
Abstract
Hereditary transthyretin-related amyloidosis (ATTRv amyloidosis) is a rare and progressively debilitating disease characterized by the deposition of transthyretin (TTR) amyloid fibrils in various organs and tissues, most commonly in the heart and peripheral nerves. This pathological deposition can lead to significant organ dysfunction and, ultimately, organ failure. ATTRv amyloidosis exhibits a broad range of clinical presentations, from purely neurological symptoms to purely cardiac manifestations, as well as mixed phenotypes which result from both neurological and cardiac implications. This wide phenotypical spectrum realistically challenges disease diagnosis and prognosis, especially in individuals without or with an unknown family history. Multiple factors are thought to contribute to this variability, including genetic, epigenetic, and even environmental influences. Understanding these factors is crucial, as they can significantly affect disease expression and progression. This review aims to summarize each of these contributing factors, to help elucidate the current knowledge on the phenotypical variability of ATTRv amyloidosis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
First described in Portugal, in 1952 [1], hereditary transthyretin-related amyloidosis (ATTRv amyloidosis—formerly “Familial Amyloid Polyneuropathy”) is a fatal, progressive, autosomal-dominant disorder caused by variants in the transthyretin (TTR) gene [2].
TTR, located on chromosome 18, encodes transthyretin (TTR—formerly “prealbumin”), a homotetrameric protein [3] produced predominantly in the liver, though small amounts are synthesized in the choroid plexus, retinal epithelium, and pancreatic islets [4,5,6]. Its primary functions include the transport of thyroxine and the retinol-binding protein bound to retinol, although other potential roles have been discovered [7].
It is widely accepted that protein destabilization is a crucial factor behind amyloid fibril formation [8]. In such case, variants in the TTR gene lead to protein destabilization and dissociation of its tetramer structure into dimers and subsequently into monomers. These monomers misfold and form the amyloidogenic intermediate of TTR that then aggregate into amyloid fibrils which deposit in several tissues and organs, mainly in nerves, heart, kidneys, and eyes (Fig. 1). The deposits in these organs and tissues lead to organ dysfunction and possible organ failure giving rise to a vast array of symptoms [8, 9].

Schematic representation of TTR amyloidogenesis. Image constructed with BioRender and adapted from [10]
However, it is important to mention that TTR proteolysis also has been extensively studied as a mechanism that can drive amyloid formation [11]. This is due, in large part, to the presence of C-terminal TTR fragments in ex vivo TTR amyloid deposits regardless of the TTR variant [12].
Importantly, wild-type TTR is also responsible for a relatively common, aging-related, nonhereditary type of amyloidosis termed senile systemic amyloidosis or wild-type TTR amyloidosis (ATTRwt amyloidosis), where the heart is the main affected organ [13, 14].
Nonetheless, regarding ATTRv amyloidosis, one of its most striking aspects is the wide range of clinical presentations that can occur. There are more than 140 TTR variants (most of which are pathogenic), and each variant can cause a different pattern of symptoms, disease severity, and age of onset [15]. The TTR Val30Met variant is one of the most prevalent variants worldwide (possibly only trumped by the Val122Ile variant and its occurrence is in 3–4% of African Americans [16]) and is responsible for the high prevalence of ATTRv amyloidosis in endemic regions such as Portugal, Sweden, and Japan [17,18,19].
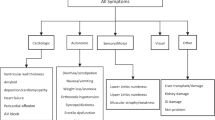
The most common symptom of ATTRv amyloidosis, its “hallmark”, is a progressive peripheral sensory–motor neuropathy, which typically begins in the feet and hands and can cause tingling, numbness, pain, and weakness. Other common manifestations include autonomic dysfunction, cardiomyopathy, nephropathy, and gastrointestinal and ophthalmological abnormalities.
Autonomic symptoms are common and can involve various systems, including the cardiovascular, gastrointestinal, and genitourinary systems and symptoms include orthostatic hypotension, erectile dysfunction, sweating abnormalities, and recurrent urinary tract infections. Cardiovascular involvement can lead to cardiomyopathy, which can cause arrhythmias, atrial fibrillation, and heart failure. Gastrointestinal dysfunction can lead to symptoms such as nausea, vomiting, diarrhea, constipation, and early satiety. These symptoms can be particularly debilitating and can contribute to malnutrition and weight loss. Renal involvement, although less common than neurological or cardiac symptoms, may also occur, causing proteinuria and kidney failure. In addition, patients with ATTRv amyloidosis may also develop ocular symptoms such as dry eyes, abnormal conjunctival vessels, papillary abnormalities, and glaucoma [20,21,22].
Given this immense phenotypical variability, patients may present symptoms that often overlap with other conditions, being frequently misdiagnosed with diseases such as idiopathic axonal polyneuropathy, chronic inflammatory demyelinating polyneuropathy (CIDP), or heart failure and might not be correctly diagnosed until later in the disease course [20]. This variability makes ATTRv amyloidosis difficult to diagnose, especially in older patients or those without a known family history [23, 24].
Understanding the biological reason behind symptom variability in ATTRv amyloidosis is extremely important since it can provide insights into the disease mechanisms and reveal potential targets for treatment.
In this review, we discuss the current knowledge on factors that might influence disease outcome and help explain phenotype variability (Fig. 2). A summarized overview of this review is presented in Table 1.

Factors that potentially influence ATTRv amyloidosis phenotypic variability
Genotype
As stated above, TTR has over 140 variants described and most of them are pathogenic. Although the same variant can be associated with an array of symptoms, some are more associated with a specific phenotype than others. This is known as the genotype–phenotype correlation. Generally, clinical presentation can be predominantly neuropathic, predominantly cardiac, or mixed (exhibiting both polyneuropathy and cardiomyopathy) [25]. The most common TTR variants and their predominant phenotype are presented in Table 2.
Despite the high prevalence of TTR V30M, data indicates that patients’ survival rate is quite high, especially when compared to other variants, such as Thr60Ala, Val122Ile, or even wild-type TTR (wtTTR), which are generally associated with poorer prognoses. Intriguingly, these variants are typically linked to a more cardiac-type disease [47].
Pathogenic TTR variants are generally amyloidogenic, though most of the variants reported were identified in a small group of patients and their pathogenicity is not clear. However, some variants have been reported to have higher or lower amyloidogenicity. For instance, although rare, TTR variant Leu55Pro is notably aggressive, exhibiting a high propensity for protein aggregation. It is associated with rapid disease progression of mixed symptoms and early onset, with some individuals experiencing initial symptoms during childhood [48,49,50,51].
In contrast, TTR variant Glu61Lys, a rare variant so far only identified in late-onset patients, has low amyloidogenicity similar to that of wild-type TTR [52]. Interestingly, in patients carrying this variant, sural nerve biopsy showed no amyloid deposits, although significant loss of myelinated fibers was detected. Deposits were, however, present in muscle, salivary gland, and heart biopsies [52,53,54].
Additionally, some non-pathogenic variants (Thr119Met and Arg104His) can have a protective effect in compound heterozygotes carrying the Val30Met variant. The simultaneous occurrence of these protective variants and Val30Met results in the absence of symptoms or a delayed form of the disease [55, 56]. It is thought that these variants increase TTR tetramer stability and thus increase its resistance to dissociation [57, 58].
The full list of all known TTR variants is presented in the “Variants in Hereditary Amyloidosis” database (http://amyloidosisvariants.com).
The specific pathogenic TTR variant is a significant factor in the clinical variability of ATTRv amyloidosis. Each variant has a unique impact on clinical presentation and disease course. However, the amino acid substitutions alone do not completely explain the variability in disease penetrance, pathology, and clinical course. This is evident especially in cases of monozygotic twins that share the same variant, but have very different clinical presentations [59,60,61,62].
The genotype of ATTRv amyloidosis significantly influences its clinical presentation, with over 140 known TTR variants showing diverse pathogenic effects. Certain variants are strongly associated with specific phenotypes—neuropathic, cardiac, or mixed. Additionally, some non-pathogenic variants can mitigate the severity of symptoms in individuals with the Val30Met variant. Despite the clear genotype–phenotype correlations, variability in disease expression remains, highlighting the complexity of ATTRv amyloidosis and the need for further research to fully understand these genotype influences.
