
Abstract
Hereditary ataxia is a heterogeneous group of complex neurological disorders. Next-generation sequencing methods have become a great help in clinical diagnostics, but it may remain challenging to determine if a genetic variant is the cause of the patient’s disease. We compiled a consecutive single-center series of 87 patients from 76 families with progressive ataxia of known or unknown etiology. We investigated them clinically and genetically using whole exome or whole genome sequencing. Test methods were selected depending on family history, clinical phenotype, and availability. Genetic results were interpreted based on the American College of Medical Genetics criteria. For high-suspicion variants of uncertain significance, renewed bioinformatical and clinical evaluation was performed to assess the level of pathogenicity. Thirty (39.5%) of the 76 families had received a genetic diagnosis at the end of our study. We present the predominant etiologies of hereditary ataxia in a Swedish patient series. In two families, we established a clinical diagnosis, although the genetic variant was classified as “of uncertain significance” only, and in an additional three families, results are pending. We found a pathogenic variant in one family, but we suspect that it does not explain the complete clinical picture. We conclude that correctly interpreting genetic variants in complex neurogenetic diseases requires genetics and clinical expertise. The neurologist’s careful phenotyping remains essential to confirm or reject a diagnosis, also by reassessing clinical findings after a candidate genetic variant is suggested. Collaboration between neurology and clinical genetics and combining clinical and research approaches optimizes diagnostic yield.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Ataxia is a neurological sign with incoordination of movements resulting from dysfunction of the cerebellum and its afferent and efferent pathways. According to the location of the underlying dysfunction, ataxias can be classified as cerebellar, sensory, and vestibular. The main manifestations of ataxia are gait impairment, limb incoordination, nystagmus, and slurred speech. Ataxia can be subdivided into sporadic, hereditary, and acquired forms [1, 2]. Monogenetic ataxias are chronically progressive neurological disorders that can be further categorized by their inheritance pattern and underlying genetic causes into autosomal dominant cerebellar ataxias (ADCAs), autosomal recessive cerebellar ataxias (ARCAs), X-linked cerebellar, and mitochondrial ataxias. The hereditary ataxias are a large and heterogeneous group of diseases with variable genetic, clinical, pathogenic, pathophysiologic, and neuropathologic features [3].
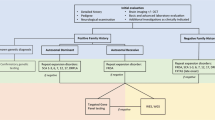
Massively parallel sequencing, also called Next-generation sequencing (NGS), is increasingly being used for clinical diagnostics and greatly facilitates the identification of the underlying genetic causes. However, the diagnostic yield of NGS for ataxia is only between 12 and 52% [4], and approximately 30% of patients with clinical suspicion of ADCAs and 50% of ARCAs remain undiagnosed [5,6,7]. In recent years, the technical methodology to identify various types of genetic variants through NGS has dramatically increased, but it can remain very challenging to firmly decide if a detected genetic variant indeed is the cause of the examined patient’s or family’s disorder. In 2015, the American College of Medical Genetics and Genomics (ACMG) developed guidelines for the interpretation of sequence variants that have become quasi-universally accepted and applied. However, it has also been pointed out that they may be suboptimal for diagnosing very rare disorders such as ataxias [8], especially when they are used in a standard clinical setting where only those results of genetic analyses that are considered highly likely disease causes are sent to the neurologists [9].
Here, we present a cohort of patients with ataxia of known or unknown etiology from southern Sweden. These patients and many of their family members have been studied clinically and radiologically, and we investigated the molecular etiology of previously undiagnosed cases. In our study, we used a collaborative approach where possible genetic disease causes were revealed to the treating neurologists and discussed between neurologists, bioinformaticians as well as medical and clinical geneticists.
Methods
Recruitment and selection of patients
Patients were identified through a search for the diagnosis of hereditary ataxia (ICD-10 Version: 2019 (who.int) G11.0, G11.1, G11.2, G11.3, G11.8 or G11.9) between the years of 2011–2020 in the diagnosis register of the department of neurology at Skåne University Hospital. We also recruited patients with the diagnosis of hereditary ataxia who were referred by other neurologists, from contact with the Swedish patient organization SCA-Network, or through their families. Patients with SCA3 were specifically targeted for a multicenter study. Patients with Friedreich ataxia from our center were already included in a previous study [10] and these were not approached again. A research nurse sent a letter with detailed information about the project, written by the research group, and a request to send back a response form (interested/not interested) by mail to each patient found to have a clinical diagnosis of hereditary ataxia. A more detailed description of patient recruitment in this study has recently been published [11].
Clinical examination
All the included patients were seen by a study doctor and a study nurse during a research visit at our clinic, or during home visits. All were interviewed following a standardized checklist for medical and family history. Results from brain imaging, nerve conduction studies, analysis of cerebral spinal fluid, and genetic examination (if available) were retrieved from the clinical records. The neurological examination was conducted using a standardized protocol for the examination of patients with ataxia, focusing on speech, eye movements, coordination of movements, and gait. To assess the disease severity, we used the Scale for the Assessment and Rating of Ataxia (SARA) [12]. Family pedigrees were drawn based on probands’ and relatives’ information.
Radiology reports and when available, original images, were reviewed by the authors for cerebellar and spinal cord atrophy. Records from nerve conduction studies, if existing, were evaluated for peripheral nerve impairment. Blood samples were collected by our research nurse from each patient after the clinical examination. In addition, cerebral spinal fluid was collected by the main author from patients with spinocerebellar ataxia type 3 (SCA3) for a multicenter study [13, 14].
