
Abstract
Background
Proliferation and migration of retinal endothelial cells (REC) are associated with the development of proliferative diabetic retinopathy. REC proliferation is stimulated by isoforms of vascular endothelial growth factor-A (i.e., VEGF121 and VEGF165), basic fibroblast growth factor (bFGF), and insulin-like growth factor (IGF-1) of which VEGF165 also enhances migration of REC. Effects induced by VEGF-A can be blocked with ranibizumab, a VEGF-binding Fab fragment used in therapy of diabetic macular edema. In this study, we investigated potential angiogenic effects of placental growth factors (PlGF-1, PlGF-2) as other members of the VEGF family and whether the primary action of VEGF165 is modulated in the presence of bFGF, IGF-1 and PlGF-1/-2. We also studied how effects of growth factor combinations can be attenuated with ranibizumab.
Methods
Effects of single growth factors or their combinations on proliferation and migration of immortalized bovine retinal endothelial cells (iBREC) were studied with or without ranibizumab or the inhibitor of VEGF receptors KRN951.
Results
Proliferation of iBREC was significantly stimulated by 1–100 ng/ml PlGF-1 or PlGF-2, but additive effects were not observed with various combinations of the tested growth factors. Ranibizumab neutralized VEGF’s effect on proliferation but was not effective when the other growth factors were used in combination with VEGF. bFGF and IGF-1 but not PlGF-1 or PlGF-2 stimulated iBREC migration as single agents, and they further enhanced VEGF-induced migration. The effects of such growth factor combinations including VEGF on migration were efficiently blocked by targeting only VEGF with ranibizumab. Migration induced by VEGF plus bFGF and IGF-1 was also almost completely inhibited by KRN951 interfering with VEGF receptor signalling.
Conclusions
Migration but not proliferation of iBREC induced by combinations of bFGF, IGF-1, PlGF-1 or PlGF-2 together with VEGF is efficiently suppressed by ranibizumab. VEGF-mediated signalling through VEGFR2 seems to control REC migration dominantly in the presence of other growth factors.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Pathogenesis of diabetic retinopathy (DR) is associated with deregulated expression of various growth factors of which elevated levels were found in the vitreous fluid of DR patients [1–4]. Appearance of diabetic macular edema (DME), observed at any stage of DR, correlates with vascular endothelial growth factor-A (VEGF) induced permeability of retinal endothelial cells (REC) [5]. DME can be treated with the recently approved VEGF-binding Fab-fragment ranibizumab or the recombinant protein VEGF-trap/aflibercept binding also to placenta growth factor (PlGF) [6–10]. VEGF inhibition might also be beneficial to patients with proliferative DR (PDR), which can be concluded from evidence indicating that proliferation and migration of REC associated with neovascularization is driven by angiogenic growth factors: Both isoforms VEGF165 and VEGF121, as well as PlGF-1 and basic fibroblast growth factor (bFGF) or insulin-like growth factor-1 (IGF-1), stimulate proliferation of primary and immortalized REC of bovine or human origin over a wide range of concentrations including physiological conditions [11–16]. Migration of immortalized and primary bovine REC ((i)BREC) is induced by VEGF165 but not by VEGF121 [11, 14]. VEGF is only very weakly expressed by immortalized or primary BREC, but its secretion can be slightly enhanced with bFGF together with IGF-1 [17, 18]. BREC also express PlGF-1 which can be stimulated by binding of VEGF to VEGF receptor (VEGFR) 2 [19, 20]. In addition to VEGFR2, VEGFR1 and neuropilin 1 (NRP-1) are REC-expressed receptors that are activated by members of the VEGF family [17, 21]. VEGF165 and VEGF121 efficiently trigger signalling through VEGFR1 and VEGFR2 [22]. NRP-1 is a potent receptor for VEGF165, and is only weakly activated by VEGF121 [23, 24]. Both isoforms PlGF-1 and PlGF-2 bind to VEGFR1, but PlGF-2 also to NRP-1 [25–27].
Based on this experimental evidence and clinical observations, we studied the effects on proliferation and migration of iBREC (and their potential inhibition by ranibizumab) of combinations of involved growth factors (VEGF121, PlGF-1, PlGF-2, bFGF, and IGF-1) in concert with VEGF165. The well-established model of iBREC was used in view of its distinct advantages over primary cells or rodent models: compared to primary cells, it is a distinct advantage of iBREC that these are free of contaminating cells of other types. Although iBREC are of bovine origin, they behave like primary human retinal EC (HREC): their proliferation can be stimulated similarly with VEGF-A, IGF-1 or bFGF [13–16]. Involved bovine proteins also show a high similarity to their human counterparts. Our systematic approach including all candidate factors was chosen to reveal their relative contributions to DR pathophysiology with immediate consequences for therapies targeting VEGF.