Age of onset
In ATTRv amyloidosis, age of onset, the age at which a person first experiences symptoms, varies greatly and can range from early adulthood to old age [63]. Carriers of TTR variants can even remain asymptomatic their whole life [64]. Age of onset is generally divided into two groups, early and late onset. Early-onset individuals exhibit their first symptoms before the age of 50, whereas late-onset patients only experience their first symptoms after that age [65, 66]
Regarding the TTR Val30Met variant, despite having the same genotype, the independent clusters of endemic regions have substantial divergence in the age of onset. In Portugal, the average age of onset is 33.5 years and 87% of patients show symptoms before the age of 40 [67]. In Japan, three endemic foci have been reported, namely, in the Nagano, Kumamoto, and Ishikawa prefectures and the mean ages of onset are 33.1, 35.6, and 62.9 years of age, respectively [18, 66, 68]. In non-endemic areas in Japan, the age of onset is typically higher [69]. The Swedish endemic foci, which are around the towns of Skellefteå and Piteå, are regarded as being of late onset, as the mean age of symptom presentation is 54.4 and 58.8 years of age, respectively [70].
Origin from endemic areas, as opposed to non-endemic regions, is generally associated with higher penetrance and lower age of onset, with the exception of Swedish endemic regions that present low penetrance and late onset [71, 72]. Noteworthy, non-Val30Met variants are generally regarded as having a later onset [73].
Still concerning the Val30Met variant, it is also noteworthy to mention the phenomenon of anticipation. Anticipation is regarded as the decrease of age of onset within each generation [74]. Parents affected with ATTR Val30Met amyloidosis and a late-onset presentation frequently have early-onset children; however, the opposite has yet to be described. Anticipation is currently considered a true biological phenomenon in ATTR Val30Met amyloidosis and has been reported in several clusters [74,75,76,77,78].
As stated in Table 1, individuals with the Val30Met variant and early onset typically have a less severe disease course with neuropathy and considerable autonomic dysfunction. In contrast, late-onset Val30Met patients have more severe symptoms with less autonomic dysfunction and moderate to severe cardiac involvement [27, 73].
Since the age of onset has a great variability and seems to influence the phenotype and disease course, it is hypothesized that genetic modifiers may explain the age of onset variability. Identifying modifier genes helps to better understand the underlying mechanisms involved in the pathophysiology of diseases and to find new targets for therapeutic intervention [79]. Thus, studies have been performed to access the existence of genetic modifiers of age of onset in ATTRv amyloidosis.
The Val30Met variant is probably the most studied regarding genetic modifiers given its wide range of age of onset and the different clinical presentations between early- and late-onset individuals. Several associations have made between genetic variants and the age of onset of ATTR Val30Met amyloidosis and these are summarized in Table 3.
The age of onset in ATTRv amyloidosis varies widely, ranging from early adulthood to old age, and significantly impacts the disease phenotype and course. The Val30Met variant displays considerable regional differences in the age of onset, potentially influenced by genetic modifiers. Despite research advances, variability in age of onset remains to be explained. Understanding the mechanisms behind this variability will help predict patients’ age of onset, aiding in genetic counseling, and can also be important in determining prognosis and in developing new therapeutic strategies.
Sex and transmitting parent
Sex is an important modifier of disease, as it plays a role in epidemiology, disease pathogenesis, clinical presentation, and therapy response. These differences are ultimately attributed to the genetic heterogeneity of sex chromosomes and the hormonal influences that create molecular differences between males and females [87, 88]. In ATTRv amyloidosis, sex has also been implicated.
According to the Transthyretin Amyloidosis Outcome Survey (THAOS), a worldwide registry for ATTR amyloidosis, the symptomatic carriers of the disease are predominantly male in both wild-type ATTR amyloidosis (ATTRwt amyloidosis or senile systemic amyloidosis) and ATTRv amyloidosis [73]. Regarding the Val30Met variant, it has also been reported that males tend to have earlier onsets and/or higher disease risks than females [77, 78, 89,90,91].
Another study from THAOS on 4050 ATTRv amyloidosis carriers (2790 symptomatic and 1260 asymptomatic) showed that a higher proportion of men had cardiac involvement and that a predominantly cardiac phenotype was more common in males than females [92]. In fact, men represented a staggering 72% of patients that had ATTRv amyloidosis with cardiomyopathy. On the other hand, women represented a higher proportion of the individuals in the asymptomatic group. Although disease prevalence in males was overall generally higher than in females, it was found to be especially high in late-onset TTR Val30Met and non-Val30Met cardiac variant carriers. The study also showed a significant association between males and an earlier disease onset.
Another study from THAOS analyzed the TTR Val122Ile carriers with cardiac amyloidosis in the USA and also found increased male prevalence (75.8%) [93]. Similarly, in the UK, a study on the Val122Ile variant also showed increased male prevalence [94]. Moreover, Batra and colleagues also studied TTR Val122Ile carriers with cardiac amyloidosis and determined that although cardiac function and mortality rates were similar between sexes, females were significantly older at the time of diagnosis which could indicate a slower disease progression [95].
A study by Rapezzi et al. examined carriers of different TTR variants and reported that men had a higher prevalence of cardiomyopathy. The authors postulated that female hormones could exert a protective effect and so, in further analysis, they found that women with higher degrees of cardiomyopathy were more likely to be menopausal, which strengthened their theory [96].
Briefly, men seem to have more cardiac involvement irrespective of TTR variant. It has been theorized that female hormones may have a protective effect; however, the role of male hormones should also be considered as it has been reported that they can also upregulate TTR [97].
Nevertheless, sex-related differences are still a rather understudied topic and reports can differ especially in those with smaller cohorts.
Besides sex of the variant carrier, the sex of the parent who transmitted the variant might also have an impact on disease presentation. This factor has been mentioned in many studies regarding the Val30Met variant.
As early as 1991, two studies of Portuguese cohorts of Val30Met carriers not only reported the phenomenon of anticipation, but also observed a larger anticipation in offspring of affected mothers [89, 98]. In either case of parent transmission, men displayed an earlier onset than women, and thus sons of affected mothers had the earliest of onsets while daughters of affected fathers had the latest onsets.
Similarly, other studies, including in Swedish and Japanese cohorts, have revealed that anticipation was especially significant when the variant was inherited maternally [76, 99] and especially in mother–son pairs [77, 78].
Noteworthy, a study on the penetrance of ATTR Val30Met amyloidosis on a Swedish cohort revealed a significant association between higher penetrance of the disease and maternal inheritance of the TTR variant [100]. These findings are supported in other studies [77, 101].
Potential mechanisms responsible for these phenomena include parental imprinting and mitochondrial involvement. The imprinting possibility is not one much explored, but the role of mitochondrial DNA has been examined, albeit vaguely.
To understand the higher mean age of onset of Swedish and French patients, Olsson et al. investigated the mitochondrial haplogroups in Swedish and French TTR Val30Met carriers and controls [102]. They found that the mitochondrial haplogroups were similar between late-onset patients and controls, an expected outcome. Notably, they found that the rare mitochondrial haplogroup K appeared recurrently in early-onset patients, indicating that this haplogroup could potentially have an impact on amyloidogenesis. A subsequent study determined that this haplogroup could explain the variability seen regarding sex of the transmitting parent [103].
Similarly, mitochondrial DNA (mtDNA) copy number was assessed in Portuguese ATTR Val30Met amyloidosis patients [104]. A significantly higher mtDNA copy number was found in the asymptomatic, early-onset and late-onset groups when compared to controls, but failed to show significant differences among each other. When parent–offspring pairs where analyzed, an important increase of mtDNA copy number was seen in early-onset children compared to their late-onset parents.
To summarize, sex can significantly influence the epidemiology, clinical presentation, and disease progression in ATTRv amyloidosis. Males tend to have an earlier onset and higher prevalence of cardiac involvement, while females are more often asymptomatic. Studies suggest that female hormones might have a protective effect against cardiomyopathy. Additionally, the sex of the parent transmitting the variant also impacts disease presentation, with maternal inheritance often associated with earlier onset and higher penetrance. Potential mechanisms for these differences include parental imprinting and mitochondrial DNA, though further research is needed to fully understand these influences.
