
Abstract
Amyotrophic lateral sclerosis (ALS) can result into an incomplete locked in state (iLIS), in which communication depends on eye tracking computer devices. Oculomotor function impairments in ALS have been reported, but there is little research, particularly with respect to patients in iLIS. In the present study, we compared reflexive and executive oculomotor function by means of an eye tracking test battery between three groups: advanced ALS patients in iLIS (n = 22), patients in early to middle ALS stages (n = 44) and healthy subjects (n = 32). Patients with ALS showed significant deteriorations in oculomotor functions, with stronger impairments in iLIS. More specifically, ALS patients produced visually guided prosaccades with longer latencies and more frequent hypometria compared to healthy subjects. Longest latencies were obtained in iLIS patients, with a stronger prolongation for vertical than for horizontal prosaccades. ALS patients made more antisaccade errors and generated antisaccades with longer latencies. Smooth pursuit was also impaired in ALS. In the earlier ALS stages, bulbar onset patients presented stronger antisaccade and smooth pursuit deficits than spinal onset patients. Our findings reveal a relevant deterioration of important oculomotor functions in ALS, which increases in iLIS. It includes impairments of reflexive eye movements to loss of executive inhibitory control, indicating a progressing pathological involvement of prefrontal, midbrain and brainstem areas. The assessment of oculomotor functions may therefore provide clinically relevant bio- and progression marker, particularly in advanced ALS.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Amyotrophic lateral sclerosis (ALS) is the most prevalent and a fatal degenerative motoneuron disease. Due to progressive muscle weakness and loss of muscle control [1, 2], ALS can lead to an incomplete locked in state (iLIS). Patients in iLIS are tetraplegic. They are immobile and unable to speak, but remain fully conscious and can control their eye movements [3, 4]. Active and complex communication therefore relies exclusively on eye tracking communication systems (ETCS), which is crucial especially in advanced ALS for maintaining patients’ quality of life and even their will to live [5,6,7].
Due to the resistance of oculomotor nuclei’s to ALS pathophysiology [8], it was assumed that eye movement control is relatively spared [9,10,11]. However, it has been shown that oculomotor functioning can be affected in ALS, primarily due to the pathological processes in non-motor brain regions [10,11,12].
Eye movement control relies on well-defined neural networks [12,13,14] and may therefore reflect alterations of the central nervous system [15]. Eye tracking technology enables the precise measurement of eye movements in ALS up to iLIS [10]. Investigating oculomotor dysfunctions can therefore expand our knowledge about neurodegenerative processes and pathophysiological mechanisms in ALS disease [12, 15]. Accordingly, oculomotor changes in ALS can potentially serve as much-needed biomarker throughout the whole disease course [10], thereby contributing to diagnostics, prognostics and monitoring [16, 17]. This is particularly relevant in iLIS, when other voluntary motor functions are lost.
Deteriorations in oculomotor function can also indicate impairment of cognitive, including executive functions [10, 18]. This is important because cognitive deficits are common, non-motor symptoms in ALS [19,20,21], but cannot be (reliably) identified by standard neuropsychological tests in advanced stages, at the latest in iLIS. Focusing on advanced ALS, reduced eye movement control can hinder the use of ETCS [5, 6] and therefore endanger the patients’ quality of life, safety and self-determination, including end-of-life-decisions. Some iLIS-patients progress into a complete locked in state with a total loss of voluntary eye movement control [3, 22].
Research so far has examined changes in oculomotor control in early to middle ALS stages. Impairments in smooth pursuit eye movements have been reported [23,24,25], whereas visually guided or reflexive saccades (henceforth prosaccades) were often not altered [18, 23, 26, 27]. However, prosaccades were slower in ALS patients with bulbar onset [10, 23] and only in the vertical direction for ALS patients with frontotemporal dementia [28].
Impaired antisaccade performance has been reported at very early stages of ALS [29] and for asymptomatic carriers of the ALS-associated SOD1 gene [26]. Further evidence revealed longer latencies [18, 23] and more errors in antisaccade tasks [12, 18, 29,30,31], indicating reduced inhibitory control (i.e. deficits in higher-level oculomotor function) [18].
A sequential classification of oculomotor deficits in ALS has been suggested [29] and considered to be in line with progression of phosphorylated TAR DNA-binding protein (pTDP-43) pathology from frontal cortex into brainstem areas [32]: antisaccade deficits occur early in the disease course (“stage 1”), followed by additional smooth pursuit dysfunctions and slowing of prosaccades (“stage 2”) [29]. In turn, a longitudinal study of oculomotor function in ALS revealed no reliable changes over a time course of 20 months [18]. However, those last studies investigated ALS patients only in early to middle disease stages. Oculomotor research in ALS is importantly limited by neglecting patients in advanced stages, including iLIS [18, 23, 29]. Consequently, there is an unmet need to investigate decline of oculomotor and cognitive function decline during the whole ALS course.
Our cross-sectional study will address this research gap. By analyzing lower-level and higher-level oculomotor functions in (i) iLIS-patients, (ii) ALS patients at the early to middle stage (henceforth referred to as “earlier ALS-patients”), and (iii) healthy controls (HC), we aim for a better understanding of oculomotor impairments over the course of the disease.
We hypothesized a decreasing performance in prosaccade, antisaccade and smooth pursuit tasks from earlier ALS to iLIS patients; HC served as baseline, where best performance for all tasks was predicted. Based on initial indications of a stronger vulnerability of vertical eye movements in ALS [28], we furthermore tested for possible differences in saccadic latencies for vertical vs. horizontal eye movements. Exploratory analyses were performed to compare oculomotor functions between ALS patients with bulbar versus spinal onset, since stronger impairments for bulbar onset were reported [10, 18, 23, 33]. In addition, we explored the progress of oculomotor changes in association with the duration of ALS and the progression of motor deficits.
Methods
Sample recruitment
Patients for the earlier ALS group were recruited from the outpatient clinic at the University Hospital in Dresden. Patients for the iLIS group were recruited from specialist outpatient clinics at the University Hospital in Dresden, the Charité Berlin, the University Hospitals in Rostock, Jena, Goettingen, Hannover, and from a patient network (ALS mobil e.V.). The two groups have been recruited sequentially (first iLIS, second earlier ALS) as convenience samples.