Materials and methods
Reagents
Recombinant human growth factors bFGF, IGF-1, PlGF-1 (all E.coli-expressed) and rhVEGF165 (SF21-expressed) were purchased from R&D Systems (Wiesbaden, Germany), rhVEGF121 (SF21-expressed) from Calbiochem (Merck, Darmstadt, Germany), and PlGF-2 (E.coli-expressed) from PeproTech (Hamburg, Germany). Recombinant viral VEGF-E binding to VEGFR2 and NRP, was from Abcam (Cambrigde, UK) [28]. The modified Fab fragment ranibizumab (Lucentis, 10 mg/ml) of a humanized VEGF-binding antibody was a gift from Novartis Pharma GmbH (Nuremberg, Germany) [6]. N-(2-Chloro-4-((6,7-dimethoxy-4-quinolyl)oxy)phenyl)-N′-(5-methyl-3-isoxazolyl)urea (KRN951), a tyrosine kinase inhibitor specific for VEGFR1 and VEGFR2, was from Calbiochem [29].
Cell cultivation
Telomerase-immortalized microvascular endothelial cells from bovine retina (iBREC) were cultivated in endothelial cell growth medium (ECGM hHC; Promocell, Heidelberg, Germany) supplemented with 0.4 % endothelial cell growth supplement/H (ECGS/H), 10 ng/ml epidermal growth factor, 2.8 μM hydrocortisone, and 5 % fetal calf serum (FCS) as previously described [13, 14]. Cells used in the experiments had a history of 20–40 passages from the stage of primary culture. Their stable expression of marker proteins specific for endothelial cells (i.e., von Willebrand factor, vascular endothelial cadherin, claudins, VEGF receptors) had been confirmed [13, 14, 17].
Cell proliferation assay
In a 96-well plate, 5 × 103 iBREC in ECGM-nHC (like ECGM-hHC but with 103 nM hydrocortisone) were allowed to attach to the fibronectin-coated (50 μg/ml; BD Biosciences, Heidelberg, Germany) bottom of a cavity overnight. The medium was then changed to EBGM-nHC (same as ECGM-nHC but 0.25 % FCS). After cultivation for 24 h, cells were exposed to growth factors VEGF165, VEGF121, VEGF-E, PlGF-1, PlGF-2, bFGF, or IGF-1, applied as single agents or in various combinations at 10 ng/ml each, and 100 μg/ml ranibizumab in serum-free medium for 24 h. Enzymatic conversion of WST-1 (Roche Diagnostics, Mannheim, Germany), indicative of proliferating cells, was then measured according to the manufacturer’s protocol in 16 replicate wells [14]. Values were normalized in relation to those obtained with control cells not treated with effectors. Results were shown only of experiments in which iBREC were treated with growth factors for up to 24 h, because additional effects were not observed during prolonged exposure.
Cell migration assay
Transmembrane cell migration assays were performed in a modified Boyden chamber consisting of 12-well cell culture plates and inserts with a porous membrane (pore size 8.0 μm, ∅1 cm; BD Biosciences). Their lower compartments were filled with EBGM-nHC without ECGS/H and FCS but containing 5 μg/ml fibronectin, growth factors (VEGF165, VEGF121, VEGF-E, PlGF-1, PlGF-2, bFGF or IGF-1, applied as single agents or in various combinations at 25 ng/ml each) and/or 60 μg/ml ranibizumab. Inserted membranes were initially incubated for 1 h at 37 °C before 400 μl EBGM-nHC (without ECGS/H and FCS) and a suspension of 105 iBREC in 100 μl of this medium were added subsequently to the upper compartment. After 20 h at 37 °C, migration of cells through the pores of the membrane was assessed as described [14]. Cells remaining on top of the membrane were scraped off, and cells which had migrated to the lower surface of the membrane were fixed in methanol at −20 °C for 15 min. Nuclei of migrated cells were stained by mounting the membrane in VectaShield-DAPI, and visualized by fluorescence microscopy. Cells were counted in triplicate wells in four randomly chosen microscopic fields. Values were normalised to those obtained with control cells not treated with effectors. To investigate specific inhibition of VEGF receptors, 10 nM KRN951 dissolved in DMSO (or only the solvent in control experiments) was added to the medium in both compartments.