Genetic analyses
Some of the patients recruited here underwent genetic analyses as part of their clinical workup, others were analyzed within this study:
-
(a)
Prior to this study: some patients had already an established genetic diagnosis. However, blood samples were collected for storage in the biobank for future analyses. Many patients had been analyzed for repeat expansions causing SCA1, 2, 3, 6 and 7 in a test package that was widely used at our hospital; others had been examined for particular genes based on their phenotype. More recently, a number of patients had undergone gene panel analyses based on targeted resequencing, Whole Exome Sequencing (WES) or Whole Genome Sequencing (WGS).
-
(b)
Within this study: the patients without a genetic diagnosis were tested using WES or WGS methods. The choice between WES and WGS was made based on the clinical presentation, family history, and availability at the time of testing. Study bioinformaticians also re-analyzed available raw data from clinical WES analyses from three patients. Most WES or WGS analyses were performed at the Center for Translational Genomics at Lund University. Additionally, WES, WGS, and genotyping of family members was performed by Centogene, Rostock, Germany, or BluePrint Genetics, Helsinki, Finland.
Bioinformatic analyses
WES and WGS data were analyzed for single nucleotide variants and short insertions or deletions, repeat expansions (short tandem repeats), and copy number variants (deletions, duplications). Variant Call Format (VCF) files were annotated with Variant Effect Predictor (VEP).
Single nucleotide variants (SNVs) and short insertions and deletions were prioritized based on their CADD score [15]. SNVs were annotated further with the dbNSFP plugin, while synonymous and intronic variants were evaluated with appropriate freely available software (Trap score [16], PredictSNP2 [17]). Splice region variants were interpreted with spliceAI [18]. Vt tool was used for calls’ decomposition [19]. Only rare variants (MAF < 0.01) in gnomAD non-Finnish Europeans, 1000 Genomes, and SweGen [20] frequency databases and with genotype quality higher than 20 were selected. Variants in genes present in an in-house gene list containing 1020 related ataxia genes compiled from the Human Phenotype Ontology (HPO) entry on ataxia (HP:0001251, accessed in 2021) and ataxia gene panels were assessed further. The variant classification was performed based on the guidelines published by the American College of Medical Genetics and Genomics (ACMG) in 2015 [21], and we only considered candidate variants those that were classified as pathogenic, likely pathogenic or of uncertain significance (VUS) by the classifiers used (Varsome [22], Franklin by Genoox Franklin (genoox.com)). We also searched the variants in ClinVar database.
Expanded short tandem repeats were detected with ExpansionHunter 5.0.0, using the bundled hg19 short tandem repeat catalog and default settings. A total of 44 known short tandem repeats that may be implied in ataxia were selected from ExpansionHunter 5.0.0 and the literature [23], and examined. The outputs of ExpansionHunter were then visualized with REViewer 0.2.7, facilitating manual curation of the reads spanning the short tandem repeat. This manual curation followed the tutorial and guidelines available at https://www.illumina.com/science/genomics-research/articles/reviewer-alignments-short-reads-long-repeat.html and REViewer/docs at master · Illumina/REViewer · GitHub.
Copy number variants (CNVs) of the WES data were searched using ExomeDepth [24]. ExomeDepth is an R package with which CNVs can be detected from targeted sequence data, typically exome sequencing.
A different software is required to search for CNVs in WGS samples. GATK was chosen for this purpose, run in 'cohort' mode with otherwise default settings as described in the official guide available here: https://gatk.broadinstitute.org/hc/en-us/articles/360035531152--How-to-Call-common-and-rare-germline-copy-number-variants. CNVs were analyzed in a manually curated in-house gene list of 320 genes, compiled from gene lists used for clinical diagnostics at the Dept. of Clinical genetics in Lund and commercially available ataxia gene panels. The gene list can be made available by the authors upon request.
“Post-NGS phenotyping”
Very rare variants in ataxia-related genes that were classified as pathogenic, likely pathogenic, or of uncertain significance with a high likelihood of pathogenicity were discussed in rounds of conferences with clinicians, bioinformaticians, and a medical geneticist. These results were then verified using post-NGS phenotyping. The first author reevaluated all new genetic findings in relationship to the presenting clinical phenotype, genetic databases, and reported cases in the literature. When the findings did match (see criteria below), orthogonal validation testing was performed if necessary and feasible.
The following criteria made us consider a variant to be compatible with the patient’s/family’s phenotype:
-
Family history is consistent with the mode of inheritance of the disorder
-
The patient and the affected family members had a well-defined syndrome; we were looking for a specific signature of neurological and/or non-neurological disease phenotypes and compared between the patient and previous publications about the particular disorder, and, in the case of families, between affected individuals of a family [21]
-
Careful re-appreciation of the genetic results and database findings (variant frequency in the population, prediction tools, genotype, quality of sequencing, validity of bioinformatic methods, etc.).
In several instances, if patients described other family members with similar symptoms, these family members were invited to participate in our study. We also tested unaffected family members for analyses of co-segregation of disease with genotype or to determine if two variants in the same gene in a proband were in cis or in trans.
Results
As described previously, 158 ataxia patients were identified and contacted [11]. In the present study, 87 patients with the diagnosis of hereditary ataxia were included. From 91 patients who had been examined within the study, four were excluded because of other diagnoses that were found after clinical examination and reevaluation of each patient’s neurological records: one patient was diagnosed with multiple system atrophy type C, one with an adult form of spinal muscular atrophy, one with functional dystonia and one with a paramalignant syndrome. Patient demographic data is presented in Table 1.