For all patients, inclusion criteria were an established diagnosis of ALS or an ALS variant excluding PLS [34]. For iLIS patients, the presence of iLIS as an inclusion criterion was defined as tetraplegia and functional anarthria with loss of mobility and at most minimal residual head or limb movement (e.g. of toes, individual fingers, face muscles). The inclusion criteria of sufficient eye movement control for ETCS use was initially screened on the basis of the patient’s own report or that of their next of kin or attending physician. The successful calibration of the ETCS-system was the second precondition for the study participation. For all patients, exclusion criteria comprised a clinically defined frontotemporal dementia or obvious severe cognitive impairment, evaluated by a psychologist or neurologist with significant experience with ALS and iLIS patients.
In the HC group, we included age-, gender- and education-matched subjects without history of neurological or psychiatric diseases. Patients or control subjects were excluded in case of a known severe horizontal or vertical gaze palsy or other severe oculomotor disturbances that impeded the use of an eye tracking device.
Sample characteristics
Sixty-five patients in early to middle stage ALS (who were able to be tested in sitting position; ALSFRS ≥ 18) were invited for participation. Five of them declined to participate and 16 were categorized as drop-outs (for details see Fig. 1). Therefore, 44 earlier ALS patients were included in the analyses.

Flowchart of patient sample recruitment and assessment: earlier ALS group
Fifty-two patients were suggested for participation in the iLIS group, of whom 45 were screened for eligibility. Twenty-eight of them were enrolled in the study, of which 22 iLIS patients were included in the analyses (for details see Fig. 2). In this group, 19 patients used invasive ventilation and all were supplied with a personal ETCS (from different manufactures).

Flowchart of patient sample recruitment and assessment: iLIS group
In addition, 34 HC were recruited. One participant had to be excluded due to difficulties in calibration, very likely due to intraocular lenses as treatment of catarcat. Another participant was excluded due to strong indications for the evidence of a psychiatric disorder. Thus, 32 HC were included in the analyses.
Eye tracking device and data collection procedure
Eye movements were recorded using a monocular Eyegaze Edge® remote infrared ETCS with a sampling rate of 50 Hz (LC Technologies). The ETCS-screen (15.4 inch, 16:9 aspect ratio) was positioned fronto-parallel in a distance of 55–60 cm to the face. An observer screen served to control for a reliable gaze control of the ETCS and to ensure that subjects read the instructions and completed the tests.
The testing procedure differed between the three groups. All iLIS patients were assessed at home or their current nursing home in a light illuminated room. They completed the eye tracking test battery either lying in bed or sitting in a wheelchair.
Earlier ALS patients and HCs were tested at the University Hospital Dresden or in the Technische Universität Dresden in a light illuminated laboratory room. They completed the test battery seated on a chair, with their head rested upon an adjustable chin rest. Earlier ALS patients who were motorically unable to bend towards the chin rest where tested without it, under the condition of minimal head movement to ensure reliable recording of eye movements.
Sociodemographic and disease or health related information were requested directly from the subject, obtained from the patient file or in case of iLIS patients requested from their next of kin or professional caregiver. Disease severity was quantified by the degree of motor impairment using the ALS Functional Rating Scale-Revised (ALSFRS-R) [35]. The scale consists of 12 items and results in a single score with a range from 0 (complete loss of movement, anarthria and dependence of invasive ventilation and invasive nutrition) to 48 (normal motor abilities). Delta ALSFRS-R as a measure of the rate of disease progression was calculated by subtracting the current ALSFRS-score from 48 and dividing it by the ALS duration in months.
All iLIS patients and part of the HC completed the oculomotor function tests as part of a larger eye tracking battery of tests and questionnaires (e.g. on quality of life), for which results have been published [36]. Importantly, the assessment always began with the oculomotor tasks that are subject of this report.
Oculomotor function measures
The oculomotor tasks were designed, implemented and executed using the software NYAN 3® (Interactive Minds GmbH). Stimuli were presented against a dark-grey background (RGB: 62, 62, 62). The protocol started with a 9-point calibration. Parameters for the automatic calibration ensured an accuracy of at least 0.5°. The subsequent assessment of the oculomotor performance began with the smooth pursuit task, followed by the prosaccade and the antisaccade tasks.
The prosaccade task consisted of two blocks (horizontal, vertical) with 48 trials each. The task always began with the horizontal block comprising 24 trials to the left/right, followed by the vertical block with 24 down/up trials. Subjects were instructed to perform a saccade to an appearing target (red dot) as quick and accurately as possible. Each trial began with the presentation of a central white fixation cross (size: 0.8°) and two white anchor dots (size: 0.54°) located at the possible positions of the target stimuli. The dots were located in 9° distance (for 55 cm distance from the screen) from the fixation cross (left and right or above and below). The fixation cross and the anchor dots were replaced (after a randomly varied duration of 1700 ± 800 ms) by a red target point (size: 0.54°), which appeared at the position of one of the two anchor points in quasirandomized order.
The design of the antisaccade task (horizontal and vertical block) was identically to the prosaccade task, thus consisting of the same stimuli with the identical temporal and spatial characteristics. In the antisaccade task though, subjects were instructed to execute a saccade in the opposite direction of the red dot (i.e. to the left of the fixation cross when the red dot appeared to the right). All subjects started with the horizontal antisaccades followed by the vertical antisaccades block.
The smooth pursuit task consisted of 48 trials (one block), in which subjects were requested to follow the horizontal or vertical movement of a white dot (size: 0.54°) as accurately as possible. The movement originated in quasirandomized order at one of four starting points (with same frequency in total), each located in 9° distance from the center of the screen. Each of the trials started with the static presentation of the dot (for varying duration: 1250 ± 250 ms). It was followed by a sinusoidal movement at f = 0.58 Hz to the position of the opposite starting point (turning point) and from there back to the starting point (maximum velocity 30°/s, reached half way through in each motion segment).
Analysis of oculomotor function
The obtained eye movement data was exported from NYAN 3® and then processed and analyzed using R 4.2.1 [37]. Occurrence of saccades in all three oculomotor tasks was determined by a velocity criterion. For the prosaccade and antisaccade tasks, the start of a saccade was identified in the data by the time of the first sample of a stream of adjacent samples with a sample-to-sample velocity larger than 30°/s. For the smooth pursuit task, the start of a saccade was identified by the threshold of a sample velocity that exceeded the smooth stimulus velocity by 30°/s at a given time, and the end of the saccade by a velocity falling below that threshold.
For the prosaccade and antisaccade task analysis, trials were considered as valid when (i) there were at least 50% of valid samples following the target onset (excluding measurement errors or blinks) and (ii) if the starting gaze position was within 2° of the center of the fixation cross when the target appeared.