General considerations and statistical analyses
Controls were treated in exactly the same way with medium lacking the effectors. All experiments were repeated several times, and in each experiment data were collected from multiple replicates. The Mann–Whitney U test was used to compare two sets of experimental data and differences resulting in p-values below 0.05 were considered significant. To compare several groups representing effector-treated cells, also the Kruskal–Wallis test was used. Shown results represent several independent experiments and are visualized as conventional box-whiskers diagrams, with the middle line representing the mean and the boundaries of the boxes the 75 % and 25 % percentiles, or the mean and standard deviations are provided.
Results
Effects of different growth factors were studied under experimental conditions which were defined by important previous observations: The concentrations of growth factors (10 to 25 ng/ml), in a range similar to that measured in the vitreous fluid of DR patients, were found to be sufficient for specific and strong stimulation of proliferation or migration of REC [1–4, 11–16]. Specific inhibition of VEGF-induced iBREC proliferation or migration without affecting basal processes was observed with (therapeutically achievable) 100 or 60 μg/ml ranibizumab respectively [7, 14].
Proliferation stimulated by VEGF in combination with other growth factors is not inhibited by ranibizumab
In this study we focused on other members of the VEGF family, PlGF-1 and PlGF-2 as well as VEGF-E which acts mainly through the receptors VEGFR2 and NRP-1. In addition to potential stimulation of iBREC proliferation, possible additive effects of the growth factors were investigated. All members of the VEGF family significantly stimulated proliferation to a similar extent at concentrations between 1 and 100 ng/ml after incubation for 24 h (Fig. 1). Proliferation was also enhanced by exposure of iBREC to various growth factor combinations applied at moderate concentrations of 10 ng/ml each (Table 1). VEGF-E showed a similarly strong effect at all concentrations tested, whereas maximum stimulation was seen below the highest concentrations with PlGF-1 (at 1 and 10 ng/ml) or PlGF-2 (at 1 ng/ml; Fig. 1). Additive effects of growth factor combinations were not observed (Table 1). To investigate whether VEGF inhibition is sufficient to block surplus iBREC proliferation caused by different growth factors, experiments were also performed in the presence of 100 μg/ml ranibizumab which binds all isoforms of VEGF-A. When single growth factors were used, ranibizumab only inhibited proliferation of iBREC induced by VEGF165 or VEGF121, but not by PlGF-1/2, bFGF or IGF-1, confirming the specificity of the Fab-fragment ([14], data not shown). Accordingly, substantial inhibition of proliferation was not achieved by treatment with ranibizumab when cells were exposed to combinations of growth factors including VEGF (Table 1).

VEGF-E, PlGF-1 and PlGF-2 stimulate proliferation of iBREC. Serum-starved iBREC were incubated with 1 to 100 ng/ml of VEGF-E (a), PlGF-1 (b) or PlGF-2 (c), and proliferation was measured as described in Materials and Methods
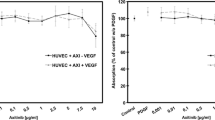
PlGF does not induce migration of iBREC
We have already demonstrated that migration of iBREC is stimulated by 10 to 100 ng/ml VEGF165 but not by VEGF121, and that VEGF-induced migration is specifically and completely inhibited by 60 μg/ml ranibizumab [14]. Similar to their reported effect on primary BREC, PlGF-1 or PlGF-2 failed to stimulate iBREC migration (Fig. 2a) [11]. Interestingly, migration stimulated by VEGF165 is further enhanced in the presence of the other growth factors, despite their inactivity as single agents. The impact of viral VEGF-E was studied because this protein activates only the VEGF receptors VEGFR2 and NRP-1, which seems to be sufficient to induce migration of iBREC, and VEGF-E was slightly more potent than VEGF165 (Fig. 2a).

