
Abstract
Cell fusions are important to fertilization, placentation, development of skeletal muscle and bone, calcium homeostasis and the immune defense system. Additionally, cell fusions participate in tissue repair and may be important to cancer development and progression. A large number of factors appear to regulate cell fusions, including receptors and ligands, membrane domain organizing proteins, proteases, signaling molecules and fusogenic proteins forming alpha-helical bundles that bring membranes close together. The syncytin family of proteins represent true fusogens and the founding member, syncytin-1, has been documented to be involved in fusions between placental trophoblasts, between cancer cells and between cancer cells and host cells. We review the literature with emphasis on the syncytin family and propose that syncytins may represent universal fusogens in primates and rodents, which work together with a number of other proteins to regulate the cell fusion machinery.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Cell fusions in the developing mammal
The fusion between the sperm and the egg marks the beginning of a new individual and ensuing development of either man or beast depends upon a number of additional cell fusion events (reviewed by Ogle et al. 2005). Thus, mononuclear cytotrophoblasts fuse to form the syncytiotrophoblast layer of the placenta in man and certain other mammals. Syncytiotrophoblasts control the exchange of gases, nutrients and waste products between the fetus and the mother, protect the fetus against the maternal immune system and are also responsible for the production of hormones, which like chorionic gonadotropin, regulate the continuation of pregnancy. In addition, in the developing individual, myoblasts fuse to form multinucleated skeletal muscle fibers, while cells of monocytic origin fuse to form osteoclasts, which participate in bone sculpturing and remodeling as well as in the regulation of serum calcium concentrations.
Cell fusions contribute to tissue repair
It is well-known that skeletal muscle is regenerated through fusion of muscle fibers with satellite cells (Bischoff 1994) and that macrophages may fuse to form multinucleated giant cells with enhanced phagocytic capabilities in response to injury and antigenic challenges (Vignery 2005). Recent data indicate that additional cell fusions may contribute to tissue repair in the adult (Vassilopoulos et al. 2003; Wang et al. 2003). Bone-marrow-derived (BMD) cells have been shown to fuse with hepatic cells, nerve cells and gastrointestinal cells and the theory has been put forward that such fusions may serve to repair damaged or corrupted cells (Alvarez-Dolado et al. 2003; Rizvi et al. 2006; Vassilopoulos et al.2003; Wang et al. 2003). However, it is still debated whether such fusions are important to tissue repair and whether they engage BMD stem cells or more differentiated fusogenic cells like monocytes/macrophages (reviewed by Vignery 2005). Intriguingly, the accumulation of macrophages in injured organs may reflect not only the need to remove debris but also to repair compromised cells through heterotypic fusions (Vignery 2005). Moreover, in recent transplant experiments, irradiated mice showed evidence of fusions between crypt stem cells and donor BMD cells (Rizvi et al. 2006). In thus transplanted mice, representatives of all cell lineages derived from intestinal crypt stem cells (absorptive columnar, enteroendocrine, goblet and Paneth cells) showed expression of chromosomal markers characterizing donor BMD cells. This phenomenon appeared to depend upon tissue injury since expression of donor cell markers never was detected in non-irradiated mice (Rizvi et al. 2006). An alternative to fusions has been suggested by Holmgren et al. (1999) and de la Taille et al. (1999), who have demonstrated that cells phagocytosing apoptotic cells may acquire functional DNA. Moreover, a mechanism of cell–cell invasion (entosis) was recently described and could potentially transfer DNA between cells (Overholtzer et al. 2007). Thus, viral as well as normal sequences from apoptotic cells have been found to be active also in phagocytosing cells (Holmgren et al. 1999). This observation challenges our understanding of apoptosis as an efficient way of clearing corrupted or alien DNA.
Cell fusions during viral infections
Infections with enveloped viruses such as HIV-1 may also lead to cell fusions. The viral genome encodes envelope (Env) proteins, which bind to cell surface receptors and assist the virus in entering the cell. The infected cell commences to synthesize Env proteins, which, upon insertion in the plasmalemma, engages receptor proteins in neighboring cells and initiate fusions. Interestingly, modifications of the cytoplasmic tail of several Env proteins appear to modify fusions. Thus, proteolytic cleavage of the cytoplasmic tail regulates fusions and also cellular signaling events, such as tyrosine kinase activity, may be involved in regulation of virally induced cell fusions (Kubo et al. 2003).
Receptor proteins may bind Env proteins from different classes of viruses. Thus, a cell infected by a specific virus becomes resistant to infections with other viruses that bind to the same receptor. This phenomenon, which probably reflects receptor saturation, has led to the grouping of retroviruses into different subtypes, which bind to the same receptor. Other types of viruses encode fusogenic proteins (like influenza hemagglutinin), which do not bind to surface receptors, but which liberate a fusion peptide in the acidic milieu of endosomes and thus initiate viral–host cell fusion following endocytic uptake of viral particles. Both these proteins and the Env proteins are referred to as class I viral fusion proteins and contain hydrophobic fusion peptides buried within their sequences. Following receptor engagement or endosomal acidification the fusion peptides are liberated by proteolytic cleavage. The cleaved peptides undergo conformational changes, resulting in hairpin-like, alpha-helical bundles, which bring the viral and cell membranes into close apposition and thereby facilitates fusion (reviewed by Chen and Olson 2005).