Performance in the prosaccade task was evaluated by latency and amplitude for valid trials with a correct response. Responses were classified as correct when a primary saccade was made in the direction of the target with a latency greater than 180 ms. Latency was calculated as the time between target onset and primary saccade onset [18, 23, 26, 29]. Amplitude was determined as the distance between the subject’s gaze position in the last sample before the target onset and the eye position at the end of the saccade.
Similarly, for the antisaccade task, we analyzed antisaccadic latency and errors for valid trials. Antisaccade latency was determined in the same way as for the prosaccades, but a correct response was defined as a primary saccade in the opposite direction of the red dot. Errors were defined as incorrect reactions in valid trials, so either (i) a primary saccadic response within 180 ms after target onset, (ii) a missing saccadic response or (iii) a primary saccadic response directed toward the target (i.e. a prosaccade) [18, 26, 29, 30]. Error rates represent the ratio between incorrect responses and valid trials (range 0–1). For all patients with error rates > 0.9 in the antisaccade task, we ensured that they corrected any of their errors (i.e. made an antisaccade after the primary prosaccade) to make sure that they had understood the task instruction.
Performance in the smooth pursuit task was evaluated by analyzing gain and the number of catch-up-saccades for valid trials. Validity of data for motion segments in each trial was determined by a visual inspection, removing trials with large amounts of missing data (i.e. eye blinks or measurement errors), noisy data or non-compliant task behavior. For the gain, only the middle 50% of the trial samples in each motion segment were used to determine the parameter. We excluded samples that were classified as saccades as well as samples adjacent to those saccade samples. Gain was then calculated as the ratio between smooth eye velocity and target velocity [15, 23, 26, 29]. Catch-up-saccades were defined as saccades towards the target and with a final position that did not exceed the current position of the stimulus.
Statistical analysis
Comparisons of demographic and clinical characteristics between groups were conducted by means of Kruskal–Wallis-test, Chi-square test or Mann–Whitney U test, respectively.
We used marginal models to test for differences in the oculomotor parameters between the three groups. These marginal models are extensions of generalized linear models that require only the mean model of the dependent variable to be correctly specified in terms of the covariates. Distributional assumptions on the dependent variable are not required and the regression parameters in such semi-parametric models can be estimated via so-called generalized estimating equations (GEE). GEE yield consistent estimators of the regression parameters of interest, even if the within-subject associations among the repeated measures have been misspecified. They also provide valid standard errors that correct for misspecifications in the within-subject correlations and for potential over- or underdispersion [38]. Models were specified with an exchangeable working correlation structure, which determines an equal correlation between all pairs of observations within each subject while observations between subjects are uncorrelated. We used a robust covariance estimator that was specifically developed to achieve close to nominal type I error rates for small sample sizes [39]. GEE were conducted using the R package glmtoolbox (version 0.1.3) and the difference of estimated marginal means (DEMM) or odds ratios (OR) were obtained for all models using the R package EMMeans (version 1.9.1-1).
Marginal models for all dependent variables included group (between subject factor, levels: iLIS patients, earlier ALS patients, HC) and direction of the demanded eye movement (within subject factor, levels: horizontal, vertical). We modelled both main effects as well as the interaction between the two factors. Age-specific effects were accounted for by including age (in years) and its interaction with group and direction in all models. All significance tests are based on the DEMM at the mean age. In the model for antisaccade errors, the dependent variable was connected with the covariates via a logit link function. Differences between patients with bulbar and spinal disease onset for each oculomotor parameter were analyzed by means of GEEs as well, separately for iLIS and earlier ALS patients. Those marginal models included the variables subgroup (between subject factor: bulbar, spinal) and direction (within subject factor: horizontal, spinal) as well as age to partial out its association with the dependent variables. All post-hoc contrasts were corrected for multiple comparisons using the Holm-Bonferroni method.
Kendall's τb correlation coefficients were calculated to determine the relationship between ALS duration (months since diagnosis), ALSFRS-R score and Delta ALSFRS-R with the oculomotor parameters, separately for the two patient groups. For correlation analyses, the mean for each direction (horizontal, vertical) was calculated using the medians for the two respective conditions (right/left or up/down) of each of the oculomotor for each subject. The significance level for all statistical tests was set at α = 0.05.
Results
Demographic and clinical sample characteristics
Results are reported for the final sample of N = 98 participants. Table 1 displays its demographic and clinical characteristics. No significant group differences for age and gender proportion between the three groups were found. The proportions of ALS onset variants (bulbar vs spinal) did not differ between earlier ALS and iLIS patients. As expected, iLIS patients presented a significantly longer ALS disease-duration and a lower ALSFRS-R score than earlier ALS patients.
Descriptive statistics for reflexive and executive oculomotor parameters and movement direction can be found in Table S.1 (supplement). The following sections outline the statistical results of the group comparisons by means of the marginal models for all the oculomotor parameters.
Prosaccades
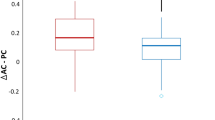
In the prosaccade task, 82.6% of all trials were defined as valid. Examining the prosaccade latency with group and direction as independent variables using GEE revealed main effects of group (χ2(2) = 89.48, p < 0.001) and direction (χ2(1) = 34.82, p < 0.001) as well as a group by direction interaction (χ2(2) = 13.68, p = 0.001; see Fig. 3A).

Prosaccade latencies (A) and amplitudes (B). Boxplots showing the medians (horizontal lines), the means (crosses), the interquartile range (length of the box), the maximum and minimum score excluding outliers (whiskers) and the outliers (dots)
Table 2 displays the results for the pairwise comparisons including confidence intervals. Prosaccade latencies were significantly longer in both directions (horizontal and vertical) in iLIS patients compared to HC and earlier ALS patients. Latencies were also significantly longer in earlier ALS patients compared to HC for both horizontal and vertical prosaccades.
Contrast analysis demonstrated stronger latency differences between iLIS patients and HC for vertical prosaccades compared to horizontal prosaccades (DEMM = − 57.0, CI [− 94.06, − 20.0], z = − 3.69, p < 0.001). This was also true for latency differences between iLIS patients and earlier ALS patients (DEMM = − 50.4, CI [− 87.58, − 13.3], z = − 3.25, p = 0.002). No reliable differences were found between horizontal and vertical prosaccade latencies for the group comparison of ALS patients and HC (DEMM = − 6.6, CI [− 21.8, 8.61], z = − 1.04, p = 0.30).