Twenty-seven patients had a genetic diagnosis before inclusion and two of their relatives were found to be presymptomatic carriers; together they came from 19 families (Online Resource 1). Fifteen patients, from 11 families, received a confirmed genetic diagnosis within our study (Table 2). At the end of this study, 44 (50.6%) of 87 individuals from 30 (39.5%) of 76 families had a confirmed genetic diagnosis. In an additional 8 probands, we remained uncertain if the variants in ataxia genes identified can explain the patient’s phenotype; we encountered different diagnostic situations as outlined in Online Resource 2.
Figure 1 shows the pedigrees of patients and families in Table 2 and Online Resource 2. Figure 2 summarizes the genetic diagnoses in this patient series. In the following paragraphs, we present clinical descriptions; additional and more detailed clinical descriptions are provided in Online Resource 3. The detailed phenotypes and pedigrees of index patients P1011 with ataxia pancytopenia syndrome and SAMD9L p.(Arg986Cys) variant and P1037 with Brown–Vialetto–Van Laere syndrome-2 have been published earlier [25, 26]. Patients were of Swedish origin unless stated otherwise.


Pedigrees of patients and families examined genetically within this study. Round symbols indicate females, square symbols males; diagonal line indicates that the individual is deceased; patient identifiers and age at onset in years are provided below symbols; solid black symbols indicate ataxia, black dots indicate possible ataxia (acc. to family history); yellow color indicates possible dementia, red color indicates dementia. CANVAS cerebellar ataxia, neuropathy and vestibular areflexia; HD Huntington's disease, het heterozygosity, hom homozygosity, SCA28 spinocerebellar ataxia 28, SCA34 spinocerebellar ataxia 34, SCA48 spinocerebellar ataxia 48, SCA5 spinocerebellar ataxia 5, SCAR16 autosomal recessive spinocerebellar ataxia 16, SPG4 spastic paraplegia 4, SPG7 spastic paraplegia 7, SPG76 spastic paraplegia 76

Subtypes of ataxia encountered in this study. Established molecular ataxia diagnoses in study participants as listed in Online Resources 1 and 2. Patients with SCA3 were recruited from a larger geographical area because they were included in multi-center studies on this disease. Nine additional patients with Friedreich ataxia from 7 families from our hospital’s uptake area had previously been included in another study and were not contacted again. ADCADN Autosomal dominant cerebellar ataxia, deafness, and narcolepsy, AT ataxia telangiectasia, ATXPC ataxia-pancytopenia syndrome, BVVLS2 Brown–Vialetto–Van Laere syndrome-2, CANVAS cerebellar ataxia, neuropathy and vestibular areflexia, EA episodic ataxia, FRDA Friedreich’s ataxia, HD Huntington's disease, SCA spinocerebellar ataxia, SCAR16 spinocerebellar ataxia autosomal recessive 16, SPG spastic paraplegia
Index patient P1017 had heterozygous SAMD9L p.(His880Gln), and this variant had previously been reported in a large family with ataxia pancytopenia syndrome [27]. A diagnosis of ataxia pancytopenia syndrome was made. Clinical details are provided in Table 2 and Online Resource 3.
Index patient P1073 was referred to our clinic for investigation of ataxia with late onset. The patient reported that his brother P1091 had similar symptoms and that their mother and one of her brothers had balance impairment as well (Fig. 1B). This information suggested a form of autosomal dominant ataxia in the family. As a first step, the patient was analyzed clinically for the most common autosomal dominant spinocerebellar ataxias: SCA1, SCA2, SCA3, SCA6, SCA7, which was negative. Then, whole exome sequencing (WES) was performed for the analyses of an ataxia genes panel within the clinical workup, which also resulted in a negative outcome. Meanwhile, another relative of our index patient was investigated in parallel at the neurology clinic. She reported that her mother, who was the cousin P1092 of our index patient, had symptoms of balance impairment. The index patient P1073, his brother P1091, and the cousin P1092 expressed interest to be included in the ataxia research project. Blood samples from the brother and the cousin were sent to a different laboratory (Centogene, Rostock, Germany) which reported a variant of uncertain significance (VUS) in the ELOVL4 gene c.511A > C, p.(Ile171Leu). No other findings were reported. Heterozygous variants in this gene are known causes of spinocerebellar ataxia type 34 (SCA34) [28, 29]. Clinical geneticists in Lund re-evaluated the existing genetic data from our index patient for the same variant that was found in his relatives, and it was confirmed positive and, in concordance with Centogene, initially classified as a VUS. After detailed post-NGS phenotyping of the three affected family members, we could conclude that all of them shared the typical clinical features for SCA34: decreased tendon reflexes, severe gait ataxia, gaze-evoked nystagmus, dysarthria, and hyperkeratotic skin manifestations (Fig. 3). Two individuals also had symptoms of cognitive decline with mildly to moderately impaired executive/visuospatial function [30]. Brain MRI showed moderate cerebellar atrophy for the index patient and severe cerebellar atrophy in his brother and cousin. Four affected family members carried the variant, and they were distant enough to each other in the pedigree to add evidence for the variant’s pathogenicity, using the modified ACMG criterion PP1_Strong for five informative meioses in the family, as suggested by Jarvik and Browning [31]. A reclassification as “likely pathogenic” according to the American College of Medical Genetics (ACMG) criteria was possible after the thorough family analyses for co-segregation of phenotype with genotype. We diagnosed SCA34.