Cancer–host cell fusions
In 1911 Aichel suggested that cancer cells might spontaneously fuse with host cells to produce hybrids with supernumerary chromosomes that could evolve into cells of increased malignancy. Subsequent studies have confirmed this prediction by demonstrating that cells with mixed phenotypes spontaneously appear in co-cultures of normal and malignant cells (Barski and Cornefert 1962; Busund et al. 2002; Mortensen et al. 2004; Wakeling et al. 1994) (Figs. 1, 2, 3). Additionally, transplanted tumor cells of animal (Busund et al. 2003; Chakraborty et al. 2000; Fortuna et al. 1990; Kerbel et al. 1983; Larizza et al. 1984a; Wiener et al. 1974a, b) or human (Goldenberg et al. 1974; Goldenberg and Pavia 1982; Mortensen et al. 2004) origin may fuse with and acquire phenotypic characteristics of normal host cells (Fig. 4). In some of the in vivo studies, data suggested that the putative fusion partner was of macrophage or endothelial origin but, in most cases, definite identification was not achieved. In a few cases, it has also been demonstrated that the fused cells expressed genetic markers of both parental cell types (Goldenberg et al. 1974; Mortensen et al. 2004; Wiener et al. 1974a, b) (Figs. 2, 3). Mortensen et al. (2004) demonstrated that, initially after fusion, bi- or multinucleated cells formed and that such cells had the parental genomes seggregated in diffferent nuclei (heterokaryons) (Fig. 2a–e). Subsequently, mitotic figures appeared showing an admixture of the parental chromosomes (Fig. 2f–j). Eventually, cells with the parental genomes mixed in a single nucleus (synkaryons) were detected (Fig. 2k–o). In agreement with the notion that synkaryons appear after mitotic divison of heterokaryons, synkaryons are usually detected in pairs (Fig. 3). A few recent studies present compelling evidence that cells with genetic characteristics of hybrid cells also may appear in human tumors (Andersen et al. 2007; Chakraborty et al. 2004; Yilmaz et al. 2005; Streubel et al. 2004). Thus, in two cases of renal carcinomas, arising in bone marrow transplant recipients, some tumor cells showed genetic markers characterizing the healthy donor (Chakraborty et al. 2004; Yilmaz et al. 2005). Moreover, in patients with multiple myelomas, osteoclasts have been shown to contain an admixture of nuclei, of which some possess tumor-specific chromosomal translocations while others are devoid of translocations (Andersen et al. 2007). Finally, lymphoma-specific genetic abnormalities were described in endothelial cells in B cell lymphomas (Streubel et al. 2004). Although these data do not formally prove that tumor–host cell hybrids did form in these patients, the combined results from studies in vitro and in vivo do present compelling evidence that cell fusions do occur in tumors (reviewed by Pawelek 2005).

Cancer–endothelial cell fusion. Co-culture of human breast cancer (MCF-7) cells and human umbilical vein endothelial cells (HUVEC). Cells were stained for the cancer cell marker cytokeratin (red, c), the endothelial cell marker vimentin (green, b) and DNA (bisbenzimide, blue, d). Note in the merged image (a) one fused, multinucleated cell reacting for both cytokeratin and vimentin (orange-yellow)

Chromosomal markers and cell fusions. Co-cultures of human breast cancer cells and bovine endothelial cells. The cultures were subjected to fluorescent in situ hybridization (FISH) with probes recognizing all bovine chromosomes (green; b, g, l) and all human chromosomes (red; c, h, m) and counter-stained for DNA with DAPI (blue; d, i, n) and observed in differential interference contrast (DIC; e, j, o). Merged red-green images are shown in a, f, k. Note that initially, binuclear cells (heterokaryons) form, having one bovine and one human nucleus (a–e). Occasionally, tri- or multinuclear cells with different admixtures of bovine and human nuclei are also detected. At longer times after mixing, mitotic figures, containing an admixture of bovine and human chromosomes appear (f–j) and, eventually, cells with a single nucleus, containing an admixture of the two genomes in mixture (synkaryons) are detected (k–o)

Chromosomal markers and cell fusions. Mixed culture of human breast cancer cells and bovine endothelial cells submitted to double FISH as in Fig. 2 (a double FISH; b combined DAPI and DIC). Low power micrograph showing a pair of synkaryons with the bovine and human genomes admixed in single nuclei (arrows). In addition, nuclei hybridizing only for the bovine (green) or only for the human (red) genome occur

Cancer–host cell fusion in vivo. a, b Section from lung of a nude mice injected with human breast cancer (MDA-MB-231) cells in the tail vein (Mortensen et al. 2004). The section underwent FISH for the mouse genome (red) and the human genome (green) (a) and DNA was counterstained with DAPI (b; blue). Note one nucleus in which the human and mouse genome co-localize (arrow). C: Similar section, stained with an antibody detecting human (but not mouse) p53 (red; p53 is mutated and overexpressed by the breast cancer cells), an antibody to beta-catenin (to mark cell membranes) and for DNA with bisbenzimide (blue). Note a micrometastasis of human breast cancer cells having violet (red + blue) fluorescent nuclei. d Section stained for human p53 (red) and the endothelial marker von Willebrand factor (green) and DNA (blue). Note a human cancer cell with a violet (red + blue) nucleus showing membrane-staining for von Willebrand factor. Since von Willebrand factor is not normally expressed by the breast cancer cells, this image is suggestive of a fusion between a human breast cancer cell and a mouse endothelial cell. Similar results were obtained using double FISH for the human and mouse genome and imunofluorescent staining for von Willebrand factor (described by Mortensen et al. 2004)
The essential question regarding cancer–host cell fusions is, of course, if they are relevant to the patient. In fact, there are two opposing views. The first is based on early experiments on fusions induced to occur between cancer cells and normal cells in culture. With few exceptions, such experiments revealed that malignancy was suppressed (Harris et al. 1969; Harris 1988; Stanbridge 1976; Wiener et al. 1974a, b). These studies were, in fact, seminal to the discovery of tumor suppressor genes (reviewed by Anderson and Stanbridge 1993). Since tumor suppressor genes, like p53 and Rb, frequently are inactivated in cancer cells, fusions would present cancer cells with unperturbed tumor suppressors from the normal fusion partner and consequently initiate cell cycle arrest or apoptosis. Although, this certainly applied for the cell types studied in the contributions cited above, it may not be a general rule. Thus, production of monoclonal antibodies depends upon the fact that it is possible to fuse antibody-producing spleen cells with myeloma cells to obtain hybridomas that retain the unlimited proliferative ability of the tumor cell partner and the antibody production of the normal cell (Kohler and Milstein 1975). In fact, several studies documented that some fusions may lead to cells of increased malignancy (Barski and Cornefert 1962; Busund et al. 2003; Chakraborty et al. 2001; De Baetselier et al. 1981; Kerbel et al. 1983; Larizza et al. 1984b; Pawelek 2000; Rachkovsky et al. 1998). Possibly, the genetic make-up of tumors may dictate the outcome of individual cancer–host cell fusions. Moreover, fusions lead to aneuploidy and chromosomal instability, which characterizes most cancers and may, by itself, stimulate carcinogenesis (Duelli et al. 2007).