The model for prosaccade amplitudes showed only a main effect of group (χ2(2) = 42.11, p < 0.001) but not of direction (χ2(1) = 3.71, p = 0.054) and no interaction (χ2(2) = 0.58, p = 0.75); see Fig. 3B).
Pairwise comparisons (Table 2) showed differences between all groups: amplitudes were shorter for iLIS patients compared to HC and earlier ALS patients for both horizontal and vertical prosaccades. Earlier ALS patients showed also significantly shorter horizontal and vertical prosaccade amplitudes than HC.
Contrast analysis revealed no differences between horizontal and vertical prosaccade amplitudes for comparisons between the groups: iLIS patients compared with HC (DEMM = 0.27, CI [− 0.87, 1.41], z = 0.57, p = 1.0), iLIS patients compared with earlier ALS patients (DEMM = 0.17, CI [− 0.99, 1.34], z = 0.35, p = 1.0), and earlier ALS patients compared with HC (DEMM = 0.098, CI [− 0.31, 0.51], z = 0.57, p = 1.0).
Antisaccades
In the antisaccade task, 83.6% of all trials were defined as valid.
The model for antisaccade latencies showed main effects of group (χ2(2) = 60.68, p < 0.001) and direction (χ2(1) = 10.13, p = 0.002). No interaction effect was observed (χ2(2) = 0.19, p = 0.91; see Fig. 4A).

Antisaccade latencies (A) and error rates (B). Boxplots showing the medians (horizontal lines), the means (crosses), the interquartile range (length of the box), the maximum and minimum score excluding outliers (whiskers) and the outliers (dots)
Pairwise comparisons (Table 2) revealed differences between all three groups: significantly longer antisaccade latencies were found for iLIS patients compared with HC and with earlier ALS patients, whereas earlier ALS patients showed longer antisaccade latencies than HC (all for horizontal and vertical).
Contrast analysis revealed no differences between horizontal and vertical antisaccade latencies for comparisons between the three groups, neither for iLIS patients compared to HC (DEMM = − 10.04, CI [− 71.0, 50.9], z = − 0.39, p = 1.0), nor for iLIS patients compared to earlier ALS patients (DEMM = − 11.27, CI [− 73.2, 50.7], z = − 0.44, p = 1.0), nor for earlier ALS patients compared to HC (DEMM = 1.22, CI [− 25.9, 28.3], z = 0.108, p = 1.0).
The model for antisaccade errors revealed only a main effect of group (χ2(2) = 35.44, p < 0.001) but not for direction (χ2(1) = 1.39, p = 0.24). There was an interaction between both factors (χ2(2) = 14.94, p < 0.001; see Fig. 4B).
Pairwise comparisons (Table 2) revealed that iLIS patients made significantly more errors than HC and also more than earlier ALS patients for antisaccades (horizontal and vertical). Earlier ALS patients showed significantly more errors than HC, but only for horizontal antisaccades.
Smooth pursuit
In the smooth pursuit task, 95.2% of all trials were defined as valid.
The model for smooth pursuit gain showed main effects of group (χ2(2) = 39.20, p < 0.001) and direction (χ2(1) = 75.49, p < 0.001) as well as an interaction effect between both factors (χ2(2) = 48.46, p < 0.001; see Fig. 5A).

Smooth pursuit gain (A) and number of catch-up saccades (B). Boxplots showing the medians (horizontal lines), the means (crosses), the interquartile range (length of the box), the maximum and minimum score excluding outliers (whiskers) and the outliers (dots)
Pairwise comparisons (Table 2) revealed significant differences between all three groups for gain of horizontal smooth pursuit: iLIS patients presented lower gains compared to HC and earlier ALS patients, while earlier ALS patients demonstrated lower gains than HC.
For vertical smooth pursuit, significantly lower gains were found in both patient groups when compared to HC, but no difference was obtained between the patient groups.
The model for the number of catch-up saccades per trial in smooth pursuit showed evidence for main effects of group (χ2(2) = 23.54, p < 0.001) and direction (χ2(1) = 174.30, p < 0.001) as well as an interaction between the two factors (χ2(2) = 11.63, p = 0.003; see Fig. 5B).
Pairwise comparisons (Table 2) revealed that iLIS patients presented significantly more catch-up saccades compared to HC in both directions. Compared with earlier ALS patients, iLIS patients showed significantly more horizontal catch-up-saccades, with no differences in vertical catch-up-saccades. There were no differences between earlier ALS patients and HC.
Associations of oculomotor function with ALS onset type, disease duration and physical impairment
Comparison of patients with bulbar and spinal ALS onset revealed differences in two oculomotor variables, but only in the earlier ALS group: bulbar onset patients compared to spinal onset patients made more errors in the horizontal antisaccade task (OR = 0.81, p = 0.034) and showed lower gains in the smooth pursuit task (horizontal: DEMM = 0.15, p = 0.006; vertical: DEMM = 0.08, p = 0.048), see Table S.2 and S.3 (Supplement) for details.
Only for earlier ALS patients, a longer disease duration was weakly correlated with lower amplitudes of horizontal prosaccades, i.e. more hypometric saccades (τb = − 0.25, p = 0.016). There was also a weak correlation between a lower ALSFRS-R score (i.e. stronger physical impairment) and lower amplitudes of horizontal prosaccades in the earlier ALS group (τb = − 0.31, p = 0.004). A lower ALSFRS-R score was weakly correlated with longer horizontal antisaccade latencies in this group (τb = − 0.23, p = 0.027). No further correlations were found, see Table S.4 (Supplement).
Discussion
The present study investigated reflexive and executive oculomotor functions in ALS-iLIS patients, ALS patients in earlier disease stages and in HC. There are three main results: Firstly, we found evidence of a deterioration in reflexive and executive oculomotor functions in ALS and over the course of the disease. Secondly, we found that this deterioration is influenced by the ALS onset variants only for horizontal antisaccade errors and smooth pursuit gain only in the earlier ALS subsample. Thirdly, the weak to none correlations between oculomotor functions and ALS duration, ALS disease severity and progression suggest that oculomotor deficits do not simply intensify in parallel to the progression of main motor symptoms.
More specifically, the deterioration in reflexive oculomotor functions was characterized by a prolongation of prosaccade latencies, as well as a significant undershoot (i.e. hypometria) and generally less accurate prosaccades (i.e. dysmetria) in patients in stage of iLIS. These results point to serious changes of reflexive saccadic function in advanced stages of ALS. There was also a slight prolongation in early stage ALS patients compared to HC, while previous work has suggested no impairment of prosaccade latencies in early to middle stages of ALS [18, 23, 26, 27].
