Gaub HE (1994) Characterization of the adhesion force between avidin-functionalized Afm tips and biotinylated agarose beads. Microsc Microanal Microstruct 5:321–328
Lyubchenko YL, Shlyakhtenko LS (1997) Visualization of supercoiled DNA with atomic force microscopy in situ. Proc Natl Acad Sci U S A 94:496–501
Madl J, Rhode S, Stangl H, Stockinger H, Hinterdorfer P, Schutz GJ, Kada G (2006) A combined optical and atomic force microscope for live cell investigations. Ultramicroscopy 106:645–651
Mathur AB, Truskey GA, Reichert WM (2000a) Atomic force and total internal reflection fluorescence microscopy for the study of force transmission in endothelial cells. Biophys J 78:1725–1735
Mathur AB, Truskey GA, Reichert WM (2000b) Total internal reflection microscopy and atomic force microscopy (TIRFM-AFM) to study stress transduction mechanisms in endothelial cells. Crit Rev Biomed Eng 28:197–202
Matzke R, Jacobson K, Radmacher M (2001) Direct, high-resolution measurement of furrow stiffening during division of adherent cells. Nat Cell Biol 3:607–610
Moy VT, Florin EL, Gaub HE (1994) Intermolecular forces and energies between ligands and receptors. Science 266:257–259
Muller DJ, Janovjak H, Lehto T, Kuerschner L, Anderson K (2002) Observing structure, function and assembly of single proteins by AFM. Prog Biophys Mol Biol 79:1–43
Nishida S, Funabashi Y, Ikai A (2002) Combination of AFM with an objective-type total internal reflection fluorescence microscope (TIRFM) for nanomanipulation of single cells. Ultramicroscopy 91:269–274
Osada T, Itoh A, Ikai A (2003) Mapping of the receptor-associated protein (RAP) binding proteins on living fibroblast cells using an atomic force microscope. Ultramicroscopy 97:353–357
Parpura V, Haydon PG, Henderson E (1993) Three-dimensional imaging of living neurons and glia with the atomic force microscope. J Cell Sci 104(Pt 2):427–432
Pfister G, Stroh CM, Perschinka H, Kind M, Knoflach M, Hinterdorfer P, Wick G (2005) Detection of HSP60 on the membrane surface of stressed human endothelial cells by atomic force and confocal microscopy. J Cell Sci 118:1587–1594
Putman CAJ, Vanderwerf KO, Degrooth BG, Vanhulst NF, Greve J (1994) Tapping mode atomic-force microscopy in liquid. Appl Phys Lett 64:454–2456
Radmacher M (2002) Atomic force microscopy in cell biology. In: Jena BP, Hörber JKH (eds) Methods cell biol. Academic, San Diego, pp 67–90
Radmacher M, Fritz M, Kacher CM, Cleveland JP, Hansma PK (1996) Measuring the viscoelastic properties of human platelets with the atomic force microscope. Biophys J 70:556–567
Reddy CV, Malinowska K, Menhart N, Wang R (2004) Identification of TrkA on living PC12 cells by atomic force microscopy. Biochim Biophys Acta 1667:15–25
Rotsch C, Jacobson K, Radmacher M (1999) Dimensional and mechanical dynamics of active and stable edges in motile fibroblasts investigated by using atomic force microscopy. Proc Natl Acad Sci U S A 96:921–926
Rotsch C, Radmacher M (2000) Drug-induced changes of cytoskeletal structure and mechanics in fibroblasts: an atomic force microscopy study. Biophys J 78:520–535
Samori B, Siligardi G, Quagliariello C, Weisenhorn AL, Vesenka J, Bustamante CJ (1993) Chirality of DNA supercoiling assigned by scanning force microscopy. Proc Natl Acad Sci U S A 90:3598–3601