Non-coding variants
Non-coding variants are single-nucleotide polymorphisms (SNPs) or structural variants (insertions, deletions, copy-number variants, and repeat expansions) that occur in the non-coding regions and that have gained growing importance being now considered contributors to disease due to their role in gene regulation and expression [105].
Depending on the localization of the variant, effects can include, but are not limited to, (i) alterations in the binding of transcription factors and RNA polymerase (in regulatory regions); (ii) alterations in splicing (in splicing sites); and (iii) alterations in the binding of miRNA and RNA-binding proteins (in untranslated regions). All these changes may result in RNA instability, aberrant protein isoforms, and an altered gene regulation and expression [106]. It is thus tempting to assume that non-coding variants might be a driving force behind the clinical variability of ATTRv amyloidosis.
Several studies that report non-coding variants in ATTRv amyloidosis are described in Table 4. Although the role of non-coding variants in this disease is still undetermined, together, these studies help shed light on the importance that this field might have in the better understanding of ATTRv amyloidosis.
Non-coding variants, such as SNPs and structural variants, influence gene regulation and expression and can alter transcription factor binding, splicing, and miRNA interactions. Studies have identified non-coding variants in the TTR gene linked to different phenotypes and disease onset, highlighting their impact on disease presentation. Further research is needed to fully understand their role in ATTRv amyloidosis.
Fibril composition
As aforementioned, TTR proteolysis has been extensively studied as a mechanism that can drive amyloid formation [11]. This theory gained force due to the presence of different types of amyloid fibrils found in deposits. In fact, a study showed that proteolysis of TTR Ser52Pro, a variant that causes a severe form of amyloidosis, leads to the formation of a 49–127 fragment, which, when released in a physiological fluid agitation, quickly forms highly stable aggregates [114].
Although still under study, some authors have reported that amyloid fibril composition may have an influence on the disease phenotype.
There are two types of amyloid fibrils, type A and type B. Type A fibrils consist of a mixture of C-terminal fragments and full-length TTR, display weak affinity to congo red, and are weakly birefringent, whereas type B fibrils are solely composed of intact full-length TTR, have high affinity for congo red and are strongly birefringent [12]. In this study, Bergström’s team found both fibril types in Swedish ATTR Val30Met amyloidosis patients, yet type A fibrils were the only type found in individuals with ATTRwt amyloidosis, an amyloidosis that is frequently observed in older individuals and is mainly cardiac in nature [12].
Later, Ihse and peers analyzed fat tissue biopsies from 33 Swedish ATTR Val30Met amyloidosis patients and found that type A fibrils were associated with late disease onset and hypertrophied myocardium whereas type B fibrils were related to an earlier onset but no cardiac involvement [115].
Shortly after, a study by Koike and colleagues strengthened these findings by examining deposits from eight autopsied patients, all TTR Val30Met carriers, three early onset from endemic parts of Japan and five late onset from non-endemic regions of Japan [116]. They discovered that amyloid deposits in early-onset cases tended to be type B fibrils while those in late-onset cases were type A fibrils. Interestingly, an analysis of the cardiac deposits of the patients showed that in early-onset individuals the deposited TTR was mostly mutated, whereas in late-onset cases the deposits presented a significant amount of wild-type TTR. The authors hypothesized that these findings could support the idea that the mechanism behind amyloid deposition is similar to the one observed in ATTRwt amyloidosis.
In another study of Japanese ATTR Val30Met amyloidosis patients, an association with fibril type and age of onset was also found [117]. However, they did find a patient with type B fibrils and age of onset of 60 years of age. Cardiac assessment for type A fibril patients was only possible in one person; nonetheless, he did show a more severe cardiac involvement than type B patients, consistent with previous findings. Intriguingly, in the same study, the authors also assessed 63 non-Val30Met patients who all, except two, had type A fibrils and were unable to find a correlation between fibril type and phenotype. The authors ultimately postulate that fibrils with fragmented TTR might be the standard amyloid composition in ATTRv amyloidosis.
These studies uncover the amyloid fibril composition as a possible determinant in age of onset for the Val30Met variant.
It has also been found that, after liver transplantation, amyloids fibrils are mostly composed of wild-type TTR [118, 119] and one study has suggested that, not only is the heart more prone to incorporate wild-type TTR than other tissues, but type A fibrils incorporate higher amounts that type B fibrils [120]. This seems to corroborate previous findings, since late-onset patients typically have type A fibrils. In fact, Gustafsson and peers analyzed and confirmed this potential link as they discovered that patients with type A fibrils develop cardiomyopathy and heart failure (or their preexisting cardiomyopathy deteriorates) after liver transplantation, while the same is not seen in patients with type B fibrils, further suggesting that type A fibrils are more susceptible to continuous amyloid deposition from wild-type TTR [121].
It is thus fair to assume that TTR fibril composition does in fact play an important role, not only in phenotypic variation of the TTR Val30Met variant, but also in the determination of survival after liver transplantation.
Succinctly, amyloid fibril composition, classified as type A or type B, plays a significant role in the phenotype of ATTRv amyloidosis. Type A fibrils, containing C-terminal fragments and full-length TTR, are linked to a later-onset disease and more cardiac involvement, while type B fibrils, composed of intact TTR, are generally more associated with early-onset disease and less or no cardiac involvement. Studies suggest that fibril type influences disease progression and survival, but further research is needed to fully understand the prognostic and diagnostic implications of fibril composition in ATTRv amyloidosis.
Epigenetic and environmental factors
Any process that alters gene expression and/or activity without modifying DNA sequences and can be inherited by daughter cells is considered an epigenetic modification. DNA methylation, histone modification, and miRNAs are three key epigenetic mechanisms [122].
Variability in phenotype and penetrance of ATTRv amyloidosis can conceivably be explained by epigenetic modifications especially when we consider monozygotic twins that carry the same variant yet display different clinical presentations [59,60,61,62]
In an epigenome-wide association study (EWAS) on 48 carriers (38 symptomatic and 10 asymptomatic) of TTR variants Val30Met, Phe64Leu, Ala120Ser, Ile68Leu, and Val122Ile and 32 controls, it was found that a CpG site in the Beta-secretase 2 (BACE2) gene was significantly hypomethylated in variant carriers [123]. BACE2 is known for cleaving APP in the brain, but is also present in peripheral tissues in inflammatory responses leading to the hypothesis that there might be a link between BACE2 and TTR-induced inflammation. They also found that the Val30Met variant disrupts a CpG site causing hypomethylation in carriers. This disrupted CpG site co-methylated with a CpG on the B4GALT6 gene, a gene that showed significantly divergent methylation levels in symptomatic patients when compared to asymptomatic carriers and encodes an enzyme also related to inflammatory processes. They also report other CpG sites possibly related to disease symptoms (in genes DSC2, DSG2 and DSC3) which the authors speculate might be involved in the formation of TTR deposits.
Posteriorly, a team performed an EWAS on TTR Val122Ile carriers of African descent to explore the possible association of methylation profile with heart disease and outpatient surgeries [124]. They found positive association between heart disease and methylation levels of genes FAM129B, SKI, WDR27 and GLS; and between outpatient surgeries and methylation levels of UBE2E3 and SEC14LS genes. Additionally, when analyzing differentially methylated functional modules, ABCA1 was found to be significantly associated with heart disease. Importantly, the genes identified are involved in transport and clearance of amyloid deposits, cardiac fibrosis, and muscle tissue regulation, pathways that are important to the ATTRv amyloidosis pathogenesis.
As for miRNA, one study reported that Swedish, but not French or Japanese, carriers of the TTR Val30Met variant contained in their haplotype the T allele of the 3′UTR polymorphism rs62093482 [125]. The authors predicted that this allele could serve as a new miRNA binding site and hypothesized that it could lead to downregulation of the variant TTR synthesis which could explain Swedish low penetrance and late-onset disease. However, this theory was later refuted when a team found that the polymorphism had no effect on the regulation of variant TTR expression via miRNA [126].
Despite epigenetics being a promising field to help unravel the reasons behind clinical variability, further studies on this subject need to be carried out to further assess the role of epigenetic factors in ATTRv amyloidosis.
Regarding environmental factors, these are often referred to as a possible contributor to ATTRv amyloidosis variability, yet not many studies have focused on this.