Images of clinical presentation in Spinocerebellar ataxia 34. A, C Hyperkeratotic skin manifestations present bilaterally on extensors and on the scalp and nail abnormalities in affected individuals which were diagnosed as psoriasis; B, D The Rey Complex Figure Test (RCFT) shows impaired visuo-spatial and executive function
Index patient P1040 had a strong family history of dementia and balance impairment (Fig. 1C). Age at onset of neurological symptoms was in the mid-60s. Clinically the patient presented with gait impairment, severe ataxia in lower limbs and moderate ataxia in upper limbs, dysarthria, hypermetric saccades, motor restlessness, disinhibition, and perseverations. However, according to the patient’s next of kin, a slowly progressing personality change and mild cognitive impairment was noticed already in the early 50s. He was evaluated at our memory clinic and the clinical impression of his cognition and behavior was that of frontotemporal dementia. Brain MRI showed severe cerebellar and hippocampal atrophy. Genetic testing within our study identified a heterozygous variant in STUB1 gene c.107T > C, p.(Leu36Pro) which was absent from population databases but had been reported to ClinVar (ClinVar ID 1297589) as a variant of uncertain significance. Heterozygous STUB1 variants had been associated with spinocerebellar ataxia type 48 (SCA48). P1040 died during the study and was examined neuropathologically. Macroscopically, the patient had cerebellar atrophy, microscopically there was subtotal loss of Purkinje cells and atrophy of the molecular layer. The cerebrum showed tau-positive neurites, neuronal bodies, and astrocytes, with degenerative changes in the cortex and more pronounced degenerative changes in the thalamus, mesencephalon, and pons. There were p62-positive intraneuronal inclusions, as previously described for SCA48. TBP repeat length was normal (37 and 38 repeats, Online Resource 4). The patient’s clinical phenotype was very similar to patients with SCA48 described in the medical literature, and the unusual and specific histopathologic features in our patient were very well compatible with those described for SCA48 [32, 33].
Index patient P1002, of German extraction, had no family history of ataxia, but an early disease onset at 18 years of age which is suggestive of an autosomal recessive form of ataxia (Fig. 1D). Genetic testing within this study identified a homozygous variant in the STUB1 gene, c.761G > A, p. (Arg254His) which previously had been reported as pathogenic. Biallelic pathogenic STUB1 variants have been associated with autosomal recessive SCAR16 [34]. The patient died at the age of 42 years due to complications of her severe neurological disease.
Index patient P1058 developed gait disturbance at 37 years of age and reported affected relatives in an autosomal dominant pattern of inheritance (Fig. 1E). At the time of the clinical examination, the patient had severe paraparesis, was wheelchair-bound, presented with lower limb ataxia (SARA score 13) and spasticity, inward rotation of the left foot, hyperreflexia, painful muscle cramps in thighs and calves. Brain CT was normal. Genetic analysis identified a heterozygous variant in the SPAST gene, c.722del, p.(His241Profs*13) previously reported as likely pathogenic. Pathogenic variants in the SPAST gene cause autosomal dominant spastic paraplegia 4 (SPG4) which we consider is in accordance with the clinical and genetic presentation of our patient.
Index patient P1048 was of Turkish origin, with a family history suggestive of an autosomal recessive genetic condition; the patient reported that his two brothers were also affected but not the parents (Fig. 1F). The initial symptom was gait and balance impairment at the age of 27. On our examination, there was ataxia and spasticity in lower limbs, hyperreflexia, clonus, and bilateral positive Babinski sign. Brain MRI was normal. Genetic testing found a homozygous variant in the CAPN1 gene, c.759 + 1G > A which has been previously described as pathogenic. Mutations in the CAPN1 gene have been associated with spastic paraplegia 76 (SPG76) which was also confirmed for this patient [35].
Index patients P1070 and P1095 are from two different families and developed symptoms at 55 and 60 years of age respectively. Both reported a family history suggestive of an autosomal recessive disease with siblings with similar symptoms (Fig. 1G/H). Analyses of WGS data for repeat expansion within our study revealed altered RFC1 pentanucleotide composition (AAGGG instead of the normal AAAAG pentanucleotides) but were not able to reliably determine the number of pentanucleotides on each allele (Online Resource 4). Targeted testing using two orthogonal methods at an external laboratory confirmed biallelic extended repeat lengths in the RFC1 gene, which has been previously described and associated with cerebellar ataxia with neuropathy and vestibular areflexia syndrome (CANVAS) [36]. CANVAS was diagnosed in both patients.
Index patient P1089 (Fig. 1I) was referred to our neurology clinic because of a unilateral intention tremor in his right upper extremity. He reported that his father had developed a marked gait disturbance in his 80s and eventually required a wheelchair. Examination of P1089 by a movement disorder specialist at age 63 confirmed this and found moderate cerebellar tremor in the right hand and mildly atactic heel shin slide bilaterally, more pronounced on the right. Further, there was action and postural tremor of the essential tremor type (Online Resource 3) and mild dysarthria of the cerebellar type. This has led to the initial diagnosis of hereditary ataxia with late onset. During our clinical examination, he presented additional clinical signs such as irritability, cognitive impairment (MoCA 22/30 at age 64), tremor, and mild involuntary body movements. Brain MRI showed mild medial temporal atrophy. Genetic testing within our study showed 36 uninterrupted CAG trinucleotide repeats in the HTT gene (Online Resource 4). Alleles with 36–39 repeats usually have a low disease penetrance, however, normally the CAG repeats are interrupted by a CAA sequence in the penultimate 3′ triplet. Interestingly studies have shown that a loss of CAA interruption in HTT is associated with an earlier age at onset [37]. The initial symptom is described usually as a progressive cognitive decline, often associated with psychiatric problems [38]. The patient in our study did not have a loss of interruption. Based on the clinical presentation, the Huntington diagnosis was confirmed.