PlGFs do not promote migration of iBREC. Migration of iBREC over 24 h towards fibronectin and VEGF165, VEGF121, PlGF-1, PlGF-2 or VEGF-E (25 ng/ml each) as single agents or in combination was measured by a modified Boyden Chamber assay in the absence (a) or presence (b) of 60 μg/ml ranibizumab. a VEGF165 and viral VEGF-E, but not PlGF-1 or PlGF-2 significantly stimulated migration. b Migration induced by treatment with a mixture of VEGF165, VEGF121, PlGF-1, and PlGF-2 was strongly inhibited by ranibizumab. GF growth factor mix containing VEGF165, VEGF121, PlGF-1, and PlGF-2
Considering the key role of VEGF165 in stimulation of iBREC migration, we hypothesized that its inhibition with ranibizumab might normalize migration even in the presence of PlGFs. Indeed, 60 μg/ml ranibizumab completely blocked effects on iBREC migration under these conditions (Fig. 2b).
bFGF and IGF-1 enhance migration of iBREC
We also investigated whether bFGF and IGF-1, angiogenic growth factors not belonging to the VEGF family, enhanced migration of iBREC. Migration of iBREC was strongly stimulated by bFGF and weaker by IGF-1, but additive effects of both factors were not observed (Fig. 3). As expected for a VEGF-binding Fab fragment, bFGF-stimulated migration was not inhibited by ranibizumab. It did, however, significantly reduce iBREC migration induced by IGF-1 (Fig. 3).

bFGF and IGF-1 enhance iBREC migration. Migration of iBREC towards fibronectin and bFGF and/or IGF-1 (25 ng/ml each) in the absence or presence of 60 μg/ml ranibizumab was measured by a modified Boyden Chamber assay. Strong stimulation of migration by bFGF was not blocked by ranibizumab, whereas the IGF-1-induced effect was inhibited
Migration stimulated by VEGF in combination with other growth factors is efficiently inhibited by ranibizumab
Migration of iBREC was measured in the presence of various combinations of the six growth factors used in this study to investigate potential additive effects of their concerted actions. bFGF but not IGF-1 significantly enhanced the stimulation of VEGF165 on iBREC migration. Further addition of IGF-1, VEGF121, and/or PlGFs did not amplify the impact of VEGF165 together with bFGF (Fig. 4a).

Migration stimulated by growth factor combinations is inhibited by ranibizumab. Migration of iBREC over 24 h towards fibronectin and VEGF165, VEGF121, PlGF-1, PlGF-2, bFGF and IGF-1 (25 ng/ml each) in various combinations was measured by a modified Boyden Chamber assay without (a) or with (b) 60 μg/ml ranibizumab. a VEGF in combination with either bFGF plus IGF-1 or PlGFs stimulated iBREC migration significantly stronger than VEGF165 alone. b Migration induced by combination of all growth factors including VEGF165 was strongly inhibited by ranibizumab. GF growth factors
Co-stimulation of migration with VEGF165 and bFGF was partly inhibited with ranibizumab targeting the VEGF-A isoforms, whereas complete inhibition by ranibizumab was seen when VEGF165 was supplemented with IGF-1 (Fig. 4b). In addition, migration induced by the collective action of all growth factors tested was almost completely restrained by ranibizumab (Fig. 4b). Even co-stimulation with bFGF and IGF-1 in the presence of VEGF121 (196 % ± 30 % relative to controls), which is inactive as a single factor, was suppressed by ranibizumab to basal migration (91 % ± 50 % relative to controls; all differences significant with p < 0.0003).
Specific blocking of VEGFR1/R2 efficiently prevents migration stimulated by combinations of VEGF with other growth factors
Our observation that VEGF-inhibition efficiently blocked iBREC migration stimulated by combined action of bFGF, IGF-1 and VEGF165 suggested a dominant role of VEGF165 which activates signalling through its receptors VEGFR1, -R2, und NRP-1. Therefore, we investigated whether blocking the tyrosine kinase activities of VEGFR1 and -R2 with the specific inhibitor KRN951 is a sufficient alternative to inhibit migration under these conditions (Fig. 5). This was confirmed by experiments showing that KRN951 almost completely inhibited migration stimulated by VEGF165 plus bFGF and IGF-1. All our results support a dominant role of VEGF signalling through the VEGF receptors in stimulation of iBREC migration.

Specific interference with VEGFR1/R2 signalling blocks stimulated iBREC migration. Migration of iBREC over 24 h towards fibronectin and growth factors (VEGF165 or VEGF165 plus bFGF and IGF-1) was measured by a modified Boyden Chamber assay with or without 10 nM KRN951. This VEGFR inhibitor almost completely inhibited migration induced by the three factors. GF growth factors
Discussion
We investigated the effects of a collection of growth factors potentially involved in the control of proliferation and migration in retinal endothelial cells, the key processes in PDR-associated neovascularization. To evaluate the potential of VEGF inhibition to counteract neovascularization even in the presence of other stimulating factors, VEGF-binding ranibizumab was included in the experiments.