Mechanisms behind cell–cell fusions
Fusions between normal cells
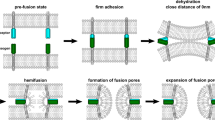
Cell fusion events must be extremely well controlled. Due to their major importance to fertilization, placentation, muscle development, bone structure, calcium homeostasis and the immune defense system, much effort has gone into elucidating mechanisms underlying cell–cell fusions. Additionally, the potential role of cell fusions as a repair mechanism and the role of cell fusions in cancer development and progression have further stimulated research in this field. In spite of this, much less is known about cell–cell fusion mechanisms than is known about how intracellular membranes fuse through v- and t-SNAREs. Interestingly, engineered flipping of the v- and t-SNARE machinery has been shown to promote cell–cell fusions (Weber et al. 1998). However, it is evident that this mechanism is not a physiological mediator of cell–cell fusions. Interestingly, v- and t-SNAREs act similar to class I viral fusion proteins in that they form bundles of alpha-helices, which result in membrane apposition and fusion (Blumenthal et al. 2003; Jahn et al. 2003). Of the many proteins, which to date have been shown (or proposed) to be involved in cell fusions in mammals, only the syncytin family appears to use a similar alpha-helical mechanism. That they do is not surprising in view of the fact that syncytins represent conserved endogenous retroviral Env sequences. The founding family member, syncytin-1, was discovered as a protein capable of mediating fusions between cytotrophoblasts into syncytiotrophoblasts (Blond et al. 2000; Mi et al. 2000). This capability may have contributed to a high degree of evolutionary conservation of the syncytin-1 sequence. Syncytin-1 represents the Env protein of the human endogenous retroviral (HERV) W sequence, which entered the primate genome 25–40 million years ago. In contrast, most other retroviral sequences inserted in our genome have been subject to inactivating changes and probably represent garbage sequences. Molecular studies have shown that syncytin-1 (Env W) shares a structure similar to class I viral fusion proteins, especially in the region of the N- and C-terminal heptad repeats (NHR and CHR), and shares a common fusion mechanism with these proteins (Chang et al. 2004; Gong et al. 2005). A synthetic peptide derived from the CHR is also capable of inhibiting syncytin-1-mediated fusions by perturbing this mechanism (Chang et al. 2004). Syncytin-1 binds to the D-type retroviral receptor ASCT-2 (Blond et al. 2000) and may use another neutral amino acid transporter (ASCT-1) as an auxiliary receptor (Lavillette et al. 2000). Syncytin antibodies, syncytin-1 downregulation through antisense oligonucleotides and the syncytin-1 CHR peptide have been shown to inhibit fusions between trophoblast-derived cells (Blond et al. 2000; Chang et al. 2004; Mi et al. 2000). Agents increasing cellular levels of cAMP or cAMP analogues have been shown to promote cytotrophoblast fusions in vitro (Keryer et al. 1998) and such agents are also known to elevate protein and mRNA levels of syncytin-1 in isolated cytotrophoblasts (Frendo et al. 2003). Also estradiol may regulate syncytin-1 expression (Carino et al. 2003) and the placenta-specific transcription factor GCMa interacts with two upstream sites in the HERV-W 5′-long terminal repeat and stimulates syncytin-1 transcription (Yu et al. 2002). Additionally, experimentally induced truncations in the cytoplasmic tail of syncytin-1 increases its fusogenicity (Drewlo et al. 2006), similar to what has been observed for some virally derived Env proteins (reviewed by Kubo et al. 2003). However, if modifications in the cytoplasmic tail of syncytin-1 are of physiological importance for regulating fusogenicity has yet to be demonstrated.
A second syncytin-family member, syncytin-2, was subsequently found also to be expressed in the placenta (Blaise et al. 2003). Syncytin-2 also represents a highly conserved endogenous retroviral envelope gene and is derived from the HERV FRD sequence (Blaise et al. 2003). Immunocytochemical studies have localized syncytin-1 primarily to syncytiotrophoblasts as well as to cytotrophoblasts (Fig. 5). The exact localization is somewhat controversial (reviewed by Potgens et al. 2004), which may reflect differences in antibodies, fixation procedures and controls. A major point of concern is also the degree of cross-reactivity to the related sequences in syncytin-2, which recently was localized to a subpopulation of cytotrophoblasts (Malassiné et al. 2008). The syncytin-1 receptor protein ASCT-2 was recently localized to cytotrophoblasts (Hayward et al. 2007). More studies on the exact distribution of syncytin expression in the placenta using syncytin-1 and -2 specific antibodies seem warranted in order to exactly localize where fusions are likely to occur.

Human term placenta immunocytochemically stained with a syncytin-1 peptide antiserum. Staining occurs in the syncytiotrophoblasts, which predominate at this stage. Nuclei are lightly counterstained with haematoxylin
Trophoblast cell fusions are, however, not unique to the primate placenta. Thus, also in the mouse placenta, trophoblasts fuse to form syncytiotrophoblasts but mice do, of course, not express HERV sequences. Amazingly, an in-silico search of the mouse genome unraveled the existence of two murine endogenous retroviral (MERV) Env genes, labeled syncytin-A and -B (Dupresssoir et al. 2005). These genes were also fusogenic and orthologous genes were present in additional species of muridae (rats, gerbils, voles and hamsters) and appear to have entered the rodent lineage some 20 million years ago (Dupresssoir et al. 2005). They were expressed in the placenta and at least syncytin-A has been shown to be involved in the formation of syncytiotrophoblasts in mice (Gong et al. 2007). This represents an amazing example of parallel acquisition of retroviral genes of importance to reproduction in primates and rodents and poses the question whether also other species may have acquired similar viral genes of importance to cell fusions. The ability of syncytins to induce cell fusions may not be their only physiologic role. Thus, recent studies have shown that while syncytin-2 and syncytin-B also possess immunosuppressive activity, syncytin-1 and syncytin-A do not (Mangeney et al. 2007). Syncytin-1 expression was originally detected in the placenta and testis (Blond et al. 2000; Mi et al. 2000) but subsequent studies have revealed the presence of syncytin-1 also in the brain (Antony et al. 2004) and in breast, colon and endometrial cancers (Bjerregaard et al. 2006; Strick et al. 2007, Larsen, Talts, Andersen, Bjerregaard and Larsson: work in progress). Syncytin-1 may also regulate production of inflammatory mediators (Antony et al. 2004).
