
Abstract
Background
The characterisation of presymptomatic disease-burden patterns in asymptomatic mutation carriers has a dual academic and clinical relevance. The understanding of disease propagation mechanisms is of considerable conceptual interests, and defining the optimal time of pharmacological intervention is essential for improved clinical trial outcomes.
Methods
In a prospective, multimodal neuroimaging study, 22 asymptomatic C9orf72 GGGGCC hexanucleotide repeat carriers, 13 asymptomatic subjects with SOD1, and 54 “gene-negative” ALS kindreds were enrolled. Cortical and subcortical grey matter alterations were systematically appraised using volumetric, morphometric, vertex, and cortical thickness analyses. Using a Bayesian approach, the thalamus and amygdala were further parcellated into specific nuclei and the hippocampus was segmented into anatomically defined subfields.
Results
Asymptomatic GGGGCC hexanucleotide repeat carriers in C9orf72 exhibited early subcortical changes with the preferential involvement of the pulvinar and mediodorsal regions of the thalamus, as well as the lateral aspect of the hippocampus. Volumetric approaches, morphometric methods, and vertex analyses were anatomically consistent in capturing focal subcortical changes in asymptomatic C9orf72 hexanucleotide repeat expansion carriers. SOD1 mutation carriers did not exhibit significant subcortical grey matter alterations. In our study, none of the two asymptomatic cohorts exhibited cortical grey matter alterations on either cortical thickness or morphometric analyses.
Discussion
The presymptomatic radiological signature of C9orf72 is associated with selective thalamic and focal hippocampal degeneration which may be readily detectable before cortical grey matter changes ensue. Our findings confirm selective subcortical grey matter involvement early in the course of C9orf72-associated neurodegeneration.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
One of the important paradigm shifts in amyotrophic lateral sclerosis (ALS) research is the departure from the concept of “one-drug for all” to the pursuit of precision, genotype-specific pharmacological interventions [1, 2]. The recognition of the fundamental heterogeneity of ALS led to the nuanced characterisation of various ALS genotypes and phenotypes [3,4,5,6]. It is increasingly recognised that symptom manifestation in ALS is preceded by a long presymptomatic phase [7] and degenerative changes may be detected decades before symptom manifestation [8, 9]. The ideal timing of therapeutic intervention should therefore be reconsidered, especially in genetically susceptible cohorts. Antisense oligonucleotide (ASO)-therapies have been approved for the treatment of spinal muscular atrophy and Duchenne muscular dystrophy [10,11,12], and also trialled in ALS [1]. Accordingly, the assessment of disease burden prior to symptom manifestation is of pressing practical relevance. Two most commonly studied genotypes in ALS are the GGGGCC hexanucleotide repeat expansions (HRE) in C9orf72 and SOD1. C9orf72 HRE may lead to a spectrum of clinical manifestations spanning from ALS to frontotemporal dementia (FTD) and clinical manifestations are thought to be closely associated with patterns of phosphorylated 43 kDa TAR DNA-binding protein (pTDP-43) burden. Striatal [13], temporal [14], frontal [15], cerebellar [16], and thalamic [13, 16] grey matter alterations have previously been described in asymptomatic C9orf72 cohorts. Presymptomatic orbitofrontal [17], corpus callosum, cingulate, uncinate [8, 13, 18], and corticospinal tract [9, 13] white matter changes have also been consistently detected and PET studies captured frontotemporal, thalamic, and basal ganglia hypometabolism [19]. Three mechanisms have been proposed for C9orf72 HRE-associated pathophysiology; loss of C9orf72 function through haploinsufficiency, toxic gain-of-function due to the generation of aberrant HRE-containing RNA, and toxic gain-of-function through the accumulation of dipeptide repeat proteins translated from hexanucleotide repeat RNA [20]. These mechanisms are thought to trigger a multitude of cellular responses, of which pTDP-43 accumulation may only be one amongst several processes. Accordingly, cerebral involvement outside the neocortex may represent a distinguishing feature of C9orf72 distinct from other genetic variants, such as SOD1 mutations [21]. The presymptomatic imaging signature of SOD1 is thought to be relatively unique; white matter changes in the posterior limb of the internal capsule [22], reduced superior spinal cord NAA/Cr and NAA/Myo ratios [23], and reduced left frontotemporal junction flumazenil binding [24], and have been reported. While structural, metabolic, and function thalamic [8, 13, 16, 18, 25,26,27,28,29,30] changes have been previously described in presymptomatic C9orf72 hexanucleotide carriers and longitudinal thalamic changes have also been explored [25, 31, 32], the predilection for specific thalamic nuclei remains poorly characterised despite the unique role of thalamic nuclei in relaying specific sensory, cognitive, and behavioural functions [33,34,35,36]. Presymptomatic amygdalar and hippocampal alterations are also under evaluated despite the selective involvement of these structures in symptomatic mutation carriers [37,38,39,40,41]. Accordingly, the principal objective of this study is the nuanced characterisation of cortical and subcortical grey matter changes in two cohorts of presymptomatic mutation carriers using a panel of supplementary imaging techniques. Our hypothesis is that presymptomatic hexanucleotide expansion carriers exhibit focal subcortical degeneration with concomitant alterations in their cortical projection areas.
Methods
The study was approved by the Ethics Committee of the University of Ulm (reference 68/19), in accordance with the ethical standards of the current version of the revised Helsinki declaration. All participants gave informed consent prior to enrolment. Recruitment strategy and genetic testing have been previously described [17].