All six growth factors tested stimulated proliferation of iBREC, but only VEGF165, bFGF, and IGF-1 also enhanced migration (see also Table 2). That PlGFs only stimulated proliferation was also observed in experiments with primary BREC [11]. However, the proposed action of PlGFs through mobilization of VEGF seems unlikely because PlGF-stimulated proliferation of iBREC was not inhibited by ranibizumab [24]. iBREC proliferation induced by co-stimulating growth factors including VEGF165 was not completely inhibited by the Fab fragment, suggesting parallel activation of independent signalling pathways. This assumption was supported by previous studies also indicating that incomplete inhibition of VEGF165-induced proliferation by the anti-VEGF antibody bevacizumab might be due to VEGF-upregulated PlGF [15, 19, 30]. Such possibly persistent induction of PlGFs or other pro-proliferative factors might play a role in PDR that weakly respond to anti-VEGF therapies. However, proliferation stimulated by PlGF in combination with VEGF can most likely be completely inhibited by VEGF trap because it also binds to PlGF [8, 9, 31]. Whether this is sufficient to block proliferation stimulated by a combination together with bFGF and IGF-1 remains to be shown.
In contrast to its co-stimulation of REC proliferation, VEGF165 seems so dominant in the regulation of migration that—despite some effects contributed by other growth factors observed in this study and by others—induced migration was almost completely suppressed by ranibizumab, even in experiments with the most complex combinations of factors. The dominant role of VEGF165 was confirmed by our observation that the inhibitor of VEGFR KRN951 also blocked iBREC migration stimulated by a mixture of VEGF165, bFGF, and IGF-1. At the concentration used in this study, KRN951 specifically inhibits the tyrosine kinase activity of VEGFR1 and -R2 without affecting other receptor tyrosine kinases [29]. Interestingly, migration stimulated by VEGF121 together with bFGF and IGF-1 was totally blocked by ranibizumab, although bFGF-induced migration was not affected, and VEGF121 by itself did not even enhance iBREC migration. There is some evidence supporting the concept that bFGF facilitates binding of VEGF121 by its induction of VEGFR2 expression [32]. Then migration may be essentially driven by VEGF121, and its removal with ranibizumab might be sufficient to normalize migration.
Migration of (i)BREC was not induced by PlGF-1/-2, which is in contrast to the observation that these growth factors strongly stimulate migration of macrovascular endothelial cells of the human umbilical vein (HUVEC) [31]. However, both isoforms stimulated proliferation of iBREC, thereby confirming that the used recombinant human polypeptides can activate the relevant bovine receptor VEGFR1. Another example for the obviously different behavior of macrovascular and microvascular EC is the observation that VEGF121 induces migration of HUVEC, whereas iBREC do not respond [14, 24]. Basic differences between HUVEC and HREC in their expression of genes important for angiogenic processes were also shown by expression profiling [33].
Neovascularization requires migration and proliferation of REC, but therapeutic interference with only one of these processes might not be enough to prevent this hallmark of PDR. Results of several studies based on in-vitro or in-vivo models indeed suggest that more than inhibition of VEGF (which is not the only factor controling REC proliferation) is needed to suppress neovascularization. In an in-vitro model, sprouting of EC was stronger inhibited when bFGF was targeted in addition to VEGF [34]. Efficient suppression of retinal neovascularization was also achieved with VEGF-trap, a chimeric recombinant protein consisting of the VEGF-binding domains of VEGFR1 and -R2 that inhibits VEGF as well as other members of the protein family such as PlGFs [8, 9]. This therapeutic effect is most likely due to inhibition of PlGF-driven proliferation of REC.
Our results also suggest that different VEGF receptors (VEGFR1, -R2, and/or NRP-1) are involved in the processes leading to altered proliferation and migration of iBREC (Table 2): all members of the VEGF family tested—VEGF165, VEGF121, PlGF-1, PlGF-2, and VEGF-E—stimulated proliferation, suggesting that activation of either receptor is sufficient, which is in accordance with other studies using EC from various sources [35]. In contrast, binding of a ligand to VEGFR2 and to NRP-1 seems to be necessary to enhance migration, because only VEGF165 and VEGF-E, but not VEGF121 were efficient stimulators. Because VEGF-E was most efficient in stimulating migration of iBREC, one could assume that VEGFR1 is a negative regulator. But combined activation of VEGFR1 and -R2 by VEGF165/121 plus PlGF-1/-2 did not result in a lower migration rate compared to stimulation with VEGF165 alone. More likely, different receptors compete for the available factors, and VEGFR2-mediated signalling is the most efficient pathway leading to enhanced migration. The results of our experiments with immortalized BREC support the assumption that VEGFR2 is crucially involved in signal transduction in EC from various species and different tissues [35]. Although VEGFR1 can play an important role in EC proliferation, its contribution to other cellular processes stimulated by VEGF is still unclear: potentiating the effect of VEGF by cross-activation of VEGFR2 through VEGFR1 in response to stimulation with PlGF has been reported thereby [35]. In contrast, activated VEGFR1 was also reported to be a negative regulator of VEGFR2 signaling in the event that VEGFR2 had been activated first [21]. However, binding of both receptors by a combination of VEGF and PlGF did not markedly affect VEGF-induced iBREC proliferation or migration.