A number of additional molecules have been shown to be important to cell fusions. In fact, in all systems studied so far, from mating yeast to man, a bewildering array of mechanisms have been identified (see Chen and Olson 2005; Chen et al. 2007; Oren-Suissa and Podbilewicz 2007 for recent reviews). Molecules potentially involved in mammalian cell fusions include ADAM (a disintegrin and metalloproteinase domain) 12 (meltrin alpha), which has been associated with myoblast and osteoclast cell fusions (Abe et al. 1999; Galliano et al. 2000; Gilpin et al. 1998; Yagami-Hiromasa et al. 1995). In addition, ADAMs 1 and 2 (fertilins) may be involved in sperm–oocyte fusions, but do not seem indispensable for this function and, in man, the fertilin alpha gene is dysfunctional (Chen and Olson 2005; Cho et al. 1997, 1998, Evans et al. 1998; Jury et al. 1997, 1998). In contrast, the tetraspanin protein CD9 is needed for sperm–oocyte fusions (Le Naour et al. 2000). CD9 is also expressed by BeWo trophoblast tumor (choriocarcinoma) cells and has been linked both to BeWo invasiveness and to invasion during mouse embryo implantation (Hirano et al. 1999; Liu et al. 2006). CD9 has, together with another tetraspanin family member, CD81, also been linked to myoblast fusion and myotube maintenance (Tachibana and Hemler 1999). Moreover, antibodies to either CD9 or CD81 have been shown to block fusions induced by Mason—Pfizer monkey virus—a D-type retrovirus (Duelli et al. 2005). Tetraspanins are known to organize other proteins into membrane microdomains and may link to the actin cytoskeleton via EWI (Glu-Trp-Ile) and ERM (ezrin–radixin–moesin) proteins (Hemler 2003; Sala-Valdés et al. 2006). Possibly, their role as organizers of other proteins into microdomains may play a role in their involvement with cell fusions (Zivyat et al. 2006). They do not express characteristics of fusogenic proteins like the syncytins, SNAREs and class I viral envelope proteins, but are coexpressed with syncytin-1, at least in BeWo cells. A macrophage fusion receptor (MFR, SIRPalpha), resembling CD4–the cell surface receptor for HIV—has been identified in macrophages (Saginario et al. 1995, 1998; van den Berg et al. 2005). MFR, which belongs to the immunoglobulin superfamily, binds another member of this family, CD47. CD47 is also structurally related to proteins expressed by Vaccinia and Variola viruses (Chen et al. 2007). Whereas expression of MFR is restricted to myeloid cells and neurons, CD47 is ubiquitously expressed. MFR is transiently induced in macrophages at the onset of fusion while CD47 expression is constant. It has been hypothesized that CD47 initially binds to a long form of MFR to secure recognition and then switches to bind a shorter form to bring the plasma membranes closer (5–10 nm) for fusion (Vignery 2005). Additionally, or alternatively, CD47 may promote calcium entry by forming a membrane pore (reviewed in Chen et al. 2007). Also the hyaluronan receptor, CD44, is induced transiently when macrophages start to fuse. CD44 and MFR are subsequently cleaved by proteases during fusion. As mentioned above, CD9 and CD81 may also contribute to macrophage fusion. Additionally, DC-STAMP seems to be required for macrophage fusions. Mice lacking DC-STAMP are osteopetrotic and lack multinucleated osteoclasts and giant cells (Yagi et al. 2005). DC-STAMP is a seven-transmembrane receptor, somewhat similar to the HIV co-receptor CXCR4, but a ligand has yet to be identified.
These and many more molecules, including integrins, vacuolar ATPase and receptors and their ligands, as well as different signaling intermediates have been associated with cell fusions. With the exception of the syncytins, most of the mammalian molecules so far studied do not fulfill strict criteria for fusogens and several are dispensable for fusions (reviewed by Chen and Olson 2005; Chen et al. 2007; Oren-Suissa and Podbilewicz 2007). However, this may reflect molecular redundancy and does not definitely exclude that these molecules may participate in fusions. It is noteworthy that, in both man and mouse, two different syncytins are expressed and are both fusogenic. It is possible that, in the mammalian system, fusion requires a flotilla of molecules organized into membrane microdomains by proteins like CD9, encompassing receptors/ligands, signaling entities, proteases and fusogens of retroviral origin that are capable of forming alpha-helical bundles, which bring membranes closer. Syncytins and related retroviral envelope sequences may, in this connection, function both as fusogens and receptor ligands. However, it seems unlikely that as irreversible an event as a cell–cell fusion should depend upon a single receptor–ligand interaction. Thus, both facilitatory and inhibitory factors are expected to be part of the flotilla.
Cancer cell fusions
Studies by Mortensen et al. (2004) documented that human breast cancer cells fused with endothelial cells in culture. Stimulated by studies showing that syncytin-1 was involved in cytotrophoblast cell fusions (Blond et al. 2000; Mi et al. 2000), we examined whether a similar mechanism could account for cancer–endothelial cell fusions. Expression of syncytin-1 was documented in the breast cancer cell lines examined (MCF-7 and MDA-MB-231 cells; Bjerregaard et al. 2006, SK-BR-3 cells: Talts, Bjerregaard and Larsson: unpublished data). Moreover, we found that both tumor cells and endothelial cells expressed the syncytin-1 receptor ASCT-2. Use of phosphorthioate-protected syncytin-1 antisense oligonucleotides downmodulated syncytin-1 expression as measured by either quantitative RT-PCR or Western blotting and inhibited breast cancer–endothelial cell fusions, whereas a scrambled oligonucleotide control was without effect. Additionally, the syncytin-1 CHR peptide, referred to above, also inhibited the fusions whereas a control peptide was without effect (Bjerregaard et al. 2006). However, neither the antisense nor the CHR peptide experiments effected a total inhibition of cancer-endothelial cell fusions. There may be several reasons to this. Thus, the antisense oligonucleotide did not totally downmodulate syncytin-1 levels and could therefore not be expected to decrease cell fusions to zero and the lack of total inhibition by the CHR peptide could potentially be ascribed to proteolytic degradation. However, we also detected that the breast cancer cells produced syncytin-2 and cannot exclude that also this molecule contributed to the cancer–host cell fusions. Further experiments using shRNA-directed downmodulation of both syncytin-1 and -2 as well as of additional putative fusogenic retroviral sequence are now underway to test this. Nevertheless, these data provide strong evidence that syncytin-1 is involved in mediating cancer–endothelial cell fusions in vitro.