The present results, particularly for iLIS patients, support and complement the proposed staging of structural and subsequent functional changes in ALS [29]—which was based on data for earlier ALS only. It suggests that a stronger slowing of prosaccades is characteristic of more advanced ALS-related oculomotor dysfunctions (oculomotor “stage 2”). According to knowledge about prosaccades [12, 13, 23], prolonged latencies can be attributed to TDP-43 pathology spreading into brainstem regions in more advanced ALS stages [12, 29], causing lesions of saccadic burst and omnipause neurons [13].
Regarding explanations for longer latencies for vertical compared to horizontal prosaccades in iLIS patients, burst neurons for horizontal saccades are located in the pons, those for vertical saccades in the midbrain [13, 14]. A post-mortem analysis revealed TDP-43 pathology in brainstem and midbrain in approximately 50% of the ALS patients [40]. A large imaging study evidenced hypermetabolism in midbrain areas in early ALS, construed as correlates of neurodegeneration [41].
The successful generation of antisaccades requires cognitive, particularly inhibitory control [42]. The error rate is the most important parameter of antisaccade performance as it reveals a failure of inhibitory control. Higher error rates for horizontal antisaccades in earlier ALS patients compared to HC confirm previous findings [18, 26, 29, 30, 43] and are also represented in the proposed oculomotor staging model [29]: the isolated increase of antisaccade errors characterize oculomotor “stage 1”. These early deficits are associated with the progression of pTDP-43 pathology into the dorsolateral prefrontal cortex (DL-PFC) [29], which is responsible for the required inhibitory of prosaccades in antisaccade tasks [13, 42, 44, 45]. It was proven that the increased error rate in ALS patients is associated with a reduced activation of the DL-PFC [43]. Consistently, pathological changes of the DL-PFC have been shown to be a structural correlate of impairments in executive functions in ALS [46]. Cognitive deficits affect 30–50% of non-demented early to middle stage ALS patients [19, 21, 47,48,49], most often the executive functions domain [19, 20, 47, 50, 51].
The increased antisaccade error rate in iLIS patients suggests a relevant deterioration in the course of ALS. This complements and challenges previous longitudinal findings of a stable cognitive function for pre-iLIS ALS patients [18, 52, 53], including antisaccade performance [18]. Antisaccade error rates correlate with performance on more complex neuropsychological tests of executive function control [45]. Hence, our findings support the view that executive dysfunctions develop very early in ALS, but challenge the conclusion drawn from longitudinal studies that such deficits remain stable throughout the disease [53].
Regarding the higher antisaccade error rates in earlier ALS patients with bulbar versus spinal onset, literature is inconsistent: two meta-analyses do not confirm an association between bulbar onset and stronger cognitive or particularly executive dysfunction [19, 20], but a recent one finds bulbar onset to be a strong predictor of cognitive impairment and frontotemporal dementia in ALS [33].
Studies of smooth pursuit in ALS are rare and mostly did not employ eye tracking [9, 10, 23,24,25]. Some of them did not prove an impaired performance [26, 29], others only in a very small proportion of the patients [25] However, deficits in horizontal smooth pursuit were also characterized as a feature of ALS [10], which is supported by our results. As we found those deficits in earlier ALS patients already but they were more pronounced in iLIS, they indicate a serious progression of oculomotor impairments.
The already mentioned staging model suggests smooth pursuit deficits to appear later in ALS [29]: frequent catch-up-saccades in smooth pursuit—in addition to antisaccade deficits (of “stage 1”)—characterize oculomotor “stage 2”. This transition is explained by the progression of pathological changes from the frontal lobe into the lower brainstem. Our finding of strong impairments in iLIS patients compared to both earlier ALS patients and HC is a supportive addition to this model. However, smooth pursuit movements are generated by a complex network [14, 54, 55]. As suggested by clinical and imaging studies, catch-up-saccades might be caused by dysfunctions of the ponto-cerebellar pathways [12, 13, 54]—while a reduced gain might reflect impaired pursuit movement control by fronto-striatal pathways [29]. Especially the frontal eye fields seem to be responsible for the reduced gain in ALS [12, 55]. On this basis, our findings may support the progression of pathological changes from frontal regions towards brainstem: iLIS patients show both increased catch-up saccades and a reduced gain, whereas earlier ALS patients show only reduced gain compared to HC. Interestingly, Poletti et al. observed that patients with ALS-specific cognitive disturbances in the ECAS were also significantly more likely to show abnormal smooth pursuit and/or prosaccade performance on clinical bedside assessment [25].
We observed less pronounced group differences for vertical smooth pursuit. Since higher speed of smooth pursuit degrades performance [55, 56] and vertical pursuit is known to be slower than horizontal in healthy subjects [56], this finding might present a floor effect due to the high velocity of the demanded pursuit movement.
A relevant implication of the oculomotor changes is their prospective negative impact on use of ETCS in iLIS. A good subjective quality of life with ALS was also reported for patients in iLIS [36, 57, 58]. However, it is strongly related to their access to ETCS [5,6,7, 59], which they use extensively in daily life [36, 60]. Since these systems require intact oculomotor and cognitive functions, any deterioration of these functions might reduce the system’s usability [61]. Problems in ETCS use are frequent, but often detected belatedly and their oculomotor or/and cognitive causes are almost impossible to identify. Our findings support the possibility and relevance of a systematic eye tracking based assessment of such potential reasons. The changes in eye movement coordination and control we observed, e.g. a slowed generation of voluntary saccades, a reduced ability to suppress visually guided eye movements and inaccurate saccades, might substantially reduce the efficiency and/or speed of ETCS use. Based on knowledge about the natural course of oculomotor and neurocognitive functions in ALS, their monitoring could allow to detect changes earlier and more precisely—and consequently to predict a deterioration that might compromise the ability to communicate via ETCS. There are cases of patients merging into a complete locked in state [3, 22] and around 10% of ALS patients develop frontotemporal dementia [19]. This means, deficits can lead to a state in which patients cannot reliably make and/or communicate their will and decisions—including end-of-life decisions. To detect such a progress would allow to prepare for it, e.g. by recording patients’ wishes as long as it is still (reliably) possible.