Whereas stimulation of migration by the concerted action of growth factors can be at least partly prevented by ranibizumab, substantial inhibition of proliferation was not observed in such experimental setting. Both proliferation and migration of REC are required for neovascularization observed in PDR, and it will be interesting to learn from the ongoing clinical trials whether inhibition of one of these processes by ranibizumab can delay or slow down progression of the disease. The possible surplus benefit of inhibiting all VEGF family members by VEGF-trap remains to be shown, and in view of our results, additional targeting of bFGF and IGF-1 might be considered a more promising approach in the therapy of PDR [9, 34]. However, these growth factors are also involved in retinal neuroprotection, and their complete inhibition might be detrimental to the function of retinal cells [36–38]. Moderate instead of complete depletion of these growth factors might therefore be more promising, although complete inhibition or reversal of pathological processes may not be achieved under these conditions.
References
Meyer-Schwickerath R, Pfeiffer A, Blum WF, Freyberger H, Klein M, Lösche C, Röllmann R, Schatz H (1993) Vitreous levels of the insulin-like growth factors I and II and the insulin-like growth factor binding proteins 2 and 3, increase in neovascular eye disease. J Clin Invest 92:2620–2625
Aiello LP, Avery RL, Arrigg PG, Keyt BA, Jampel HD, Shah ST, Pasquale LR, Thieme H, Iwamoto MA, Park JE, Nguyen HV, Aiello LM, Ferrara N, King GL (1994) Vascular endothelial growth factor in ocular fluid of patients with diabetic retinopathy and other retinal disorders. N Engl J Med 331:1480–1487
Boulton M, Gregor Z, McLeod D, Charteris D, Jarvis-Evans J, Moriarty P, Khaliq A, Foreman D, Allamby D, Bardsley B (1997) Intravitreal growth factors in proliferative diabetic retinopathy: correlation with neovascular activity and glycaemic management. Br J Ophthalmol 81:228–233
Khaliq A, Foreman D, Ahmed A, Weich H, Gregor Z, McLeod D, Boulton M (1998) Increased expression of placenta growth factor in proliferative diabetic retinopathy. Lab Invest 78:109–115
Nguyen QD, Tatlipinar S, Shah SM, Haller JA, Quinlan E, Sung J, Zimmer-Galler I, Do DV, Campochiaro PA (2006) Vascular endothelial growth factor is a critical stimulus for diabetic macular edema. Am J Ophthalmol 142:961–969
Ferrara N, Damico L, Shams N, Lowman H, Kim R (2006) Development of ranibizumab, an anti-vascular endothelial growth factor antigen binding fragment, as therapy for neovascular age-related macular degeneration. Retina 26:859–870
Mitchell P, Bandello F, Schmidt-Erfurth U, Lang GE, Massin P, Schlingemann RO, Sutter F, Simader C, Burian G, Gerstner O, Weichselberger A, RESTORE study group (2011) RESTORE study: ranibizumab monotherapy or combined with laser versus laser monotherapy for diabetic macular edema. Ophthalmology 118:615–625
Holash J, Davis S, Papadopoulos N, Croll SD, Ho L, Russell M, Boland P, Leidich R, Hylton D, Burova E, Ioffe E, Huang T, Radzierjewski C, Bailey K, Fandl JP, Daly T, Wiegand SJ, Yancopoulos GD, Rudge JS (2002) VEGF-Trap: a VEGF blocker with potent antitumor effects. Proc Natl Acad Sci USA 99:11393–11398
Aiello LP, Pierce EA, Foley ED (1995) Suppression of retinal neovascularization in vivo by inhibition of vascular endothelial growth factor (VEGF) using soluble VEGF-receptor chimeric proteins. Proc Natl Acad Sci USA 92:10457–10461
Do DV, Nguyen QD, Boyer D, Schmidt-Erfurth U, Brown DM, Vitti R, Berliner AJ, Gao B, Zeitz O, Ruckert R, Schmelter T, Sandbrink R, Heier JS, DA VINCI Study Group (2012) One-year outcomes of the DA VINCI Study of VEGF Trap-Eye in eyes with diabetic macular edema. Ophthalmology 119:1658–1665