We next examined two series of human breast cancer patients for tumoral expression of syncytin-1 using a polyclonal antiserum raised to a synthetic nonapeptide derived from the syncytin-1 sequence. In addition, tumors were also screened for expression of ASCT-2 using a peptide antiserum. Preabsorption of the antisera with the corresponding peptides, but not with irrelevant peptides, abolished staining (Larsson et al. 2007b) (Fig. 6). The results showed that 38% of all breast cancer samples showed detectable staining for syncytin and that endothelial cells expressed ASCT-2 (Fig. 6). Moreover, significant expression of ASCT-2 was detected also in many tumor cells (Fig. 6). The degree of syncytin immunostaining was visually graded using coded specimens and statistical analysis showed that it correlated positively with disease-free survival of the patients (Larsson et al. 2007b). Multivariate analysis included age dichotomized at 40 years, tumor size dichotomized at 20 mm, grade and adjuvant therapy and identified syncytin expression as an independent prognostic indicator of increased disease-free survival Also when used as a continuous variable, syncytin expression emerged as a significant prognostic indicator for disease-free survival in the Cox model (P = 0.02) (Larsson et al. 2007b).

Human breast cancers immunocytochemically stained for syncytin-1 (a) or with antigen-preabsorbed antiserum (b) and for ASCT-2 (c, e) or with antigen-preabsorbed antiserum (d). Nuclei are lightly counterstained with haematoxylin. Note presence of syncytin immunoreactivity in tumor cells and of ASCT-2 immunoreactivity in endothelial cells (arrows) and in tumor cells
The involvement of syncytin-1 in tumor cell fusion events was subsequently confirmed by Strick et al. (2007), working with endometrial carcinomas. In their study, downmodulation of synctytin-1 expression also inhibited fusions between endometrial tumor cells. In agreement with findings on placental cells (vide supra), both cAMP elevating agents and estrogens upregulated syncytin-1 expression. However, only cAMP elevating agents stimulated cell fusions (Strick et al. 2007). This apparent discrepancy may possibly be ascribed to the fact that estrogen treatment upregulated TGFbeta, which interfered with syncytin-induced fusions. Thus, estrogen did, in fact, stimulate cell fusions if TGFbeta was immunoneutralized. Conversely, additions of TGFbeta 1 or 3 reduced fusions induced by cAMP-elevating agents. This effect was observed both in endometrial carcinoma cells and in trophoblast-derived cells. These results show that both cAMP and estrogens positively may regulate syncytin-1 expression in tumor cells and that the TGFbeta family may negatively regulate the fusogenic effects of syncytin-1 in both trophoblasts and cancer cells (Strick et al. 2007).
The role of syncytin-1 and cell fusions in cancer needs further study. Thus, our data show that syncytin-1 is not the only fusogenic protein expressed by breast cancer cells and the results presented by Strick et al. (2007) show that additional regulators, such as TGFbeta isoforms may be important modulators of cell fusions. So far, our breast cancer data indicate that syncytin-1 expression constitutes a positive prognostic factor. This is not the same as to say that cell fusions may be universally beneficial to cancers. First, syncytins may have additional effects within the tumor environment (Larsson et al. 2007a, b). Secondly, it seems likely that several factors within the cancer and its stroma (inactivated tumor suppressor genes, activated oncogenes, expression of fusogens and of CD9 and CD81 as well as TGFbeta) may act together to bring about a tumor profile that may be as diverse as the one demonstrated by the cell fusion experiments referred to above. Interestingly, however, expression of CD9 has been analyzed in a number of tumors and seems, like syncytin expression, to predict a good prognosis (Funakoshi et al. 2003; Hashida et al. 2002; Higashiyama et al. 1997; Houle et al. 2002; Miyake et al. 1995; Uchida et al. 1999). We propose that syncytins may be fusogens of importance to both trophoblast and cancer cell fusions and that they possibly also may mediate additional cell fusion events, acting in concert with other molecules with both enhancing and inhibitory regulatory effects.
References
Abe E, Mocharla H, Yamate T, Taguchi Y, Manolagas SC (1999) Meltrin-alpha, a fusion protein involved in multinucleated giant cell and osteoclast formation. Calcif Tiss Int 64:508–515
Aichel O (1911) Über Zellverschmelzung mit qualitative abnormer chromosomenverteilung. In: Roux W (eds) Vorträge und Aufsätze über Entwicklungsmechanik der Organismen. Wilhelm Engelmann, Leipzig pp 1–115
Alvarez-Dolado M, Pardal R, Garcia-Verdugo JM, Fike JR, Lee HO, Pfeffer K, Lois C, Morrison SJ, Alvarez-Buylla A (2003) Fusion of bone-marrow-derived cells with Purkinje neurons, cardiomyocytes and hepatocytes. Nature 425:968–973
Andersen TL, Boissy P, Sondergaard TE, Kupsiewicz K, Plesner T, Rasmussen T, Haaber J, Kølvraa S, Delaissé JM (2007) Osteoclast nuclei of myeloma patients show chromosome translocations specific for the myeloma cell clone: a new type of cancer–host partnership? J Pathol 211:10–17
Anderson MJ, Stanbridge EJ (1993) Tumor suppressor genes studied by cell hybridization and chromosome transfer. FASEB J 7:826–833
Antony JM, van Marle G, Opii W, Butterfield DA, Mallet F, Yong VW Wallace JL, Deacon RM, Warren K, Power C (2004) Human endogenous retrovirus glycoprotein-mediated induction of redox reactants causes oligodendrocyte death and demyelination. Nature Neurosci 7:1088–1095