A main limitations of the present study is its small sample size, which is particularly limiting regarding the heterogeneity of ALS pathology and its clinical presentation [1, 62]. Moreover, a probable selection bias must be considered, especially for the iLIS group (e.g. regarding the willingness to participate in a demanding eye tracking based study, use of personal ETCS). In support of this bias, screening failures and drop-outs were mostly due to severe oculomotor and cognitive deficits. At the same time, those frequent drop-outs emphasize the relevance of oculomotor functioning for ETCS use. Inevitable differences in the test settings between iLIS patients and all other participants limit comparability between the groups. A possible influence of apathy on, for example, the initiation of eye movements was not examined but must be considered, since apathy is the most prevalent symptom of behavioral impairment in ALS [63].
The patients in our study were not systematically screened for genetic variations, which should be considered in future studies especially including UNC13A snips [64]. We did not subject the patients in our study to (classic) neuropsychological testing (e.g. using the ECAS), which would have been possible only for the earlier ALS and HC group. In future research, a comprehensive eye-tracking based battery of cognitive function could provide differential insights into neurocognitive consequences of ALS, also in these patients with strongly impaired motor functions and thus up to iLIS. This future research should also assess the negative effects of oculomotor changes on ETCS use in order to identify early and sensitive marker of such restraints. Moreover, imaging evidence (PEG, MRI) would be needed to verify hypotheses about neuronal correlates of changes in oculomotor performance. Overall, our results emphasize the need for longitudinal studies with longer follow-up periods than previous ones. Such studies could verify the suggested potential of oculomotor functions as biomarker of pathological processes in advanced ALS stages up to and within iLIS.
Taken together, the presented study evidences relevant and progressing deficits of reflexive and executive oculomotor functions in ALS. Antisaccade error rates might be a valuable, easily assessable measure of executive function and frontal lobe impairment in all ALS stages and can be interpreted without the confounding effects of pure motor slowing of gaze. Our findings indicate a deterioration that successively leads to extensive, global dysfunctions in iLIS, which would correspond to the typical concepts of disease spreading in neurodegenerative diseases.
Data availability
Data not provided in the article may be shared (anonymized) at the request of any qualified investigator for purposes of replicating procedures and results.
References
Feldman EL, Goutman SA, Petri S et al (2022) Amyotrophic lateral sclerosis. The Lancet 400:1363–1380. https://doi.org/10.1016/S0140-6736(22)01272-7
Brown RH, Phil D (2017) Amyotrophic lateral sclerosis. N Engl J Med 377(2):162–172
Murguialday AR, Hill J, Bensch M et al (2011) Transition from the locked in to the completely locked-in state: a physiological analysis. Clin Neurophysiol 122:925–933. https://doi.org/10.1016/j.clinph.2010.08.019
Smith E, Delargy M (2005) Locked-in syndrome. BMJ 330:406–409. https://doi.org/10.1136/bmj.330.7488.406
Linse K, Aust E, Joos M, Hermann A (2018) Communication matters—pitfalls and promise of hightech communication devices in palliative care of severely physically disabled patients with amyotrophic lateral sclerosis. Front Neurol. https://doi.org/10.3389/fneur.2018.00603
Rosa Silva JP, Santiago Júnior JB, dos Santos EL et al (2020) Quality of life and functional independence in amyotrophic lateral sclerosis: a systematic review. Neurosci Biobehav Rev 111:1–11. https://doi.org/10.1016/j.neubiorev.2019.12.032
Corallo F, Bonanno L, Lo Buono V et al (2017) Augmentative and alternative communication effects on quality of life in patients with locked-in syndrome and their caregivers. J Stroke Cerebrovasc Dis 26:1929–1933. https://doi.org/10.1016/j.jstrokecerebrovasdis.2017.06.026
Nijssen J, Comley LH, Hedlund E (2017) Motor neuron vulnerability and resistance in amyotrophic lateral sclerosis. Acta Neuropathol (Berl) 133:863–885. https://doi.org/10.1007/s00401-017-1708-8
Kang BH, Kim JI, Lim YM, Kim KK (2018) Abnormal oculomotor functions in amyotrophic lateral sclerosis. J Clin Neurol 14:464–471. https://doi.org/10.3988/jcn.2018.14.4.464
Sharma R, Hicks S, Berna CM et al (2011) Oculomotor dysfunction in amyotrophic lateral sclerosis: a comprehensive review. Arch Neurol 68:857–861. https://doi.org/10.1001/archneurol.2011.130
Tokushige S, Terao Y, Matsuda S et al (2016) Motor neuron disease with saccadic abnormalities similar to progressive supranuclear palsy. Neurol Clin Neurosci 4:146–152. https://doi.org/10.1111/ncn3.12062
Donaghy C, Thurtell MJ, Pioro EP et al (2011) Eye movements in amyotrophic lateral sclerosis and its mimics: a review with illustrative cases. J Neurol Neurosurg Psychiatry 82:110–116. https://doi.org/10.1136/jnnp.2010.212407
Leigh RJ, Zee DS (2015) The neurology of eye movements. Oxford University Press, Oxford
Horn AKE, Straka H (2021) Functional organization of extraocular motoneurons and eye muscles. Annu Rev Vis Sci 7:793–825. https://doi.org/10.1146/annurev-vision-100119-125043
Gorges M, Pinkhardt EH, Kassubek J (2014) Alterations of eye movement control in neurodegenerative movement disorders. J Ophthalmol. https://doi.org/10.1155/2014/658243
Rojas P, Ramírez AI, Fernández-Albarral JA et al (2020) Amyotrophic lateral sclerosis: a neurodegenerative motor neuron disease with ocular involvement. Front Neurosci 14:976. https://doi.org/10.3389/fnins.2020.566858
FDA-NIH Biomarker Working Group (2016) BEST (Biomarkers, EndpointS, and other Tools) Resource. Food and Drug Administration (US), Silver Spring
Proudfoot M, Menke RAL, Sharma R et al (2015) Eye-tracking in amyotrophic lateral sclerosis: a longitudinal study of saccadic and cognitive tasks. Amyotroph Lateral Scler Front Degener 17:101–111. https://doi.org/10.3109/21678421.2015.1054292