Castellon R, Hamdi HK, Sacerio I, Aoki AM, Kenney MC, Ljubimov AV (2002) Effects of angiogenic growth factor combinations on retinal endothelial cells. Exp Eye Res 74:523–535
Yan Q, Ki Y, Hendrickson A, Sage EH (2001) Regulation of retinal capillary cells by basic fibroblast growth factor, vascular endothelial growth factor, and hypoxia. In Vitro Cell Dev Biol-Animal 37:45–49
Deissler H, Deissler H, Lang GK, Lang GE (2005) Generation and characterization of iBREC: novel hTERT-immortalized bovine retinal endothelial cells. Int J Mol Med 15:65–70
Deissler H, Deissler H, Lang S, Lang GE (2008) VEGF-induced effects on proliferation, migration and tight junctions are restored by ranibizumab (Lucentis®) in microvascular retinal endothelial cells. Br J Ophthalmol 92:839–843
Stewart EA, Samaranayake GJ, Browning AC, Hopkinson A, Amoaku WM (2011) Comparison of choroidal and retinal endothelial cells: Characteristics and response to VEGF-isoforms and anti-VEGF treatment. Exp Eye Res 93:761–766
Giannini S, Cresci B, Pala L, Ciucci A, Franchini A, Manuelli C, Fujita-Yamaguchi Y, Cappugi P, Zonefrati R, Rotella CM (2001) IGFBPs modulate IGF-I- and high glucose-controlled growth of human retinal endothelial cells. J Endocrinol 171:273–284
Deissler H, Deissler H, Lang GE (2011) Inhibition of VEGF is sufficient to completely restore barrier malfunction induced by growth factors in microvascular retinal endothelial cells. Br J Ophthalmol 95:1151–1156
Simorre-Pinatel V, Guerrin M, Chollet P, Penary M, Clamens S, Malecaze F, Plouet J (1994) Vasculotropin-VEGF stimulates retinal capillary endothelial cells through an autocrine pathway. Invest Ophthalmol Vis Sci 35:3393–3400
Zhao B, Cai J, Boulton M (2004) Expression of placenta growth factor is regulated by both VEGF and hyperglyceamia via VEGFR-2. Microvasc Res 68:239–246
Yao YG, Hoseong SY, Zhiming C, Danielsson J, Duh EJ (2005) Upregulation of placental growth factor by vascular endothelial growth factor via a post-transcrptional mechanism. FEBS Lett 579:1227–1234
Cai J, Wu L, Qi X, Shaw L, Li Calzi S, Caballero S, Jiang WG, Vinores SA, Antonetti D, Ahmed A, Grant MB, Boulton ME (2011) Placenta growth factor-1 exerts time-dependent stabilization of adherens junctions following VEGF-induced vascular permeability. PLoS One 6:e18076
Ferrara N (2004) Vascular endothelial growth factor: basic science and clinical progress. Endocr Rev 25:581–611
Oh H, Takagi H, Otani A, Koyama S, Kemmochi S, Uemura A, Honda Y (2002) Selective induction of neuropilin-1 by vascular endothelial growth factor (VEGF): a mechanism contributing to VEGF-induced angiogenesis. Proc Natl Acad Sci USA 99:383–388
Pan Q, Chathery Y, Wu Y, Rathore N, Tong RK, Peale F, Bagri A, Tessier-Lavigne M, Koch AW, Watts RJ (2007) Neuropilin-1 binds to VEGF121 and regulates endothelial cell migration and sprouting. J Biol Chem 282:24049–24056
Park JE, Chen HH, Winer J, Houck KA, Ferrara N (1994) Placenta growth factor. Potentiation of vascular endothelial growth factor bioactivity, in vitro and in vivo, and high affinity binding to Flt-1 but not to Flk-1/KDR. J Biol Chem 269:25646–25654
Sawano A, Takahashi T, Yamaguchi S, Aonuma M, Shibuya M (1996) Flt-1 but not KDR/Flk-1 tyrosine kinase is a receptor for placental growth factor, which is related to vascular endothelial growth factor. Cell Growth Differ 7:213–221
Migdal M, Huppertz B, Tessler S, Comforti A, Shibuya M, Reich R, Baumann H, Neufeld G (1998) Neuropilin-1 is a placenta growth factor-2 receptor. J Biol Chem 273:22272–22278