Barski G, Cornefert F (1962) Characteristics of “hybrid”-type clonal cell lines obtained from mixed cultures in vitro. J Natl Cancer Inst 28:801–821
Bischoff R (1994) The satellite cell and muscle regeneration. In: Engel AG (eds) Myogenesis. Franszini-Armstrong, McGraw-Hill, New York, pp 97–118
Bjerregaard B, Holck S, Christensen IJ, Larsson L-I (2006) Syncytin is involved in breast cancer–endothelial cell fusions. Cell Mol Life Sci 63:1906–1911
Blaise S, de Parseval N, Benit L, Heidmann T (2003) Genomewide screening for fusogenic human endogenous retrovirus envelopes identifies syncytin 2, a gene conserved on primate evolution. Proc Natl Acad Sci USA 100:13013–13018
Blond JL, Lavillette D, Cheynet V, Bouton O, Oriol G, Chapel-Fernandes S, Mandrand B, Mallet F, Cosset FL (2000) An envelope glycoprotein of the human endogenous retrovirus HERV-W is expressed in the human placenta and fuses cells expressing the type D mammalian retrovirus receptor. J Virol 74:3321–3329
Blumenthal R, Clague MJ, Durell SR, Epand RM (2003) Membrane fusion. Chem Rev 103:53–69
Busund LTR, Killie MK, Bartnes K, Olsen R Seljelid R (2002) Spontaneous hybridization of macrophages and Meth A sarcoma cells. Int J Cancer 98:573–581
Busund LTR, Killie MK, Bartnes K Seljelid R (2003) Spontaneously formed tumorigenic hybrids of Meth A sarcoma cells and macrophages in vivo. Int J Cancer 106:153–159
Carino C, Hirchenhain J, Krussel JS, Schanz A, Hess A, Larrea F (2003) Estradiol stimulation increases syncytin expression in normal trophoblasts in vitro. Placenta 24:A58
Chakraborty A, Lazova R, Davies S, Backvall H, Ponten F, Brash D, Pawelek J ((2004) Donor DNA in a renal cell carcinoma metastasis from a bone marrow transplant recipient. Bone Marrow Transplant 34:183–186
Chakraborty AK, Pawelek J, Ikeda Y, Miyoshi E, Kolesnikova N, Funasaka Y, Ichihashi M, Taniguchi N (2001) Fusion hybrids with macrophage and melanoma cells up-regulate N-acetylglucosaminyltransferase V, beta1-6 branching, and metastasis. Cell Growth Differ 12:623–30
Chakraborty AK, Sodi S, Rachkovsky M, Kolesnikova N, Platt JT, Bolognia JL Pawelek JM (2000) A spontaneous murine melanoma lung metastasis comprised of host × tumor hybrids. Cancer Res 60:2512–2519
Chang C, Chen PT, Chang GD, Huang CJ, Chen H (2004) Functional characterization of the placental fusogenic membrane protein syncytin. Biol Reprod 71:1956–1962
Chen EH, Grote E, Mohler W, Vignéry A (2007) Cell–cell fusion. FEBS Lett 581:2181–2193
Chen EH, Olson EN (2005) Unveiling the mechanisms of cell–cell fusion. Science 308:369–373
Cho C, Turner L, Primakoff P, Myles DG (1997) Genomic organization of the mouse fertilin beta gene that encodes an ADAM family protein active in sperm–egg fusion. Dev Genet 20:320–328
Cho C, Bunch DO, Faure JE, Goulding EH, Eddy EM, Primakoff P, Myles DG (1998) Fertilization defects in sperm from mice lacking fertilin beta. Science 281:1857–1859
De Baetselier P, Gorelik E, Eshhar Z, Ron Y, Katzav S, Feldman M, Segal S. (1981) Metastatic properties conferred on nonmetastatic tumors by hybridization of spleen B-lymphocytes with plasmacytoma cells. J Natl Cancer Inst 67:1079–1087
de la Taille A, Chen M-W, Burchardt M, Chopin DK, Buttyan R (1999) Apoptotic conversion: evidence for exchange of genetic information between prostate cancer cells mediated by apoptosis. Cancer Res 59:5461–5463
Drewlo S, Leyting S, Kokozidou M, Mallet F, Potgens AJG (2006) C-terminal truncations of syncytin-1 (ERVWE1 envelope) that increase its fusogenicity. Biol Chem 387:1113–1120
Duelli DM, Hearn S, Myers MP, Lazebnik Y (2005) A primate virus generates transformed human cells by fusion. J Cell Biol 171:493–503
Duelli DM, Padilla_Nash HM, Berman D, Murphy KM, Ried T, Lazebnik Y (2007) A virus causes cancer by inducing massive chromosoma instability through cell fusion. Curr Biol 17:431–437
Dupressoir A, Marceau G, Vermochet C, Benit L, Kanellopoulos C, Sapin V, Heidmann T (2005) Syncytin-A and syncytin-B, two fusogenic placenta-specific murine envelope gtenes of retroviral origin conserved in Muridae. Proc Natl Acad Sci USA 102:725–730
Evans JP, Schultz RM, Kopf GS (1998) Roles of the disintegrin domains of mouse fertilins alpha and beta in fertilization. Biol Reprod 59:145–152
Fortuna MB, Dewey MJ, Furmanski P (1990) Enhanced lung colonization and tumorigenicity of fused cells isolated from primary MCA tumors. Cancer Lett 55:109–114
Frendo JL, Olivier D, Cheynet V, Blond JL, Bouton O, Vidaud M, Rabreau M, Evain-Brion D, Mallet F (2003) Direct involvement of HERV-W Env glycoprotein in human trophoblast cell fusion and differentiation. Mol Cell Biol 23:3566–3574
Funakoshi T, Tachibana I, Hoshida Y, Kimura H, Takeda Y, Kijima T, Nishino K, Goto H, Yoneda T, Kumagai T, Osaki T, Hayashi S, Aozasa K, Kawase I (2003) Expression of tetraspanins in human lung cancer cells: frequent downregulation of CD9 and its contribution to cell motility in small cell lung cancer. Oncogene 22:674–687
Galliano MF, Huet C, Frygelius J, Polgren A, Wever UM, Engvall E (2000) Binding of ADAM12, a marker of skeletal muscle regeneration, to the muscle-specific actin-binding protein, alpha-actinin-2, is required for myoblast fusion. J Biol Chem 275:13933–13939
Gilpin BJ, Loechel F, Mattei MG, Engvall E, Albrechtsen R, Wewer UM (1998) A novel secreted form of human ADAM 12 (meltrin alpha) provokes myogenesis in vivo. J Biol Chem 273:157–167
Goldenberg DM, Pavia RA (1982) In vivo horizontal oncogenesis by a human tumor in nude mice. Proc Natl Acad Sci USA 79:2389–2392
Goldenberg DM, Pavia RA, Tsao MC (1974) In vivo hybridisation of human tumour and normal hamster cells. Nature 250:649–651