Beeldman E, Raaphorst J, Twennaar MK et al (2016) The cognitive profile of ALS: a systematic review and meta-analysis update. J Neurol Neurosurg Psychiatry 87:611–619. https://doi.org/10.1136/jnnp-2015-310734
Raaphorst J, de Visser M, Linssen WHJP et al (2010) The cognitive profile of amyotrophic lateral sclerosis: a meta-analysis. Amyotroph Lateral Scler 11:27–37. https://doi.org/10.3109/17482960802645008
Montuschi A, Iazzolino B, Calvo A et al (2015) Cognitive correlates in amyotrophic lateral sclerosis: a population-based study in Italy. J Neurol Neurosurg Psychiatry 86:168–173. https://doi.org/10.1136/jnnp-2013-307223
Bensch M, Martens S, Halder S et al (2014) Assessing attention and cognitive function in completely locked-in state with event-related brain potentials and epidural electrocorticography. J Neural Eng 11:026006
Donaghy CG, Thurtell M, Pioro E, Leigh R (2010) Using eye movements to distinguish amyotrophic lateral sclerosis (ALS) from its variants and mimics. In: Neurology, pp A179
Moss HE, McCluskey L, Elman L et al (2012) Cross-sectional evaluation of clinical neuro-ophthalmic abnormalities in an amyotrophic lateral sclerosis population. J Neurol Sci 314:97–101. https://doi.org/10.1016/j.jns.2011.10.016
Poletti B, Solca F, Carelli L et al (2021) Association of clinically evident eye movement abnormalities with motor and cognitive features in patients with motor neuron disorders. Neurology 97:e1835–e1846. https://doi.org/10.1212/WNL.0000000000012774
Behler A, Knehr A, Finsel J et al (2021) Eye movement alterations in presymptomatic C9orf72 expansion gene carriers. J Neurol. https://doi.org/10.1007/s00415-021-10510-z
Burrell JR, Hornberger M, Carpenter RHS et al (2012) Saccadic abnormalities in frontotemporal dementia. Neurology 78:1816. https://doi.org/10.1212/WNL.0b013e318258f75c
Moon SY, Lee BH, Seo SW et al (2008) Slow vertical saccades in the frontotemporal dementia with motor neuron disease. J Neurol 255:1337. https://doi.org/10.1007/s00415-008-0890-y
Gorges M, Müller H-P, Lulé D et al (2015) Eye movement deficits are consistent with a staging model of pTDP-43 pathology in amyotrophic lateral sclerosis. PLoS ONE 10:e0142546. https://doi.org/10.1371/journal.pone.0142546
Becker W, Gorges M, Lulé D et al (2019) Saccadic intrusions in amyotrophic lateral sclerosis (ALS). J Eye Mov Res. https://doi.org/10.16910/jemr.12.6.8
Evdokimidis I, Constantinidis TS, Gourtzelidis P et al (2002) Frontal lobe dysfunction in amyotrophic lateral sclerosis. J Neurol Sci 195:25–33. https://doi.org/10.1016/S0022-510X(01)00683-9
Brettschneider J, Tredici KD, Toledo JB et al (2013) Stages of pTDP-43 pathology in amyotrophic lateral sclerosis. Ann Neurol 74:20–38. https://doi.org/10.1002/ana.23937
Yang T, Hou Y, Li C et al (2021) Risk factors for cognitive impairment in amyotrophic lateral sclerosis: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry 92:688–693. https://doi.org/10.1136/jnnp-2020-325701
Ludolph A, Drory V, Hardiman O et al (2015) A revision of the El Escorial criteria—2015. Amyotroph Lateral Scler Front Degener 16:291–292. https://doi.org/10.3109/21678421.2015.1049183
Cedarbaum JM, Stambler N, Malta E et al (1999) The ALSFRS-R: a revised ALS functional rating scale that incorporates assessments of respiratory function. BDNF ALS Study Group (Phase III). J Neurol Sci 169:13–21. https://doi.org/10.1016/s0022-510x(99)00210-5
Aust E, Linse K, Graupner S-T et al (2022) Quality of life and mental health in the locked-in-state—differences between patients with amyotrophic lateral sclerosis and their next of kin. J Neurol. https://doi.org/10.1007/s00415-022-11238-0
R Core Team (2017) R: a language and environment for statistical computing. R Foundation for Statistical Computing. R Foundation for Statistical Computing, Vienna
Fitzmaurice GM, Laird NM, Ware JH (2011) Applied longitudinal analysis, 2nd edn. Wiley, Hoboken
Mancl LA, DeRouen TA (2001) A covariance estimator for GEE with improved small-sample properties. Biometrics 57:126–134. https://doi.org/10.1111/j.0006-341X.2001.00126.x
Geser F, Brandmeir NJ, Kwong LK et al (2008) Evidence of multisystem disorder in whole-brain map of pathological TDP-43 in amyotrophic lateral sclerosis. Arch Neurol 65:636–641. https://doi.org/10.1001/archneur.65.5.636
Pagani M, Chio A, Valentini MC et al (2014) Functional pattern of brain FDG-PET in amyotrophic lateral sclerosis. Neurology 83:1067–1074. https://doi.org/10.1212/WNL.0000000000000792
Munoz DP, Everling S (2004) Look away: the anti-saccade task and the voluntary control of eye movement. Nat Rev Neurosci 5:218–228. https://doi.org/10.1038/nrn1345
Witiuk K, Fernandez-Ruiz J, McKee R et al (2014) Cognitive deterioration and functional compensation in ALS measured with fMRI using an inhibitory task. J Neurosci 34:14260–14271. https://doi.org/10.1523/JNEUROSCI.1111-14.2014
Ford KA, Goltz HC, Brown MRG, Everling S (2005) Neural processes associated with antisaccade task performance investigated with event-related FMRI. J Neurophysiol 94:429–440. https://doi.org/10.1152/jn.00471.2004
Hutton SB, Ettinger U (2006) The antisaccade task as a research tool in psychopathology: a critical review. Psychophysiology 43:302–313. https://doi.org/10.1111/j.1469-8986.2006.00403.x
Lulé D, Böhm S, Müller H-P et al (2018) Cognitive phenotypes of sequential staging in amyotrophic lateral sclerosis. Cortex 101:163–171. https://doi.org/10.1016/j.cortex.2018.01.004
Phukan J, Elamin M, Bede P et al (2011) The syndrome of cognitive impairment in amyotrophic lateral sclerosis: a population-based study. J Neurol Neurosurg Psychiatry. https://doi.org/10.1136/jnnp-2011-300188
Murphy J, Factor-Litvak P, Goetz R et al (2016) Cognitive-behavioral screening reveals prevalent impairment in a large multicenter ALS cohort. Neurology 86:813–820. https://doi.org/10.1212/wnl.0000000000002305
Bersano E, Sarnelli MF, Solara V et al (2020) Decline of cognitive and behavioral functions in amyotrophic lateral sclerosis: a longitudinal study. Amyotroph Lateral Scler Front Degener 21:373–379. https://doi.org/10.1080/21678421.2020.1771732