Meyer M, Clauss M, Lepple-Wienhues A, Waltenberger J, Augustin HG, Ziche M, Lanz C, Buttner M, Rziha HJ, Dehio C (1999) A novel endothelial growth factor encoded by Orf virus, VEGF-E, mediates angiogenesis via signaling through VEGFR-2 (KDR) but not VEGFR-1 (Flt-1) receptor kinases. EMBO J 18:363–374
Nakamura K, Taguchi E, Miura T, Yamamoto A, Takahashi K, Bichat F, Guilbaud N, Hasegawa K, Kubo K, Fujiwara Y, Suzuki R, Kubo K, Shibuya M, Isae T (2006) KRN951, a highly potent inhibitor of vascular endothelial growth factor receptor tyrosine kinases, has antitumor activities and affects functional vascular properties. Cancer Res 66:9134–9142
Deissler H, Deissler H, Lang GE (2012) Actions of bevacizumab and ranibizumab on microvascular retinal endothelial cells: similarities and differences. Br J Ophthalmol 96:1023–1028
Papadopoulos N, Martin J, Ruan Q, Rafique A, Rosconi MP, Shi E, Pyles EA, Yancopoulos GD, Stahl N, Wiegand SJ (2012) Binding and neutralization of vascular endothelial growth fact (VEGF) and related ligands by VEGF Trap, ranibizumab and bevacizumab. Angiogenesis 15:171–185
Hata Y, Rook SL, Aiello LP (1999) Basic fibroblast growth factor induces expression of VEGF receptor KDR through a protein kinase C and p44/p42 mitogen-activated protein kinase-dependent pathway. Diabetes 48:1145–1155
Browning AC, Halligan EP, Stewart EA, Swan DC, Dove R, Samaranayake GJ, Amoaku WM (2012) Comparative gene expression profiling of human umbilical vein endothelial cells and ocular vascular endothelial cells. Br J Ophthalmol 96:128–132
Stahl A, Paschek L, Martin G, Feltgen N, Hansen LL, Agostini HT (2009) Combinatory inhibition of VEGF and FGF2 is superior to solitary VEGF inhibition in an in vitro model of RPE-induced angiogenesis. Graefes Arch Clin Exp Ophthalmol 247:767–773
Koch S, Tugues S, Li X, Gualandi L, Claesson-Welsh L (2011) Signal transduction by vascular endothelial growth factor receptors. Biochem J 437:169–183
O’Driscoll C, Wallace D, Cotter TG (2007) bFGF promotes photoreceptor cell survival in vitro by PKA-mediated inactivation of glycogen synthase kinase 3 beta and CREB-dependent Bcl-2 up-regulation. J Neurochem 103:860–870
Nishijima K, Ng YS, Zhong L, Bradley J, Schubert W, Jo N, Akita J, Samuelsson SJ, Robinson GS, Adamis AP, Shima DT (2007) Vascular endothelial growth factor-A is a survival factor for retinal neurons and a critical neuroprotectant during the adaptive response to ischemic injury. Am J Pathol 171:53–67
Seigel GM, Chiu L, Paxhia A (2000) Inhibition of neuroretinal cell death by insulin-like growth-1 and its analogs. Mol Vis 6:157–163
Acknowledgments
The authors thank Susanne Denning, Nadine Gubernath and Anita Ruepp for expert technical assistance and Novartis Pharma GmbH for a generous gift of ranibizumab.
This study was supported by a research grant by Novartis Pharma GmbH, Nuremberg, Germany to Heidrun L. Deissler and Gabriele E. Lang.
Author information
Authors and Affiliations
Corresponding author
Additional information
The authors have full control of all primary data, and they agree to allow Graefe’s Archive for Clinical and Experimental Ophthalmology to review their data upon request.
This study was presented at the ARVO 2012 Annual Meeting, May 6–10, in Fort Lauderdale, Florida.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Deissler, H.L., Deissler, H., Lang, G.K. et al. Ranibizumab efficiently blocks migration but not proliferation induced by growth factor combinations including VEGF in retinal endothelial cells. Graefes Arch Clin Exp Ophthalmol 251, 2345–2353 (2013). https://doi.org/10.1007/s00417-013-2393-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00417-013-2393-5