One study reported an association between ATTRv amyloidosis and organic solvent exposure as well as with being a dressmaker, although these findings could be due to chance [127]. They also stated that history of prostatic hyperplasia, cholecystic disease, or appendectomy was a risk factor and hypothesized it could be due to anesthesia. It has also been suggested that gut microbiota could possibly play a role in amyloidogenesis [128]. Studies on mice carrying the TTR Val30Met variant further implied environmental factors in amyloidogenesis, as they reported that mice kept in specific pathogen-free conditions (i.e., minimum environmental interaction) exhibited substantially less amyloid deposition than mice maintained under conventional conditions [129, 130].
Epigenetic modifications, such as DNA methylation and miRNAs, can potentially impact the phenotype and penetrance of ATTRv amyloidosis. Studies have found differential methylation in genes associated with inflammation and amyloid deposition among variant carriers. Environmental factors, though less studied, may also contribute to disease variability and animal studies even suggest environmental conditions can affect amyloid deposition, indicating a complex interplay between genetics and environment in ATTRv amyloidosis.
Other factors
Importantly, there are some factors that have been described in the literature that, despite being isolated reports, deserve to be mentioned.
The traditional model for gene expression in a diploid cell is by biallelic expression, where both alleles of a gene are expressed. On the other hand, monoallelic gene expression occurs when only one allele is actively transcribed while the other is silent [131]. Yordanova and colleagues analyzed the plasma and urine of 13 TTR Glu89Gln positive patients, with the aim to assess TTR transcription profile [132]. After analysis, the authors found a monoallelic expression signature in their cohort and proposed that this expression is age-related with wild-type TTR being expressed at a younger age subsequently shifting to variant TTR expression as the individual ages. This model could potentially explain why ATTRv amyloidosis is a disease of adult onset.
Somatic mosaicism, a phenomenon that has been implicated in disease states, is the presence of two genetically different populations of cells within a single individual as a result of a postzygotic variant [133]. In an interesting article, a large family with both healthy individuals and TTR Glu89Gln carriers was studied [134]. Analysis of peripheral blood, exfoliative buccal cells, and hair bulb cells from the TTR Glu89Gln carriers revealed some to have somatic mosaicism with reversion to normality in two cell types. Specifically, the TTR variant was found in the DNA of peripheral blood but not in that of buccal or hair bulb cells. This study raises the concern of testing for TTR variants in only one cell type since a false negative could easily be reported. It is unknown if this phenomenon has any impact in ATTRv amyloidosis severity or variability.
Monoallelic gene expression and somatic mosaicism are additional factors that may influence ATTRv amyloidosis. Monoallelic expression, where only one allele is transcribed, may shift from wild-type to variant TTR with age, potentially explaining adult-onset disease. Somatic mosaicism, the presence of different genetic populations within an individual, was observed in TTR Glu89Gln carriers, suggesting that variant testing in multiple cell types is necessary to avoid false negatives. The impact of these phenomena on disease severity and variability remains to be fully understood.
Conclusions
Hereditary transthyretin-related amyloidosis (ATTRv amyloidosis) represents a complex and heterogeneous disease with significant phenotypic variability influenced by multiple factors. The genotype–phenotype correlation plays a crucial role in determining the clinical presentation and disease progression. Key variants like Val30Met, Val122Ile, and are associated with distinct phenotypes, affecting specific organs and systems. Age of onset further complicates the clinical landscape, with early- and late-onset presentations showing substantial differences in disease severity and affected organ systems.
Sex of the affected individual also impacts disease expression, with males often exhibiting earlier onset and more severe cardiomyopathy compared to females, potentially due to hormonal differences. The phenomenon of anticipation, particularly pronounced in maternal transmission of the variant, underscores the importance of parental inheritance patterns in disease progression.
Emerging evidence also suggests that non-coding variants, fibril composition, and epigenetic factors contribute to the phenotypic diversity observed in ATTRv amyloidosis. The distinction between type A and type B amyloid fibrils, and their respective impacts on disease manifestation and progression, highlights the importance of amyloid composition in understanding disease pathology. Epigenetic modifications, such as DNA methylation and miRNA interactions, and non-coding genetic variants can affect gene expression and/or regulation, potentially influencing the severity and presentation of the disease. These factors offer promising avenues for further research into the underlying mechanisms of this variability.
Environmental factors and isolated reports of phenomena like monoallelic expression and somatic mosaicism add additional layers of complexity to the disease.
In conclusion, all these findings underscore the necessity for comprehensive and multifaceted approaches in studying ATTRv amyloidosis. The intricate interplay between genetic, epigenetic, and environmental factors in ATTRv amyloidosis necessitates continued research to fully elucidate the mechanisms driving phenotypic variability. Such understanding is vital for improving diagnosis, genetic counseling, and the development of targeted therapeutic strategies, ultimately enhancing patient outcomes in this challenging disease.
Future perspectives
The clinical variability of ATTRv amyloidosis presents a significant challenge to clinicians and researchers alike. Diagnosis, prognosis, and treatment of this disease, as well as its pathophysiology are complex. As such, ongoing research efforts are focused on improving our understanding of the underlying mechanisms that contribute to clinical variability in ATTRv amyloidosis, as well as developing new prognostic and therapeutic approaches. Hopefully, growing knowledge of the intricacies of ATTRv amyloidosis will lead to a better understanding of pathogenesis and disease course, as well as earlier and more accurate diagnosis and treatment.
Data availability
Not applicable.
References
Andrade C (1952) A peculiar form of peripheral neuropathy; familiar atypical generalized amyloidosis with special involvement of the peripheral nerves. Brain 75:408–427. https://doi.org/10.1093/BRAIN/75.3.408
Mascarenhas Saraiva MJ, Birken S, Costa PP, Goodman DS (1984) Amyloid fibril protein in familial amyloidotic polyneuropathy, Portuguese type: definition of molecular abnormality in transthyretin (prealbumin). J Clin Invest 74:104–119. https://doi.org/10.1172/JCI111390
Tsuzuki T, Mita S, Maeda S et al (1985) Structure of the human prealbumin gene. J Biol Chem 260:12224–12227. https://doi.org/10.1016/S0021-9258(17)39013-0