Genetic studies are still ongoing for some variants that were not yet confirmed as pathologic for specific cases, or it has remained impossible to fully elucidate pathogenicity. See Online Resource 2 and Fig. 1.
Index patient P1008 reported no family history of ataxia or other neurologic disease (Fig. 1P). At age 30 years he started to experience impaired balance, impaired gait, and muscle weakness. Brain MRI showed signs of moderate cerebellar and lower brainstem atrophy. During the clinical examination within our study the following clinical signs were found: the patient was in a wheelchair, there were fasciculations of face muscles, moderate dysarthria, saccadic smooth pursuit, gaze-evoked nystagmus, general muscle atrophy, and distal weakness in upper and lower extremities, gynecomastia, myoclonus, hyperreflexia, and spasticity, bilateral positive Babinski signs, weight loss. Genetic analysis within our study identified a heterozygous variant in the POLR3B gene, c.1568 T > A, p.(Val523Glu). This variant was described as pathogenic in ClinVar. Pathogenic POLR3B variants are associated with autosomal dominant demyelinating Charcot-Marie-Tooth type 1 disease and autosomal recessive hypomyelinating leukodystrophy [39]. However, the patient’s particular variant has so far only been described in homozygous or compound heterozygous form in recessive disease. Our patient’s MRI images did not show any signs of hypomyelination and the patient did not report any family history of demyelinating polyneuropathy. The patient’s clinical phenotype was not well compatible with the POLR3B-associated diseases. We conclude that the disease’s cause remains unknown.
Index patient P1086 had a family history of autosomal dominant motor polyneuropathy and an age at onset of 60 years. (Fig. 1Q). The patient had gait and limb ataxia, sensorimotor polyneuropathy, impaired vibration sense, hypometric saccades, hypoactive vestibulo-ocular reflex, hyperreflexia, and tremor of the head and left hand. Brain MRI showed mild cerebellar atrophy and white matter lesions. Similar to P1070 and P1095 described above, extended repeat lengths were found in the RFC1 gene. Nevertheless, we remain uncertain about this finding since CANVAS is an autosomal recessive disease and this patient has a family history of autosomal dominant motor polyneuropathy. The evaluation of genes known to cause hereditary polyneuropathies by our bioinformaticians was negative. The vestibular signs and the polyneuropathy are compatible with the CANVAS diagnosis, but the hyperreflexia is not easily explained. Our present hypothesis is that this patient may have CANVAS but also an additional dominantly inherited disorder.
Discussion
This study describes a series of 87 patients with progressive ataxia actively recruited within the uptake area of Skåne University Hospital over a period of 8 years and illustrates the process of examining patients with progressive ataxia with or without a confirmed genetic diagnosis. Our results show the high variability between the phenotypes of different forms of ataxia and the complexity of the interpretation of genetic findings.
Our study sheds some light on the presence of genetic forms of ataxia in the Swedish population. The most common form of autosomal dominant ataxia in our patient series was SCA3, which may be explained by the fact that we actively searched for and recruited these patients for a multi-centre study on biomarkers in SCA3 [13, 14]. Another more common form of autosomal dominant ataxias in our series was SCA2. These findings are similar to what was previously reported for European populations [6, 40, 41]. We found CANVAS to be the most common autosomal recessive form of ataxia among our patients, in line with other studies confirming that biallelic AAGGG expansion in RFC1 is a frequent cause of late-onset ataxia in Europeans [36]. Friedreich ataxia may be more prevalent, but most patients had been included in an earlier research study. Several rarer genetic forms of ataxia were also encountered, as expected with the increasingly large number of internationally known diseases with ataxia. Additionally, we found diagnoses that are not classically counted as genetic ataxias; two patients had hereditary spastic paraplegia, one Huntington’s disease and one Brown–Vialetto–Van Laere syndrome-2. Although this might be explained by the fact that clinicians registered the “wrong” ICD-10 code for these patients, all patients did have ataxia on examination, and hereditary ataxia and hereditary spastic paraplegias share not only overlapping phenotypes and underlying genes but also common disease mechanisms and cellular pathways [42]. Ataxia has been reported as an initial finding in 8.3% of patients with Huntington’s disease, and in over 70% during later disease stages [43]. The presence of these disorders among patients with a clinical diagnosis of ataxia shows that grouping neurological disorders with combined symptomatology can be difficult, or impossible, as the clinically most prominent symptom or finding may change over time or vary between individuals with the same genetic disease cause.