Schwesinger F, Ros R, Strunz T, Anselmetti D, Guntherodt HJ, Honegger A, Jermutus L, Tiefenauer L, Pluckthun A (2000) Unbinding forces of single antibody–antigen complexes correlate with their thermal dissociation rates. Proc Natl Acad Sci U S A 97:9972–9977
Seelert H, Poetsch A, Dencher NA, Engel A, Stahlberg H, Muller DJ (2000) Structural biology. Proton-powered turbine of a plant motor. Nature 405:418–419
Sen S, Subramanian S, Discher DE (2005) Indentation and adhesive probing of a cell membrane with AFM: theoretical model and experiments. Biophys J 89:3203–3213
Simons K, Toomre D (2000) Lipid rafts and signal transduction. Nat Rev Mol Cell Biol 1:31–39
Steiner P, Alberi S, Kulangara K, Yersin A, Sarria JC, Regulier E, Kasas S, Dietler G, Muller D, Catsicas S, Hirling H (2005) Interactions between NEEP21, GRIP1 and GluR2 regulate sorting and recycling of the glutamate receptor subunit GluR2. EMBO J 24:2873–2884
Steyer JA, Almers W (2001) A real-time view of life within 100 nm of the plasma membrane. Nat Rev Mol Cell Biol 2:268–275
Stroh C, Wang H, Bash R, Ashcroft B, Nelson J, Gruber H, Lohr D, Lindsay SM, Hinterdorfer P (2004) Single-molecule recognition imaging microscopy. Proc Natl Acad Sci U S A 101:2503–12507
Trache A, Trzeciakowski JP, Gardiner L, Sun Z, Muthuchamy M, Guo M, Yuan SY, Meininger GA (2005) Histamine effects on endothelial cell fibronectin interaction studied by atomic force microscopy. Biophys J 89:888–2898
van Noort SJ, van der Werf KO, Eker AP, Wyman C, de Grooth BG, van Hulst NF, Greve J (1998) Direct visualization of dynamic protein–DNA interactions with a dedicated atomic force microscope. Biophys J 74:840–2849
Viani MB, Pietrasanta LI, Thompson JB, Chand A, Gebeshuber IC, Kindt JH, Richter M, Hansma HG, Hansma PK (2000) Probing protein–protein interactions in real time. Nat Struct Biol 7:44–647
Willemsen OH, Snel MM, Cambi A, Greve J, De Grooth BG, Figdor CG (2000) Biomolecular interactions measured by atomic force microscopy. Biophys J 79:267–3281
Wu HW, Kuhn T, Moy VT (1998) Mechanical properties of L929 cells measured by atomic force microscopy: effects of anticytoskeletal drugs and membrane crosslinking. Scanning 20:89–397
Yersin A, Hirling H, Kasas S, Roduit C, Kulangara K, Dietler G, Lafont F, Catsicas S, Steiner P (2007) Elastic properties of the cell surface and trafficking of single AMPA receptors in living hippocampal neurons. Biophys J 92:482–4489
Yersin A, Hirling H, Steiner P, Magnin S, Regazzi R, Huni B, Huguenot P, De los Rios P, Dietler G, Catsicas S, Kasas S (2003) Interactions between synaptic vesicle fusion proteins explored by atomic force microscopy. Proc Natl Acad Sci U S A 100:736–8741
Yersin A, Osada T, Ikai A (2008) Exploring transferrin–receptor interactions at the single molecule level. Biophys J 94 (in press)
Acknowledgements
We address a special thank to Stefan Catsicas for his support and advices. We thank Christoph Bernoulli, Brett Carter, Andrew Giessel, Mike Higley, Tom Nielsen, and Fitz Sturgill for helpful discussions and suggestions. Alexandre Yersin is supported by the Japan Society for the Promotion of Science (P06711) and Pascal Steiner by the Swiss National Science Foundation (no. PA00A-113192/1).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Yersin, A., Steiner, P. Receptor trafficking and AFM. Pflugers Arch - Eur J Physiol 456, 189–198 (2008). https://doi.org/10.1007/s00424-007-0380-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-007-0380-6