Martone RL, Herbert J, Dwork A, Schon EA (1988) Transthyretin is synthesized in the mammalian eye. Biochem Biophys Res Commun 151:905–912. https://doi.org/10.1016/S0006-291X(88)80367-X
Dickson PW, Schreiber G (1986) High levels of messenger RNA for transthyretin (prealbumin) in human choroid plexus. Neurosci Lett 66:311–315. https://doi.org/10.1016/0304-3940(86)90037-6
Jacobsson B, Collins VP, Grimelius L et al (1989) Transthyretin immunoreactivity in human and porcine liver, choroid plexus, and pancreatic islets. J Histochem Cytochem 37:31–37. https://doi.org/10.1177/37.1.2642294
Liz MA, Coelho T, Bellotti V et al (2020) A narrative review of the role of transthyretin in health and disease. Neurol Ther 9:395. https://doi.org/10.1007/S40120-020-00217-0
Quintas A, Vaz DC, Cardoso I et al (2001) Tetramer dissociation and monomer partial unfolding precedes protofibril formation in amyloidogenic transthyretin variants. J Biol Chem 276:27207–27213. https://doi.org/10.1074/jbc.M101024200
Bezerra F, Saraiva MJ, Almeida MR (2020) Modulation of the mechanisms driving transthyretin amyloidosis. Front Mol Neurosci 13:234. https://doi.org/10.3389/FNMOL.2020.592644/BIBTEX
Gertz MA (2017) Hereditary ATTR amyloidosis: burden of illness and diagnostic challenges. Am J Manag Care 23:S107–S112
Marcoux J, Mangione PP, Porcari R et al (2015) A novel mechano-enzymatic cleavage mechanism underlies transthyretin amyloidogenesis. EMBO Mol Med 7:1337–1349. https://doi.org/10.15252/emmm.201505357
Bergström J, Gustavsson Å, Hellman U et al (2005) Amyloid deposits in transthyretin-derived amyloidosis: cleaved transthyretin is associated with distinct amyloid morphology. J Pathol 206:224–232. https://doi.org/10.1002/PATH.1759
Ueda M, Horibata Y, Shono M et al (2011) Clinicopathological features of senile systemic amyloidosis: an ante- and post-mortem study. Mod Pathol 24:1533–1544. https://doi.org/10.1038/MODPATHOL.2011.117
Cornwell GG, Murdoch WL, Kyle RA et al (1983) Frequency and distribution of senile cardiovascular amyloid: a clinicopathologic correlation. Am J Med 75:618–623. https://doi.org/10.1016/0002-9343(83)90443-6
Rowczenio DM, Noor I, Gillmore JD et al (2014) Online registry for mutations in hereditary amyloidosis including nomenclature recommendations. Hum Mutat. https://doi.org/10.1002/HUMU.22619
Obi CA, Mostertz WC, Griffin JM, Judge DP (2022) ATTR epidemiology, genetics, and prognostic factors. Methodist Debakey Cardiovasc J 18:17. https://doi.org/10.14797/MDCVJ.1066
Holmgren G, Costa PMP, Andersson C et al (1994) Geographical distribution of TTR met30 carriers in northern Sweden: discrepancy between carrier frequency and prevalence rate. J Med Genet 31:351–354. https://doi.org/10.1136/JMG.31.5.351
Kato-Motozaki Y, Ono K, Shima K et al (2008) Epidemiology of familial amyloid polyneuropathy in Japan: identification of a novel endemic focus. J Neurol Sci 270:133–140. https://doi.org/10.1016/J.JNS.2008.02.019
Inês M, Coelho T, Conceição I et al (2018) Epidemiology of transthyretin familial amyloid polyneuropathy in Portugal: a nationwide study. Neuroepidemiology 51:177–182. https://doi.org/10.1159/000490553
Nativi-Nicolau JN, Karam C, Khella S et al (2021) Screening for ATTR amyloidosis in the clinic: overlapping disorders, misdiagnosis, and multiorgan awareness. Heart Fail Rev 1:3. https://doi.org/10.1007/s10741-021-10080-2
Luigetti M, Romano A, Di PA et al (2020) Diagnosis and treatment of hereditary transthyretin amyloidosis (hATTR) polyneuropathy: current perspectives on improving patient care. Ther Clin Risk Manag 2020:109–123. https://doi.org/10.2147/TCRM.S219979
Gertz M, Adams D, Ando Y et al (2020) Avoiding misdiagnosis: expert consensus recommendations for the suspicion and diagnosis of transthyretin amyloidosis for the general practitioner. BMC Fam Pract 21:1–12. https://doi.org/10.1186/S12875-020-01252-4/FIGURES/3
Ando Y, Coelho T, Berk JL et al (2013) Guideline of transthyretin-related hereditary amyloidosis for clinicians. Orphanet J Rare Dis 8:1–18. https://doi.org/10.1186/1750-1172-8-31/TABLES/2
Adams D, Lozeron P, Lacroix C (2012) Amyloid neuropathies. Curr Opin Neurol 25:564–572. https://doi.org/10.1097/WCO.0B013E328357BDF6
Benson MD, Kincaid JC (2007) The molecular biology and clinical features of amyloid neuropathy. Muscle Nerve 36:411–423. https://doi.org/10.1002/MUS.20821
Pinto MV, Pinto LF, Dias M et al (2019) Late-onset hereditary ATTR V30M amyloidosis with polyneuropathy: characterization of Brazilian subjects from the THAOS registry. J Neurol Sci 403:1–6. https://doi.org/10.1016/J.JNS.2019.05.030
Waddington-Cruz M, Wixner J, Amass L et al (2021) Characteristics of patients with late- vs. early-onset Val30Met transthyretin amyloidosis from the transthyretin amyloidosis outcomes survey (THAOS). Neurol Ther 10:753–766. https://doi.org/10.1007/S40120-021-00258-Z
Koike H, Misu KI, Ikeda SI et al (2002) Type I (transthyretin Met30) familial amyloid polyneuropathy in Japan: early- vs late-onset form. Arch Neurol 59:1771–1776. https://doi.org/10.1001/ARCHNEUR.59.11.1771
Parcha V, Malla G, Ivin MR et al (2022) Association of transthyretin Val122Ile variant with incident heart failure among black individuals. JAMA - J Am Med Assoc 327:1368–1378. https://doi.org/10.1001/JAMA.2022.2896
Chandrashekar P, Alhuneafat L, Mannello M et al (2021) Prevalence and outcomes of p.Val142Ile TTR amyloidosis cardiomyopathy: a systematic review. Circ Genom Precis Med. https://doi.org/10.1161/CIRCGEN.121.003356
Buxbaum JN, Ruberg FL (2017) Transthyretin V122I (pV142I)* cardiac amyloidosis: an age-dependent autosomal dominant cardiomyopathy too common to be overlooked as a cause of significant heart disease in elderly African Americans. Genet Med 19:733–742. https://doi.org/10.1038/GIM.2016.200
Reilly MM, Staunton H, Harding AE et al (1995) Familial amyloid polyneuropathy (TTR ala 60) in north west Ireland: a clinical, genetic, and epidemiological study. J Neurol Neurosurg Psychiatry 59:45–49. https://doi.org/10.1136/jnnp.59.1.45
Sattianayagam PT, Hahn AF, Whelan CJ et al (2012) Cardiac phenotype and clinical outcome of familial amyloid polyneuropathy associated with transthyretin alanine 60 variant. Eur Heart J 33:1120–1127. https://doi.org/10.1093/EURHEARTJ/EHR383
Gospodinova M, Sarafov S, Chamova T et al (2020) Cardiac involvement, morbidity and mortality in hereditary transthyretin amyloidosis because of p.Glu89Gln mutation. J Cardiovasc Med (Hagerstown) 21:688–695. https://doi.org/10.2459/JCM.0000000000001036
Gentile L, Tournev I, Amass L et al (2021) Phenotypic differences of Glu89Gln genotype in ATTR amyloidosis from endemic loci: update from THAOS. Cardiol Ther 10:481–490. https://doi.org/10.1007/S40119-021-00226-6/TABLES/5
González-Duarte A, Cárdenas-Soto K, Bañuelos CE et al (2018) Amyloidosis due to TTR mutations in Mexico with 4 distincts genotypes in the index cases. Orphanet J Rare Dis. https://doi.org/10.1186/S13023-018-0801-Y
González-Duarte A, Soto KC, Martínez-Baños D et al (2012) Familial amyloidosis with polyneuropathy associated with TTR Ser50Arg mutation. Amyloid 19:171–176. https://doi.org/10.3109/13506129.2012.712925
Russo M, Mazzeo A, Stancanelli C et al (2012) Transthyretin-related familial amyloidotic polyneuropathy: description of a cohort of patients with Leu64 mutation and late onset. J Peripher Nerv Syst 17:385–390. https://doi.org/10.1111/J.1529-8027.2012.00436.X