NGS technology has seen a rapid development and refinement, also during the time of our study. Sophisticated algorithms have been developed for the bioinformatic filtering of the ample sequencing data that result from NGS methods. Increasingly accurate indirect bioinformatic methods also detect copy number variants or short tandem repeats in new-generation sequencing datasets; these types of genetic variants are not directly assessed by sequencing. In this study, we used WES and WGS and analyzed the data for single nucleotide variants and small insertions or deletions, for copy number variants and short tandem repeats. We chose WES initially for reasons of cost and availability at the time, but transitioned to WGS, in part motivated by the discovery that intronic RFC1 variants were a relatively common cause of recessive ataxia with a particular combination of signs and symptoms [36]. The bioinformatic analyses detected an expansion of the HTT gene (36 trinucleotide repeats, total length 108 nucleotides) and unambiguously determined the repeat expansion’s length. Our methodology failed to reliably determine the length of the RFC1 pentanucleotide repeats based on our short-read WGS data. However, we detected the known abnormality in the base sequence of the pentanucleotides associated with RFC1 expansions and in all cases where we continued to analyze the patients by additional methods, expansions were identified. Long-read sequencing is likely to become more widely available in the near future and is expected to accurately determine the length of longer repeat expansions with thousands of repeat units [23]. Clearly, the use of short read WES rather than WGS and/or long-read sequencing is a limitation of our study, but our study used the methods that are presently available for clinical diagnostics in many healthcare settings. It is difficult to estimate how many additional patients may have received a diagnosis had they been examined by the more advanced NGS methods rather than short-read WES.
Eleven families received a genetic diagnosis during our study (Table 2). We considered two variants to be the disease cause that according to ACMG guidelines were classified only as variants of uncertain significance when they were first detected: Heterozygous ELOVL4 NM_022726.4 c.511A > C p.(Ile171Leu) in Family P1073_P1091_P1093 (Fig. 1B) and heterozygous STUB1 NM_005861.2 c.107 T > C p.(Leu36Pro) in familial proband P1040 (Fig. 1C). The ELOVL4 variant was present in four affected family members; two siblings, their first-degree cousin and that cousin’s daughter, which increased the likelihood for its pathogenicity according to predefined criteria [31]. Also, there was a characteristic combination of ataxia and skin and nail changes (“psoriasis” and hyperkeratotic nails) as well as a mild visuospatial and executive function deficit, of the same type as previously describe in this disease [28,29,30]. The patient with the heterozygous STUB1 variant showed an unusual clinical combination of ataxia with frontotemporal cognitive dysfunction, and the histo-neuro-pathological examination showed unusual inclusions that are very well compatible with the diagnosis SCA48 [32]. The PP4 criterium in the ACMG guidelines can be used when “the patient has a well-defined syndrome with little overlap with other clinical presentations”, such as in these two cases, but PP4 only counts as “supporting” the pathogenicity. For very rare and almost pathognomonic combinations of clinical features, as in these very rare neurological disorders, the weight of this criterium may be increased. However, there have also been concerns that the presence of both genetic and phenotypic diversity and the general “narrative potential” of a human genome [44] may lead to overinterpretation and wrong conclusions regarding variants of uncertain significance, especially in a post-NGS phenotyping scenario.
Recent work reported di-genic inheritance of intermediate length TBP expansions (40–49 repeats) and heterozygous STUB1 variants [45]. In almost all families, only individuals who carried variants in both these genes developed the clinical phenotype, that was named SCA17-DI (for di-genic) and who were observed to share certain clinical features [46]. This observation was partially replicated in a larger case series but only about half the patients with SCA17 and intermediate length TBP expansions had STUB1 variants, and more than a third of patients with SCA48 had normal TBP repeat size [47], as our patient with SCA48 (P1040). We have unfortunately been unable to examine STUB1 in our patient with SCA17 (P1047) in whom an intermediate length TBP allele was found several years ago during clinical testing.
For 8 patients, we had seemingly relevant genetic findings but were not able to set a clear diagnosis for variable reasons (Online Resource 2). In some of these families, examination of the proband’s relatives may lead to a diagnosis. We also encountered the situation that a patient (P1086, Online Resource 2) had a confirmed, clearly pathogenic variant (RFC1 repeat expansion), but we remained doubtful if this could explain the full clinical picture of this patient and the family.
Most of the patients included in our study are being followed at our clinic and we thus routinely re-evaluate new ways to provide a diagnosis. This includes testing for newly discovered genetic causes for ataxia that so far evade detection in WES or WGS analyses, re-running bioinformatic analyses with updated gene lists, and perhaps in the near future also new methods such as long-read sequencing, considering di-genic inheritance. We try to expand family pedigrees and test additional informative family members, when contact with these can be established. Likely, the majority of patients have a genetic cause for their ataxia, but non-genetic causes that have not been assessed or that may not yet be known cannot entirely be excluded.
Additional limitations of our study include the relatively small number of participants, which however is a common challenge for research on rare diseases. A proportion of the 158 contacted were not included in the present analyses, because they declined participation or because of scheduling difficulties. Our results are based on a selected patient cohort, and thus may not represent the true prevalence of genetic ataxia subtypes in Sweden. The study design reflects that of a real-life diagnostic clinic and neurologists were not blinded for the genetic results but were also informed about potentially relevant variants of uncertain significance. This could lead to false-positive results, but we consider the risk for this low, at least in the setting of a research study at a highly specialized center, and rather see the advantages of multi-disciplinary discussions to obtain a diagnosis, as is common practice in many other (non-genetic) diagnostic situations.