Gong R, Huang L, Shi J, Luo K, Qiu G, Feng H, Tien P, Xiao G (2007) Syncytin-A mediates the formation of syncytiotrophoblast involved in mouse placental development. Cell Physiol Biochem 20:517–526
Gong R, Peng X, Kang S, Feng H, Huang J, Zhang W, Lin D, Tien P, Xiao G (2005) Structural characterization of the fusion core in syncytin, envelope protein of human endogenous retrovirus family W. Biochem Biophys Res Commun 331:1193–1200
Harris H, Miller OJ, Klein G, Worst P, Tachibana T (1969) Suppression of malignancy by cell fusion. Nature 223:363–368
Harris H (1988) The analysis of malignancy by cell fusion: the position in 1988. Cancer Res 48:3302–3306
Hashida H, Takabayashi A, Tokuhara T, Taki T, Kondo K, Kohno N, Yamaoka Y, Miyake M (2002) Integrin alpha3 expression as a prognostic factor in colon cancer: association with MRP-1/CD9 and KAI1/CD82. Int J Cancer 97:518–525
Hayward MD, Potgens AJG, Drewlo S, Kaufmann P, Rasko JEJ (2007) Distribution of human endogenous retrovirus type W receptor in normal human villous placenta. Pathol 39:406–412
Hemler ME (2003) Tetraspanin proteins mediate cellular penetration, invasion and fusion events and define a novel type of membrane microdomain. Annu Rev Cell Dev Biol 19:397–422
Higashiyama M, Doi O, Kodama K, Yokouchi H, Adachi M, Huang CL, Taki T, Kasugai T, Ishiguro S, Nakamori S, Miyake M (1997) Immunohistochemically detected expression of motility-related protein-1 (MRP-1/CD9) in lung adenocarcinoma, its relation to prognosis. Int J Cancer 74:205–211
Hirano T, Higuchi T, Katsuragawa H, Inoue T, Kataoka N, Park KR, Ueda M, Maeda M, Fujiwara H, Fuji S (1999) CD9 is involved in invasion of human trophoblast-like choriocarcinoma cell line, BeWo cells. Mol Human Reprod 5:168–174
Holmgren L, Szeles A, Rajnavolgyi E, Folkman J, Klein G, Ernberg I, Falk KI (1999) Horizontal transfer of DNA by the uptake of apoptotic bodies. Blood 93:3956–3963
Houle CD, Ding XY, Foley JF, Afshari CA, Barrett JC, Davis BJ (2002) Loss of expression and altered localization of KAI1 and CD9 protein are associated with epithelial ovarian cancer progression. Gynecol Oncol 86:69–78
Jahn R, Lang T, Sudhof TC (2003) Membrane fusion. Cell 112:519–533
Jury JA, Frayne J, Hall L (1997) The human fertilin alpha gene is non-functional: implications for its proposed role in fertilization. Biochem J 321:577–581
Jury JA, Frayne J, Hall L (1998) Sequence analysis of a variety of primate fertilin alpha genes: evidence for non-functional genes in the gorilla and man. Mol Reprod Dev 51:92–97
Kerbel RS, Lagarde AE, Dennis JW, Donaghue TP (1983) Spontaneous fusion between normal host and tumor cells: possible contribution to tumor progression and metastasis studied with a lectin-resistant mutant tumor. Mol Cell Biol 3:523–538
Keryer G, Alsat E, Taskén K, Evain-Brion D (1998) Cyclic AMP-dependent protein kinasees and human trophoblast cell differentiation in vitro. J Cell Sci 11:995–1004
Kohler G, Milstein C (1975) Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 256:495–497
Kubo Y, Ishimoto A, Amanuma H (2003) Genistein, a protein tyrosine kinase inhibitor, suppresses the fusogenicity of Moloney murine leukemia virus envelope protein in XC cells. Arch Virol 148:1899–1914
Larizza L, Schirrmacher V, Graf L, Pfluger E, Peres-Martinez M, Stohr M (1984a) Suggestive evidence that the highly metastatic variant ESb of the T-cell lymphoma Eb is derived from spontaneous fusion with a host macrophage. Int J Cancer 34:699–702
Larizza L, Schirrmacher V, Pfluger E (1984b) Acquisition of high metastatic capacity after in vitro fusion of a nonmetastatic tumor line with a bone marrow-derived macrophage. J Exp Med 160:1579–1584
Larsson L-I, Bjerregaard B, Wulf-Andersen L, Talts J F (2007a) Syncytin and cancer cell fusions. World Sci J 7:1193–1197
Larsson L-I, Holck S, Christensen IJ (2007b) Prognostic role of syncytin expression in breast cancer. Human Pathol 38:726–731
Lavillette D, Marin M, Ruggieri A, Mallet F, Cosset FL, Kabat D (2000) The envelope glycoprotein of human endogenous retrovirus type W uses a divergent family of amino acid transporters/cell surface receptors. J Virol 76:6442–6452
Le Naour F, Rubinstein E, Jasmin C, Prenant M, Boucheix C (2000) Severely reduced female fertility in CD9-deficient mice. Science 287:319–321
Liu WM, Cao YJ, Yang YJ, Li J, Hu Z, Duan EK (2006) Tetraspanin CD9 regulates invasion during mouse embryo implantation. J Mol Endocrinol 36:121–130
Malassiné A, Frendo JL, Blaise S, Handschuh K, Gerbaud P, Tsatsaris V, Heidmann T, Evain-Brion D (2008) Human endogenous retrovirus-FRD envelope protein (syncytin 2) expression in normal and trisomy 21-affected placenta. Retrovirology 5:1–10
Mangeney M, Renard M, Schlecht-Louf G, Bouallaga I, Heidmann O, Letzelter C, Richaud A, Ducos B, Heidmann T (2007) Placental syncytins: genetic disjunction between the fusogenic and immunosuppressive activity of retroviral envelope proteins. Proc Natl Acad Sci USA 104:20534–20539
Mi S, Lee X, Li X, Veldman G M, Finnerty H, Racie L, LaVallie E, Tang XY, Edouard P, Howes S, Keith Jr JC, McCoy JM (2000) Syncytin is a captive retroviral envelope protein involved in human placental morphogenesis. Nature 403:785–789
Miyake M, Nakano K, Ieki Y, Adachi M, Huang CL, Itoi S, Koh T, Taki T (1995) Motility related protein 1 (MRP-1/CD9) expression: inverse correlation with metastases in breast cancer. Cancer Res 55:4127–4131
Mortensen K, Lichtenberg J, Thomsen PD, Larsson LI (2004) Spontaneous fusion between cancer cells and endothelial cells. Cell Mol Life Sci 61:2125–2131