Mazzeo A, Russo M, Di Bella G et al (2015) Transthyretin-related familial amyloid polyneuropathy (TTR-FAP): a single-center experience in Sicily, an Italian Endemic Area. J Neuromuscul Dis 2:39–48. https://doi.org/10.3233/JND-150091
Gagliardi C, Perfetto F, Lorenzini M et al (2018) Phenotypic profile of Ile68Leu transthyretin amyloidosis: an underdiagnosed cause of heart failure. Eur J Heart Fail 20:1417–1425. https://doi.org/10.1002/ejhf.1285
Pastorelli F, Fabbri G, Rapezzi C et al (2021) Amyloid Neurological involvement in Ile68Leu (p.Ile88Leu) ATTR amyloidosis: not only a cardiogenic mutation Neurological involvement in Ile68Leu (p.Ile88Leu) ATTR amyloidosis: not only a cardiogenic mutation. Amyloid 28:173–181. https://doi.org/10.1080/13506129.2021.1917357
Damy T, Kristen AV, Suhr OB et al (2019) Transthyretin cardiac amyloidosis in continental Western Europe: an insight through the transthyretin amyloidosis outcomes survey (THAOS). Eur Heart J 43:391–400. https://doi.org/10.1093/EURHEARTJ/EHZ173
Hastrup Svendsen I, Steensgaard-Hansen F, Nordvåg BY (1998) A clinical, echocardiographic and genetic characterization of a Danish kindred with familial amyloid transthyretin methionine 111 linked cardiomyopathy. Eur Heart J 19:782–789. https://doi.org/10.1053/EUHJ.1997.0841
Dori A, Arad M, Wasserstrum Y et al (2023) Ser77Tyr transthyretin amyloidosis in Israel: initial manifestations and diagnostic features. Ann Clin Transl Neurol 10:553–567. https://doi.org/10.1002/ACN3.51741
Davion JB, Bocquillon P, Cassim F et al (2021) Electro-clinical presentation of hereditary transthyretin related amyloidosis when presenting as a polyneuropathy of unknown origin in northern France. Rev Neurol (Paris) 177:1160–1167. https://doi.org/10.1016/J.NEUROL.2021.02.392
Mariani LL, Lozeron P, Théaudin M et al (2015) Genotype-phenotype correlation and course of transthyretin familial amyloid polyneuropathies in France. Ann Neurol 78:901–916. https://doi.org/10.1002/ANA.24519
Antonopoulos AS, Panagiotopoulos I, Kouroutzoglou A et al (2022) Prevalence and clinical outcomes of transthyretin amyloidosis: a systematic review and meta-analysis. Eur J Heart Fail 24:1677–1696. https://doi.org/10.1002/EJHF.2589
Xu J, Yang M, Pan X et al (2017) Transthyretin-related hereditary amyloidosis with recurrent vomiting and renal insufficiency as the initial presentation: a case report. Medicine 96:e5737. https://doi.org/10.1097/MD.0000000000005737
Jacobson DR, McFarlin DE, Kane I, Buxbaum JN (1992) Transthyretin Pro55, a variant associated with early-onset, aggressive, diffuse amyloidosis with cardiac and neurologic involvement. Hum Genet 89:353–356. https://doi.org/10.1007/BF00220559
Yamamoto K, Hsu S-P, Yoshida K et al (1994) Familial amyloid polyneuropathy in Taiwan: identification of transthyretin variant (Leu55–>Pro). Muscle Nerve 17:637–641. https://doi.org/10.1002/MUS.880170611
Lee YJ, Oh J, Hwang SK et al (2019) Extremely early onset transthyretin familial amyloid polyneuropathy with a Leu55Pro mutation: a pediatric case report and literature review. Neuropediatrics 50:322–326. https://doi.org/10.1055/S-0039-1693145
Murakami T, Yokoyama T, Mizuguchi M et al (2021) A low amyloidogenic E61K transthyretin mutation may cause familial amyloid polyneuropathy. J Neurochem 156:957–966. https://doi.org/10.1111/JNC.15162
Chu X, Wang M, Tang R et al (2022) Clinical and biochemical characterization of hereditary transthyretin amyloidosis caused by E61K mutation. Front Mol Neurosci 15:1003303. https://doi.org/10.3389/FNMOL.2022.1003303
Murakami T, Nishimura H, Nagai T et al (2017) Clinical and pathological findings in familial amyloid polyneuropathy caused by a transthyretin E61K mutation. J Neurol Sci 381:55–58. https://doi.org/10.1016/J.JNS.2017.08.017
Terazaki H, Ando Y, Misumi S et al (1999) A novel compound heterozygote (FAP ATTR Arg104His/ATTR Val30Met) with high serum transthyretin (TTR) and retinol binding protein (RBP) levels. Biochem Biophys Res Commun 264:365–370. https://doi.org/10.1006/BBRC.1999.1514
Coelho T, Chorão R, Sousa A et al (1996) Compound heterozygotes of transthyretin Met30 and transthyretin Met119 are protected from the devastating effects of familial amyloid polyneuropathy. Neuromuscul Disord 6:S20. https://doi.org/10.1016/0960-8966(96)88826-2
Sekijima Y, Dendle MT, Wiseman RL et al (2006) R104H may suppress transthyretin amyloidogenesis by thermodynamic stabilization, but not by the kinetic mechanism characterizing T119 interallelic trans-suppression. Amyloid 13:57–66. https://doi.org/10.1080/13506120600722449
Hammarström P, Jiang X, Hurshman AR et al (2002) Sequence-dependent denaturation energetics: a major determinant in amyloid disease diversity. Proc Natl Acad Sci U S A 99(Suppl 4):16427–16432. https://doi.org/10.1073/PNAS.202495199
Ruzhansky K, Scoon J, Weimer LH et al (2014) Discordant phenotype in monozygotic female twins with Lys35Thr TTR familial amyloidotic polyneuropathy. J Clin Neuromuscul Dis 16:1–6. https://doi.org/10.1097/CND.0000000000000040
Da Cunha Saporta MA, Plante-Bordeneuve V, Misrahi M, Cruz MW (2009) Discordant expression of familial amyloid polyneuropathy in monozygotic Brazilian twins. Amyloid 16:38–41. https://doi.org/10.1080/13506120802676955
Munar-Qués M, Pedrosa JL, Coelho T et al (1999) Two pairs of proven monozygotic twins discordant for familial amyloid neuropathy (FAP) TTR Met 30. J Med Genet 36:629–632
Holmgren G, Wikström L, Lundgren HE, Suhr OB (2004) Discordant penetrance of the trait for familial amyloidotic polyneuropathy in two pairs of monozygotic twins. J Intern Med 256:453–456. https://doi.org/10.1111/J.1365-2796.2004.01399.X
Hou X, Aguilar MI, Small DH (2007) Transthyretin and familial amyloidotic polyneuropathy. FEBS J 274:1637–1650. https://doi.org/10.1111/J.1742-4658.2007.05712.X
Schmidt HHJ, Barroso F, González-Duarte A et al (2016) Management of asymptomatic gene carriers of transthyretin familial amyloid polyneuropathy. Muscle Nerve 54:353–360. https://doi.org/10.1002/MUS.25210
Parman Y, Adams D, Obici L et al (2016) Sixty years of transthyretin familial amyloid polyneuropathy (TTR-FAP) in Europe: where are we now? A European network approach to defining the epidemiology and management patterns for TTR-FAP. Curr Opin Neurol 29(Suppl 1):S3–S13. https://doi.org/10.1097/WCO.0000000000000288
Sekijima Y, Ueda M, Koike H et al (2018) Diagnosis and management of transthyretin familial amyloid polyneuropathy in Japan: red-flag symptom clusters and treatment algorithm. Orphanet J Rare Dis 13:1–17. https://doi.org/10.1186/S13023-017-0726-X/FIGURES/5
Sousa A, Coelho T, Barros J, Sequeiros J (1995) Genetic epidemiology of familial amyloidotic polyneuropathy (FAP)-type I in Póvoa do Varzim and Vila do Conde (north of Portugal). Am J Med Genet 60:512–521. https://doi.org/10.1002/AJMG.1320600606
Ikeda SI, Hanyu N, Hongo M et al (1987) Hereditary generalized amyloidosis with polyneuropathy: clinicopathological study of 65 Japanese patients. Brain 110(Pt 2):315–337. https://doi.org/10.1093/BRAIN/110.2.315
Misu K-I, Hattori N, Nagamatsu M et al (1999) Late-onset familial amyloid polyneuropathy type I (transthyretin Met30-associated familial amyloid polyneuropathy) unrelated to endemic focus in Japan Clinicopathological and genetic features. Brain 122:1951–1962
Sousa A, Anderson R, Drugge U et al (1993) Familial amyloidotic polyneuropathy in Sweden: geographical distribution, age of onset, and prevalence. Hum Hered 43:288–294. https://doi.org/10.1159/000154146