Conclusion and outlook
Analysis of our case series confirms that progressive ataxias have many different causes even when patients come from a relatively small geographic area. NGS sequencing is a powerful tool in clinical diagnostics; the absence of family history should not exclude genetic testing in patients with progressive ataxia. Re-examination of NGS datasets, as new diseases are being described, leads to more diagnoses. Closer clinical examination of patients and families with high-suspicion variants of uncertain significance may lead to additional correct diagnoses [48, 49], but clinical findings need to be re-assessed critically to confirm or reject a diagnosis [44]. The task lies ahead to define if and how this workflow can broadly be implemented in clinical diagnostics in healthcare settings, or if our approach will need to remain confined to research studies such as this one.
The ACMG diagnostic criteria [21] are helpful and, however, have their own limitations when applied for very rare diseases. Further studies may evolve these criteria that have remained unchanged since 2015 and determine new ways to combine neurological and genetic experience and knowledge from both clinical and research analyses, so that more patients will receive a genetical diagnosis within their workup in a healthcare setting.
Data availability
According to Swedish law and national, regional and institutional regulations, individual-level genetic data can only under very special circumstances be made available to collaborators. Reasonable requests can be directed to the corresponding author (SG).
References
Lieto M et al (2019) Degenerative and acquired sporadic adult onset ataxia. Neurol Sci 40(7):1335–1342
Hadjivassiliou M et al (2017) Causes of progressive cerebellar ataxia: prospective evaluation of 1500 patients. J Neurol Neurosurg Psychiatry 88(4):301–309
Pandolfo M, Manto M (2013) Cerebellar and afferent ataxias. Continuum (Minneap Minn) 19:1312–1343
Gorcenco S et al (2020) New generation genetic testing entering the clinic. Parkinsonism Relat Disord 73:72–84
Coutinho P et al (2013) Hereditary ataxia and spastic paraplegia in Portugal: a population-based prevalence study. JAMA Neurol 70(6):746–755
Ruano L et al (2014) The global epidemiology of hereditary ataxia and spastic paraplegia: a systematic review of prevalence studies. Neuroepidemiology 42(3):174–183
Synofzik M, Németh AH (2018) Recessive ataxias. In: Manto M, Huisman TAGM (eds) Handbook of clinical neurology. Elsevier, pp 73–89
Coutelier M et al (2017) A panel study on patients with dominant cerebellar ataxia highlights the frequency of channelopathies. Brain 140(6):1579–1594
Gannamani R et al (2021) Challenges in clinicogenetic correlations: one phenotype - many genes. Mov Disord Clin Pract 8(3):311–321
Ygland E et al (2014) Atypical Friedreich ataxia in patients with FXN p.R165P point mutation or comorbid hemochromatosis. Parkinsonism Relat Disord 20(8):919–923
Gorcenco S, Karremo C, Puschmann A (2022) Patients’ perspective in hereditary ataxia. Cerebellum. https://doi.org/10.1007/s12311-022-01505-1
Schmitz-Hübsch T et al (2006) Scale for the assessment and rating of ataxia: development of a new clinical scale. Neurology 66(11):1717–1720
Prudencio M et al (2020) Toward allele-specific targeting therapy and pharmacodynamic marker for spinocerebellar ataxia type 3. Sci Transl Med 12(566):eabb7086
Garcia-Moreno H et al (2022) Tau and neurofilament light-chain as fluid biomarkers in spinocerebellar ataxia type 3. Eur J Neurol 29(8):2439–2452
Kircher M et al (2014) A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 46(3):310–315
Gelfman S et al (2017) Annotating pathogenic non-coding variants in genic regions. Nat Commun 8(1):236
Bendl J et al (2016) PredictSNP2: a unified platform for accurately evaluating SNP effects by exploiting the different characteristics of variants in distinct genomic regions. PLoS Comput Biol 12(5):e1004962
de Sainte Agathe JM et al (2023) SpliceAI-visual: a free online tool to improve SpliceAI splicing variant interpretation. Hum Genom 17(1):7
Tan A, Abecasis GR, Kang HM (2015) Unified representation of genetic variants. Bioinformatics 31(13):2202–2204
Ameur A et al (2017) SweGen: a whole-genome data resource of genetic variability in a cross-section of the Swedish population. Eur J Hum Genet 25(11):1253–1260
Richards S et al (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17(5):405–424
Kopanos C et al (2019) VarSome: the human genomic variant search engine. Bioinformatics 35(11):1978–1980
Chintalaphani SR et al (2021) An update on the neurological short tandem repeat expansion disorders and the emergence of long-read sequencing diagnostics. Acta Neuropathol Commun 9(1):98
Plagnol V et al (2012) A robust model for read count data in exome sequencing experiments and implications for copy number variant calling. Bioinformatics 28(21):2747–2754
Gorcenco S et al (2017) Ataxia-pancytopenia syndrome with SAMD9L mutations. Neurol Genet 3(5):e183
Gorcenco S et al (2019) Oral therapy for riboflavin transporter deficiency - What is the regimen of choice? Parkinsonism Relat Disord 61:245–247
Chen DH et al (2016) Ataxia-pancytopenia syndrome is caused by missense mutations in SAMD9L. Am J Hum Genet 98(6):1146–1158
Cadieux-Dion M et al (2014) Expanding the clinical phenotype associated with ELOVL4 Mutation: study of a large French–Canadian family with autosomal dominant spinocerebellar ataxia and erythrokeratodermia. JAMA Neurol 71(4):470–475