Kasper E, Schuster C, Machts J et al (2015) Dysexecutive functioning in ALS patients and its clinical implications. Amyotroph Lateral Scler Front Degener 16:160–171. https://doi.org/10.3109/21678421.2015.1026267
Taylor LJ, Brown RG, Tsermentseli S et al (2013) Is language impairment more common than executive dysfunction in amyotrophic lateral sclerosis? J Neurol Neurosurg Psychiatry 84:494–498. https://doi.org/10.1136/jnnp-2012-303526
Kilani M, Micallef J, Soubrouillard C et al (2004) A longitudinal study of the evolution of cognitive function and affective state in patients with amyotrophic lateral sclerosis. Amyotroph Lateral Scler Mot Neuron Disord 5:46–54. https://doi.org/10.1080/14660820310017560
Kasper E, Zydatiss K, Schuster C et al (2016) No change in executive performance in ALS patients: a longitudinal neuropsychological study. Neurodegener Dis 16:184–191. https://doi.org/10.1159/000440957
Lencer R, Trillenberg P (2008) Neurophysiology and neuroanatomy of smooth pursuit in humans. Brain Cogn 68:219–228. https://doi.org/10.1016/j.bandc.2008.08.013
Fukushima K, Fukushima J, Warabi T, Barnes GR (2013) Cognitive processes involved in smooth pursuit eye movements: behavioral evidence, neural substrate and clinical correlation. Front Syst Neurosci. https://doi.org/10.3389/fnsys.2013.00004
Ke SR, Lam J, Pai DK, Spering M (2013) Directional asymmetries in human smooth pursuit eye movements. Invest Ophthalmol Vis Sci 54:4409–4421. https://doi.org/10.1167/iovs.12-11369
Kuzma-Kozakiewicz M, Andersen PM, Ciecwierska K et al (2019) An observational study on quality of life and preferences to sustain life in locked-in state. Neurology 93:e938–e945. https://doi.org/10.1212/WNL.0000000000008064
Linse K, Rüger W, Joos M et al (2017) Eye-tracking–based assessment suggests preserved well-being in locked-in patients. Ann Neurol 81:310–315. https://doi.org/10.1002/ana.24871
Hwang C-S, Weng H-H, Wang L-F et al (2014) An eye-tracking assistive device improves the quality of life for ALS patients and reduces the caregivers’ burden. J Mot Behav 46:233–238. https://doi.org/10.1080/00222895.2014.891970
Linse K, Rüger W, Joos M et al (2018) Usability of eyetracking computer systems and impact on psychological wellbeing in patients with advanced amyotrophic lateral sclerosis. Amyotroph Lateral Scler Front Degener 19:212–219. https://doi.org/10.1080/21678421.2017.1392576
Nakayama Y, Shimizu T, Mochizuki Y et al (2016) Predictors of impaired communication in amyotrophic lateral sclerosis patients with tracheostomy-invasive ventilation. Amyotroph Lateral Scler Front Degener 17:38–46. https://doi.org/10.3109/21678421.2015.1055276
Swinnen B, Robberecht W (2014) The phenotypic variability of amyotrophic lateral sclerosis. Nat Rev Neurol 10:661–670. https://doi.org/10.1038/nrneurol.2014.184
Lillo P, Savage S, Mioshi E et al (2012) Amyotrophic lateral sclerosis and frontotemporal dementia: A behavioural and cognitive continuum. Amyotroph Lateral Scler 13:102–109. https://doi.org/10.3109/17482968.2011.639376
Willemse SW, Harley P, van Eijk RPA et al (2023) UNC13A in amyotrophic lateral sclerosis: from genetic association to therapeutic target. J Neurol Neurosurg Psychiatry 94:649–656. https://doi.org/10.1136/jnnp-2022-330504
Acknowledgements
We express our deepest gratitude to all the participants in this study. We thank Maarten Jung very much for his great support in the statistical analyses. We acknowledge the patient screening by Susanne Petri, Jan Koch and Ulf Bodechtel.
Funding
Open Access funding enabled and organized by Projekt DEAL. The work was funded in part by a grant of the German ministry for Education and Research (BMBF, FKZ 13GW0482) to Elisa Aust, René Günther, Markus Joos, Andreas Hermann, the “Innovationsausschuss beim Gemeinsamen Bundesausschuss,” Germany (FKZ 01VSF16026) to Elisa Aust, Sven-Thomas Graupner, Markus Joos, Sebastian Pannasch, Andreas Hermann, and the Hermann und Lilly Schilling-Stiftung für medizinische Forschung im Stifterverband to Andreas Hermann.
Author information
Authors and Affiliations
Contributions
Conceptualization: EA, KL, S-TG, MJ, SP, AH; Data curation: EA, S-TG; Formal analysis: EA; Funding acquisition: AH, KL; Investigation: EA, S-TG, MJ; Methodology: EA, KL, S-TG, AH; Project administration: AH; Resources: RG, MJ, JG, JP, TM; Software: S-TG, MJ; Supervision: SP, AH; Visualization: EA; Writing—original draft: EA; Writing—review and editing: EA, S-TG, RG, KL, JG, JP, SP, AH. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Conflicts of interest
Andreas Hermann has received funding from the European Social Fonds, the Federal Ministry of Education and Research and the Hermann und Lilly Schilling-Stiftung für medizinische Forschung im Stifterverband. He has received honoraria for presentations/advisory boards/ from Amylyx, Desitin and IFT Pharma. He has received royalties from Elsevier Press and Kohlhammer. Makus Joos’ affiliation “Interactive Minds” is a provider of ETCS in the region of Dresden. He was not involved and had no influence in the blinded data analysis. The other authors declare that they have no conflict of interest. Funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or the decision to publish the results.
Ethics approval
The study was approved by the institutional review board at the Technische Universität Dresden (EK 329082017, EK 393122012).
Informed consent
In accordance with the Declaration of Helsinki, all subjects gave informed consent. For patients who were not able to write, consent was confirmed in writing through a witness in the presence of the patient after reading and explaining the protocol and giving the patient the opportunity to ask questions via ETCS.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Aust, E., Graupner, ST., Günther, R. et al. Impairment of oculomotor functions in patients with early to advanced amyotrophic lateral sclerosis. J Neurol 271, 325–339 (2024). https://doi.org/10.1007/s00415-023-11957-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-023-11957-y