Rapezzi C, Quarta CC, Riva L et al (2010) Transthyretin-related amyloidoses and the heart: a clinical overview. Nat Rev Cardiol 7:398–408. https://doi.org/10.1038/NRCARDIO.2010.67
Yamashita T, Ueda M, Misumi Y et al (2018) Genetic and clinical characteristics of hereditary transthyretin amyloidosis in endemic and non-endemic areas: experience from a single-referral center in Japan. J Neurol 265:134–140. https://doi.org/10.1007/S00415-017-8640-7/FIGURES/6
Dispenzieri A, Coelho T, Conceição I et al (2022) Clinical and genetic profile of patients enrolled in the transthyretin amyloidosis outcomes survey (THAOS): 14-year update. Orphanet J Rare Dis 17:1–12. https://doi.org/10.1186/S13023-022-02359-W/TABLES/3
Cisneros-Barroso E, González-Moreno J, Rodríguez A et al (2020) Anticipation on age at onset in kindreds with hereditary ATTRV30M amyloidosis from the Majorcan cluster. Amyloid 27:254–258. https://doi.org/10.1080/13506129.2020.1789580
Andreou S, Panayiotou E, Michailidou K et al (2018) Epidemiology of ATTRV30M neuropathy in Cyprus and the modifier effect of complement C1q on the age of disease onset. Amyloid 25:220–226. https://doi.org/10.1080/13506129.2018.1534731
Yamamoto K, Ikeda SI, Hanyu N et al (1998) A pedigree analysis with minimised ascertainment bias shows anticipation in Met30-transthyretin related familial amyloid polyneuropathy. J Med Genet 35:23–30. https://doi.org/10.1136/JMG.35.1.23
Gorram F, Olsson M, Alarcon F et al (2021) New data on the genetic profile and penetrance of hereditary Val30Met transthyretin amyloidosis in Sweden. Amyloid 28:84–90. https://doi.org/10.1080/13506129.2020.1841623
Lemos C, Coelho T, Alves-Ferreira M et al (2014) Overcoming artefact: anticipation in 284 Portuguese kindreds with familial amyloid polyneuropathy (FAP) ATTRV30M. J Neurol Neurosurg Psychiatry 85:326–330. https://doi.org/10.1136/JNNP-2013-305383
McCabe ERB (2017) Modifier genes: moving from pathogenesis to therapy. Mol Genet Metab 122:1–3. https://doi.org/10.1016/J.YMGME.2017.05.018
Nylander PO, Beckman L, Holmgren G, Steen L (1990) Association of C3 and C4A complement types with familial amyloidotic polyneuropathy. Hum Hered 40:272–277. https://doi.org/10.1159/000153944
Soares ML, Coelho T, Sousa A et al (2005) Susceptibility and modifier genes in Portuguese transthyretin V30M amyloid polyneuropathy: complexity in a single-gene disease. Hum Mol Genet 14:543–553. https://doi.org/10.1093/HMG/DDI051
Dardiotis E, Koutsou P, Zamba-Papanicolaou E et al (2009) Complement C1Q polymorphisms modulate onset in familial amyloidotic polyneuropathy TTR Val30Met. J Neurol Sci 284:158–162. https://doi.org/10.1016/J.JNS.2009.05.018
Santos D, Coelho T, Alves-Ferreira M et al (2016) Variants in RBP4 and AR genes modulate age at onset in familial amyloid polyneuropathy (FAP ATTRV30M). Eur J Hum Genet 24:756–760. https://doi.org/10.1038/ejhg.2015.180
Santos D, Coelho T, Alves-Ferreira M et al (2017) Familial amyloid polyneuropathy in Portugal: new genes modulating age-at-onset. Ann Clin Transl Neurol 4:98–105. https://doi.org/10.1002/acn3.380
Santos D, Coelho T, Alves-Ferreira M et al (2019) Large normal alleles of ATXN2 decrease age at onset in transthyretin familial amyloid polyneuropathy Val30Met patients. Ann Neurol 85:251–258. https://doi.org/10.1002/ANA.25409
Dias A, Santos D, Coelho T et al (2019) C1QA and C1QC modify age-at-onset in familial amyloid polyneuropathy patients. Ann Clin Transl Neurol 6:748–754. https://doi.org/10.1002/ACN3.748
Arnold AP (2017) A general theory of sexual differentiation. J Neurosci Res 95:291–300. https://doi.org/10.1002/JNR.23884
Mauvais-Jarvis F, Bairey Merz N, Barnes PJ et al (2020) Sex and gender: modifiers of health, disease, and medicine. The Lancet 396:565–582. https://doi.org/10.1016/S0140-6736(20)31561-0
Sousa A, Coelho T, Sequeiros J (1991) Parental transmission and age-of-onset in familial amyloidotic polineuropathy (Portuguese Type). Amyloid Amyloidosis 1990:691–693. https://doi.org/10.1007/978-94-011-3284-8_170
Planté-Bordeneuve V, Carayol J, Ferreira A et al (2003) Genetic study of transthyretin amyloid neuropathies: carrier risks among French and Portuguese families. J Med Genet. https://doi.org/10.1136/JMG.40.11.E120
Saporta MAC, Zaros C, Cruz MW et al (2009) Penetrance estimation of TTR familial amyloid polyneuropathy (type I) in Brazilian families. Eur J Neurol 16:337–341. https://doi.org/10.1111/J.1468-1331.2008.02429.X
Caponetti AG, Rapezzi C, Gagliardi C et al (2021) Sex-related risk of cardiac involvement in hereditary transthyretin amyloidosis: insights from THAOS. JACC Heart Fail 9:736–746. https://doi.org/10.1016/J.JCHF.2021.05.005
Maurer MS, Hanna M, Grogan M et al (2016) Genotype and phenotype of transthyretin cardiac amyloidosis: THAOS (transthyretin amyloid outcome survey). J Am Coll Cardiol 68:161–172. https://doi.org/10.1016/J.JACC.2016.03.596
Lane T, Fontana M, Martinez-Naharro A et al (2019) Natural history, quality of life, and outcome in cardiac transthyretin amyloidosis. Circulation 140:16–26. https://doi.org/10.1161/CIRCULATIONAHA.118.038169
Batra J, Rosenblum H, Defilippis EM et al (2021) Sex differences in the phenotype of transthyretin cardiac amyloidosis due to Val122Ile mutation: insights from noninvasive pressure-volume analysis. J Card Fail 27:67–74. https://doi.org/10.1016/J.CARDFAIL.2020.08.007
Rapezzi C, Riva L, Quarta CC et al (2008) Gender-related risk of myocardial involvement in systemic amyloidosis. Amyloid 15:40–48. https://doi.org/10.1080/13506120701815373
Gonçalves I, Alves CH, Quintela T et al (2008) Transthyretin is up-regulated by sex hormones in mice liver. Mol Cell Biochem 317:137–142. https://doi.org/10.1007/S11010-008-9841-2/FIGURES/2
Sousa A, Coelho T, Lobato L, Sequeiros J (1991) Anticipation of age-of-onset in familial amyloidotic polineuropathy (Portuguese type). Amyloid Amyloidosis 1990:694–697. https://doi.org/10.1007/978-94-011-3284-8_171
Drugge U, Andersson R, Chizari F et al (1993) Familial amyloidotic polyneuropathy in Sweden: a pedigree analysis. J Med Genet 30:388. https://doi.org/10.1136/JMG.30.5.388
Hellman U, Alarcon F, Lundgren HE et al (2008) Heterogeneity of penetrance in familial amyloid polyneuropathy, ATTR Val30Met, in the Swedish population. Amyloid 15:181. https://doi.org/10.1080/13506120802193720
Bonaïti B, Alarcon F, Bonaïti-Pellié C, Planté-Bordeneuve V (2009) Parent-of-origin effect in transthyretin related amyloid polyneuropathy. Amyloid 16:149–150. https://doi.org/10.1080/13506120903093944
Olsson M, Hellman U, Planté-Bordeneuve V et al (2009) Mitochondrial haplogroup is associated with the phenotype of familial amyloidosis with polyneuropathy in Swedish and French patients. Clin Genet 75:163–168. https://doi.org/10.1111/J.1399-0004.2008.01097.X
Bonaïti B, Olsson M, Hellman U et al (2010) TTR familial amyloid polyneuropathy: does a mitochondrial polymorphism entirely explain the parent-of-origin difference in penetrance? Eur J Hum Genet 18:948–952. https://doi.org/10.1038/EJHG.2010.36
Santos D, Santos MJ, Alves-Ferreira M et al (2018) mtDNA copy number associated with age of onset in familial amyloid polyneuropathy. J Neurol Neurosurg Psychiatry 89:300–304. https://doi.org/10.1136/JNNP-2017-316657
Pagni S, Mills JD, Frankish A et al (2022) Non-coding regulatory elements: potential roles in disease and the case of epilepsy. Neuropathol Appl Neurobiol. https://doi.org/10.1111/NAN.12775