Ozaki K et al (2015) A novel mutation in ELOVL4 leading to spinocerebellar ataxia (SCA) with the hot cross bun sign but lacking erythrokeratodermia: a broadened spectrum of SCA34. JAMA Neurol 72(7):797–805
Beaudin M et al (2020) Characterization of the phenotype with cognitive impairment and protein mislocalization in SCA34. Neurol Genet 6(2):e403
Jarvik GP, Browning BL (2016) Consideration of cosegregation in the pathogenicity classification of genomic variants. Am J Hum Genet 98(6):1077–1081
Mol MO et al (2020) Clinical and pathologic phenotype of a large family with heterozygous STUB1 mutation. Neurol Genet 6(3):e417
Chen DH et al (2020) Heterozygous STUB1 missense variants cause ataxia, cognitive decline, and STUB1 mislocalization. Neurol Genet 6(2):1–13
Ravel J-M et al (2021) Expanding the clinical spectrum of STIP1 homology and U-box containing protein 1-associated ataxia. J Neurol 268(5):1927–1937
Gan-Or Z et al (2016) Mutations in CAPN1 cause autosomal-recessive hereditary spastic paraplegia. Am J Hum Genet 98(6):1271
Cortese A et al (2019) Biallelic expansion of an intronic repeat in RFC1 is a common cause of late-onset ataxia. Nat Genet 51(4):649–658
Findlay Black H et al (2020) Frequency of the loss of CAA interruption in the HTT CAG tract and implications for Huntington disease in the reduced penetrance range. Genet Med 22(12):2108–2113
Stoker TB et al (2022) Huntington’s disease: diagnosis and management. Pract Neurol 22(1):32–41
Djordjevic D et al (2021) De novo variants in POLR3B cause ataxia, spasticity, and demyelinating neuropathy. Am J Hum Genet 108(1):186–193
Salman MS (2018) Epidemiology of cerebellar diseases and therapeutic approaches. The Cerebellum 17(1):4–11
Lipponen J et al (2021) Molecular epidemiology of hereditary ataxia in Finland. BMC Neurol 21(1):382
Synofzik M, Schüle R (2017) Overcoming the divide between ataxias and spastic paraplegias: shared phenotypes, genes, and pathways. Mov Disord 32(3):332–345
Franklin GL et al (2020) Is ataxia an underestimated symptom of Huntington’s disease? Front Neurol 11:571843
Goldstein DB et al (2013) Sequencing studies in human genetics: design and interpretation. Nat Rev Genet 14(7):460–470
Magri S et al (2022) Digenic inheritance of STUB1 variants and TBP polyglutamine expansions explains the incomplete penetrance of SCA17 and SCA48. Genet Med 24(1):29–40
Nanetti L et al (2023) Complex ataxia-dementia phenotype in patients with digenic TBP/STUB1 spinocerebellar ataxia. Mov Disord 38(4):665–675
Barbier M et al (2023) Intermediate repeat expansions of TBP and STUB1: genetic modifier or pure digenic inheritance in spinocerebellar ataxias? Genet Med 25(2):100327
Moghadasi S et al (2016) Classification and clinical management of variants of uncertain significance in high penetrance cancer predisposition genes. Hum Mutat 37(4):331–336
Morales A, Hershberger RE (2018) Variants of uncertain significance: should we revisit how they are evaluated and disclosed? Circ Genom Precis Med 11(6):e002169
Tesi B et al (2017) Gain-of-function SAMD9L mutations cause a syndrome of cytopenia, immunodeficiency, MDS, and neurological symptoms. Blood 129(16):2266–2279
Acknowledgements
We wish to thank all patients and their relatives who participated in this study. We thank SCA Network Sweden for informing their members about our research.
Funding
Open access funding provided by Lund University. This study was funded by the Swedish government (through the ALF agreement), Region Skåne, Skåne University Hospital research funds and donations, MultiPark – a Strategic research area at Lund University, The Swedish Parkinson Academy, Bundy Academy, and SCA network, all in Sweden.
Author information
Authors and Affiliations
Contributions
SG: study conception and design, acquisition, and documentation of clinical data, analysis, and interpretation of data, drafting and revising the manuscript. EK: study conception and design, analysis, and interpretation of genetic data, drafting the manuscript. JW: analysis and interpretation of genetic data, drafting the manuscript. CK: acquisition of clinical data and blood samples for genetic analyses, documenting clinical data. EA: acquisition of clinical data. SD: acquisition of clinical data. MLW: acquisition of clinical data. EE: acquisition of neuropathology data. HE: classification of candidate genetic variants, analysis and interpretation of data, revising the manuscript. KW: analysis and interpretation of data. KK: classification of candidate genetic variants, analysis and interpretation of data, revising the manuscript. AP: study conception and design, acquisition of data, analysis and interpretation of data, drafting and revising the manuscript. All authors will be asked to revise the manuscript critically for important intellectual content and to read and approve the final manuscript before its submission for publication.
Corresponding author
Ethics declarations
Conflicts of interest
The authors have no relevant financial or non-financial interests to disclose.
Ethical approval
Written and informed consent was obtained from all individual participants included in the study. Our study was ethically approved by the Regional Ethics Review Board responsible for Region Skåne (Dnr.2013/516) and the Swedish Ethical Review Authority (Dnr. 2021–00884).
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gorcenco, S., Kafantari, E., Wallenius, J. et al. Clinical and genetic analyses of a Swedish patient series diagnosed with ataxia. J Neurol 271, 526–542 (2024). https://doi.org/10.1007/s00415-023-11990-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-023-11990-x