Ogle BM, Cascalho M, Platt JF (2005) Biological implications of cell fusion. Nat Rev Mol Cell Biol 6:567–575
Oren-Suissa M, Podbilewicz B (2007) Cell fusion during development. Trends Cell Biol 17:537–546
Overholtzer M, Mailleux AA, Mouneimne G, Normand G, Schnitt SJ, King RW, Cibas ES, Brugge JS (2007) A nonapoptotic cell death process, entosis, that occurs by cell-in-cell invasion. Cell 131:966–979
Pawelek JM (2000) Tumour cell hybridization and metastasis revisited. Melanoma Res 10:507–514
Pawelek JM (2005) Tumor–cell fusion as a source of myeloid traits in cancer. Lancet Oncol 6:988–993
Potgens AJ, Drewlo S, Kokozidou M, Kaufmann P (2004) Syncytin: the major regulator of trophoblast fusion? Recent developments and hypotheses on its action. Hum Reprod Update 10:487–496
Rachkovsky M, Sodi S, Chakraborty A, Avissar Y, Bolognia J, McNiff JM, Platt J, Bermudes D, Pawelek J (1998) Melanoma × macrophage hybrids with enhanced metastatic potential. Clin Exp Metastasis 16:299–312
Rizvi AZ, Swain JR, Davies PS, Bailey AS, Decker AD, Willenbring H, Grompe M, Fleming WH, Wong MH (2006) Bone marrow-derived cells fuse with normal and transformed intestinal stem cells. Proc Natl Acad Sci USA 103:6321–6325
Saginario C, Qian HY, Vignéry A (1995) Identification of an inducible surface molecule specific to fusing macrophages. Proc Natl Acad Sci USA 92:12210–12214
Saginario C, Sterling H, Beckers C, Kobayashi RJ, Solimena M, Ullu E, Vignéry A (1998) MFR a putative receptor mediating the fusion of macrophages. Mol Cell Biol 18:6213–6223
Sala-Valdés M, Ursa A, Charrin S, Rubinstein E, Hemler ME, Sánchez-Madrid F, Yánez-Mo M (2006) EWI-2 and EWI-F link the tetraspanin web to the actin cytoskeleton through direct association with ezrin–radixin–moesin proteins. J Biol Chem 281:19665–19675
Stanbridge EJ (1976) Suppression of malignancy in human cells. Nature 260:17–20
Streubel B, Chott A, Huber D, Exner M, Jager U, Wagner O, Schwarzinger I (2004) Lymphoma-specific genetic abberations in microvascular endothelial cells in B-cell lymphomas. New Engl J Med 351:250–259
Strick R, Ackermann S, Langbein M, Swiatek J, Schubert SW, Hashemolhosseini S, Koscheck T, Fasching PA, Schild RL, Beckmann MW, Strissel PL (2007) Proliferation and cell–cell fusion of endometrial carcinoma are induced by the human endogenous retroviral syncytin-1 and regulated by TGF-beta. J Mol Med 85:23–38
Tachibana I, Hemler ME (1999) Role of transmembrane 4 superfamily (TM4SF) proteins CD9 and CD81 in muscle cell fusion and myotube maintenance. J Cell Biol 146:893–904
Uchida S, Shimada Y, Watanabe G, Li ZG, Hong T, Miyake M, Imamura M (1999) Motility-related protein (MRP-1/CD9) and KAI1/CD82 expression inversely correlate with lymph node metastasis in oesophageal squamous cell carcinoma. Br J Cancer 79:1168–1173
Van den Berg TK, van Beek EM, Buhring HJ, Colonna M, Hamaguchi M, Howard CJ, Kasuga M, Liu Y, Matozaki T, Neel BG, Parkos CA, Sano S, Vignéry A, Vivier E, Wright M, Zawatsky R, Barclay RN (2005) A nomenclature for signal regulatory protein family members. J Immunol 175:7788–7789
Vassilopoulos G, Wang PR, Russell DW (2003) Transplanted bone marrow regenerates liver by cell fusion. Nature 422:901–904
Vignery A (2005) Macrophage fusion: are somatic and cancer cells possible partners? Trends Cell Biol 15:188–193
Wakeling WF, Greetham J, Bennett DC (1994) Efficient spontaneous fusion between some co-cultured cells, especially murine melanoma cells. Cell Biol Int 18:207–210
Wiener F, Fenyo EM, Klein G (1974a) Tumor–host cell hybrids in radiochimeras. Proc Natl Acad Sci 71:148–152
Wang X, Willenbring H, Akkari Y, Torimaru Y, Foster M, Al-Dhalimy M, Lagasse E, Finegold M, Olson S, Grompe M (2003) Cell fusion is the principal source of bone marrow-derived hepatocytes. Nature 422:897–901
Wiener F, Klein G, Harris H (1974b) The analysis of malignancy by cell fusion. V. Further evidence of the ability of normal diploid cells to suppress malignancy. J Cell Sci 15:177–83
Weber T, Zemelman BV, McNew JA, Westermann B, Gmachl M, Parlati F, Söllner TH, Rothman JE. (1998) SNAREpins: minimal machinery for membrane fusion. Cell 92:759–772
Yagami-Hiromasa T, Sato T, Kurisaki T, Kamijo K, Nabeshima Y, Fujisawa-Sehara A (1995) A metalloproteinase-disintegrin participating in myoblast fusion. Nature 377:652–656
Yagi M, Miyamoto T, Sawatani Y, Iwamoto K, Hosogane N, Fujita N, Morita K, Ninomiya K, Suzuki T, Miyamoto K, Oike Y, Takeya M, Toyama Y, Suda T (2005) DC-STAMP is essential for cell–cell fusion in osteoclasts and foreign body giant cells. J Exp Med 202:345–351
Yilmaz Y, Lazova R, Qumsiyeh M, Cooper D, Pawelek J (2005) Donor Y chromosome in renal carcinoma cells of a female BMT recipient: visualization of putative BMT-tumor hybrids by FISH. Bone Marrow Transplant 35:1021–1024
Yu C, Shen K, Lin M, Chen P, Lin C, Chang GD, Chen H (2002) GCMa regulates the syncytin-mediated trophoblast fusion. J Biol Chem 277:50062–50068
Zivyat A, Rubinstein E, Monier-Gavelle F, Barraud V, Kulski O, Prenant M, Boucheix C, Bomsel M, Wolf JP (2006) CD9 controls the formation of clusters that contain tetraspanins and the integrin alpha6beta1, which are involved in human and mouse gamete fusion. J Cell Sci 119:416–424
Acknowledgments
Work by the authors reported herein was supported by the Lundbeck Foundation, the Danish Medical Research Council and the Danish Cancer Society.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Larsson, LI., Bjerregaard, B. & Talts, J.F. Cell fusions in mammals. Histochem Cell Biol 129, 551–561 (2008). https://doi.org/10.1007/s00418-008-0411-1
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00418-008-0411-1