French JD, Edwards SL (2020) The role of noncoding variants in heritable disease. Trends Genet 36:880–891. https://doi.org/10.1016/J.TIG.2020.07.004
Soares ML, Coelho T, Sousa A et al (2004) Haplotypes and DNA sequence variation within and surrounding the transthyretin gene: genotype-phenotype correlations in familial amyloid polyneuropathy (V30M) in Portugal and Sweden. Eur J Hum Genet 12:225–237. https://doi.org/10.1038/SJ.EJHG.5201095
Polimanti R, Di Girolamo M, Manfellotto D, Fuciarelli M (2013) Functional variation of the transthyretin gene among human populations and its correlation with amyloidosis phenotypes. Amyloid 20:256–262. https://doi.org/10.3109/13506129.2013.844689
Polimanti R, Di Girolamo M, Manfellotto D, Fuciarelli M (2014) In silico analysis of TTR gene (coding and non-coding regions, and interactive network) and its implications in transthyretin-related amyloidosis. Amyloid 21:154–162. https://doi.org/10.3109/13506129.2014.900487
Iorio A, De Lillo A, De Angelis F et al (2017) Non-coding variants contribute to the clinical heterogeneity of TTR amyloidosis. Eur J Hum Genet 25:1055–1060. https://doi.org/10.1038/EJHG.2017.95
De Lillo A, De Angelis F, Di Girolamo M et al (2019) Phenome-wide association study of TTR and RBP4 genes in 361,194 individuals reveals novel insights in the genetics of hereditary and wildtype transthyretin amyloidoses. Hum Genet 138:1331–1340. https://doi.org/10.1007/s00439-019-02078-6
Alves-Ferreira M, Coelho T, Santos D et al (2018) A trans-acting factor may modify age at onset in familial amyloid polyneuropathy ATTRV30M in Portugal. Mol Neurobiol 55:3676–3683. https://doi.org/10.1007/S12035-017-0593-4
Alves-Ferreira M, Azevedo A, Coelho T et al (2021) Beyond Val30Met transthyretin (TTR): variants associated with age-at-onset in hereditary ATTRv amyloidosis. Amyloid 28:100–106. https://doi.org/10.1080/13506129.2020.1857236
Mangione PP, Porcari R, Gillmore JD et al (2014) Proteolytic cleavage of Ser52Pro variant transthyretin triggers its amyloid fibrillogenesis. Proc Natl Acad Sci 111:1539–1544. https://doi.org/10.1073/pnas.1317488111
Ihse E, Ybo A, Suhr OB et al (2008) Amyloid fibril composition is related to the phenotype of hereditary transthyretin V30M amyloidosis. J Pathol 216:253–261. https://doi.org/10.1002/PATH.2411
Koike H, Ando Y, Ueda M et al (2009) Distinct characteristics of amyloid deposits in early- and late-onset transthyretin Val30Met familial amyloid polyneuropathy. J Neurol Sci 287:178–184. https://doi.org/10.1016/J.JNS.2009.07.028
Ihse E, Rapezzi C, Merlini G et al (2013) Amyloid fibrils containing fragmented ATTR may be the standard fibril composition in ATTR amyloidosis. Amyloid 20:142–150. https://doi.org/10.3109/13506129.2013.797890
Liepnieks JJ, Benson MD (2007) Progression of cardiac amyloid deposition in hereditary transthyretin amyloidosis patients after liver transplantation. Amyloid 14:277–282. https://doi.org/10.1080/13506120701614032
Yazaki M, Tokuda T, Nakamura A et al (2000) Cardiac amyloid in patients with familial amyloid polyneuropathy consists of abundant wild-type transthyretin. Biochem Biophys Res Commun 274:702–706. https://doi.org/10.1006/bbrc.2000.3203
Ihse E, Suhr OB, Hellman U, Westermark P (2011) Variation in amount of wild-type transthyretin in different fibril and tissue types in ATTR amyloidosis. J Mol Med (Berl) 89:171–180. https://doi.org/10.1007/S00109-010-0695-1
Gustafsson S, Ihse E, Henein MY et al (2012) Amyloid fibril composition as a predictor of development of cardiomyopathy after liver transplantation for hereditary transthyretin amyloidosis. Transplantation 93:1017–1023. https://doi.org/10.1097/TP.0B013E31824B3749
Moosavi A, Ardekani AM (2016) Role of epigenetics in biology and human diseases. Iran Biomed J 20:246. https://doi.org/10.22045/IBJ.2016.01
De Lillo A, Pathak GA, De Angelis F et al (2020) Epigenetic profiling of Italian patients identified methylation sites associated with hereditary transthyretin amyloidosis. Clin Epigenetics 12:1–12. https://doi.org/10.1186/S13148-020-00967-6/TABLES/2
Pathak GA, Wendt FR, De Lillo A et al (2021) Epigenomic profiles of African–American transthyretin Val122Ile carriers reveals putatively dysregulated amyloid mechanisms. Circ Genom Precis Med. https://doi.org/10.1161/CIRCGEN.120.003011
Olsson M, Norgren N, Obayashi K et al (2010) A possible role for miRNA silencing in disease phenotype variation in Swedish transthyretin V30M carriers. BMC Med Genet. https://doi.org/10.1186/1471-2350-11-130
Norgren N, Hellman U, Ericzon BG et al (2012) Allele specific expression of the transthyretin gene in Swedish patients with hereditary transthyretin amyloidosis (ATTR V30M) is similar between the two alleles. PLoS ONE. https://doi.org/10.1371/JOURNAL.PONE.0049981
Hardell L, Holmgren G, Steen L et al (1995) Occupational and other risk factors for clinically overt familial amyloid polyneuropathy. Epidemiology 6:598–601. https://doi.org/10.1097/00001648-199511000-00006
Noguchi H, Ohta M, Wakasugi S et al (2002) Effect of the intestinal flora on amyloid deposition in a transgenic mouse model of familial amyloidotic polyneuropathy. Exp Anim 51:309–316. https://doi.org/10.1538/EXPANIM.51.309
Inoue S, Ohta M, Li Z et al (2008) Specific pathogen free conditions prevent transthyretin amyloidosis in mouse models. Transgenic Res 17:817–826. https://doi.org/10.1007/S11248-008-9180-9
Petrakis I, Mavroeidi V, Stylianou K et al (2013) Human TTRV30M localization within podocytes in a transgenic mouse model of transthyretin related amyloidosis: does the environment play a role? Transgenic Res 22:101–116. https://doi.org/10.1007/S11248-012-9632-0
Gui B, Slone J, Huang T (2017) Perspective: Is random monoallelic expression a contributor to phenotypic variability of autosomal dominant disorders? Front Genet 8:191. https://doi.org/10.3389/FGENE.2017.00191/BIBTEX
Yordanova I, Pavlova Z, Kirov A et al (2019) Monoallelic expression of the TTR gene as a contributor to the age at onset and penetrance of TTR-related amyloidosis. Gene 705:16–21. https://doi.org/10.1016/J.GENE.2019.04.030
Freed D, Stevens EL, Pevsner J (2014) Somatic mosaicism in the human genome. Genes 5:1064–1094. https://doi.org/10.3390/GENES5041064
Federico C, Dugo K, Bruno F et al (2017) Somatic mosaicism with reversion to normality of a mutated transthyretin allele related to a familial amyloidotic polyneuropathy. Hum Genet 136:867–873. https://doi.org/10.1007/S00439-017-1810-Y
Acknowledgements
This work was supported by Fundação para a Ciência e a Tecnologia (FCT) via research grant 2022.01656.PTDC. Estefânia Carvalho was financed by a fellowship grant UI/BD/154392/2023 and Mariana Santos was supported by the program DL 57/2016 – Norma Transitória, both from FCT.
Funding
Open access funding provided by FCT|FCCN (b-on). Fundação para a Ciência e a Tecnologia, 2022.01656.PTDC, PI: Carolina Lemos.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors have no competing interests to declare that are relevant to the content of this article. The authors have no relevant financial or non-financial interests to disclose.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Carvalho, E., Dias, A., Coelho, T. et al. Hereditary transthyretin amyloidosis: a myriad of factors that influence phenotypic variability. J Neurol (2024). https://doi.org/10.1007/s00415-024-12509-8
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00415-024-12509-8