Neuroimaging
T1-weighted data were acquired on a 1.5 Tesla Magnetom Symphony (Siemens Medical) with a 12-channel head coil. Acquisition parameters have been described previously [42]. T2-weighted and fluid-attenuated inversion recovery (FLAIR) images were systematically reviewed for confounding vascular or neuroinflammatory pathologies. Raw T1-weighted MR data were screened for artifacts, developmental malformations, arachnoid or porencephalic cysts, hydrocephalus, or other pathologies that could impact on quantitative morphometric analyses prior to pre-processing.
Volumetric analyses
The standard pre-processing steps of the FreeSurfer image analysis suite [43] were first implemented, including removal of non-brain tissue, segmentation of the subcortical white matter and deep grey matter structures, intensity normalization, tessellation of the grey matter–white matter boundary, and automated topology correction. Following quality control steps for segmentation accuracy, overall volumes of subcortical structures and total intracranial volume estimates (eTIV) were retrieved from each subject. Total volume estimates of the following structures were generated in the left and right hemispheres separately: thalamus, caudate, putamen, pallidum, hippocampus, amygdala, and nucleus accumbens.
Nuclear segmentation of the thalamus and amygdala
The thalamus was segmented into 25 subregions using Bayesian inference based on a probabilistic atlas developed upon histological data [44]. The thalamus was first parcellated into the following nuclei in each hemisphere: anteroventral (AV), laterodorsal (LD), lateral posterior (LP), ventral anterior (VA), ventral anterior magnocellular (VA mc), ventral lateral anterior (VLa), ventral lateral posterior (VLp), ventral posterolateral (VPL), ventromedial (VM), central medial (CeM), central lateral (CL), paracentral (Pc), centromedian (CM), parafascicular (Pf), paratenial (Pt), reuniens/medial ventral (MV-re), mediodorsal medial magnocellular (MDm), mediodorsal lateral parvocellular (MDl), lateral geniculate (LGN), medial geniculate (MGN), limitans/suprageniculate (L-SG), pulvinar anterior (PuA), pulvinar medial (PuM), pulvinar lateral (PuL), and pulvinar inferior (PuI). The raw volume estimates of the above nuclei were averaged between left and right and merged into the following 10 core group of nuclei defined based on their distinctive physiological function: “anteroventral”, “lateral geniculate”, “medial geniculate”, “pulvinar-limitans” (PuA, PuM, PuL, PuI, L-SG), “laterodorsal”, “lateroposterior”, “mediodorsal-paratenial-reuniens” (MDm, MDl, MV-re, Pt), “motor nuclei” (VA, VAmc, VLa, VLp), “sensory nuclei” (VPL, VM), and “intralaminar” (CeM, CL, Pc, CM, Pf). “Total thalamic volume” was defined as the mean of the left and right thalamus volume estimates and used as a covariate in the relevant statistical models. Post hoc statistics were corrected for demographic variables, total thalamic volume, and multiple testing.
A Bayesian inference was also used to parcellate the amygdala into nine subregions using a probabilistic atlas developed based on histological data [44, 45]. The amygdala was segmented into the following nuclei in each hemisphere: lateral nucleus (LN), basal nucleus (BN), accessory basal nucleus (ABN), anterior amygdaloid area (AAA), central nucleus (CN), medial nucleus (MN), cortical nucleus (CN), cortico-amygdaloid transition (CAT), and paralaminar nucleus (PN). The raw volume estimates of the above nuclei were averaged between left and right. “Total amygdala volume” was defined as the mean of the left and right total amygdala volume estimates. Post hoc statistics were corrected for demographic variables, total amygdala volume, and multiple testing.
Hippocampal subfield parcellation
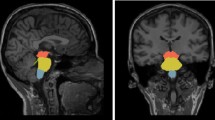
The hippocampus was segmented into cytologically-defined subfields (Fig. 1A) using the FreeSurfer image analysis suite [43]. The pre-processing pipeline included the removal of non-brain tissue, segmentation of the subcortical white matter and deep grey matter structures, intensity normalization, tessellation of the grey matter–white matter boundary, and automated topology correction. The hippocampal stream of the FreeSurfer package was used for the delineation of the following hippocampal subfields: CA1, CA2/3, CA4, fimbria, hippocampal fissure, presubiculum, subiculum, hippocampal tail, parasubiculum, molecular layer; granule cell layer of the dentate gyrus (GC-DG), and hippocampal–amygdala transition area (HATA) [46].

Effect-size differences in thalamic nuclei volumes between C9orf72 hexanucleotide repeat expansion carriers, gene-negative controls and SOD1 mutation carriers in the two thalami. AV anteroventral nuclei, C9orf72 chromosome 9 open-reading frame 72, LD laterodorsal nuclei, LGN lateral geniculate, LP lateral posterior nuclei, MGN medial geniculate nuclei
Vertex analyses
Surface projected patterns of atrophy were evaluated using vertex analyses. As described previously [47], FMRIB’s subcortical segmentation and registration tool FIRST [48] was utilised to characterise focal thalamic shape deformations. Vertex locations of each participant were projected on the surface of an average thalamic shape template as scalar values, positive value being outside the surface, and negative values inside. Using study-specific design matrices specifying group membership and covariates, permutation-based non-parametric inference was implemented for group comparisons using FMRIB’s ‘RANDOMISE’ module [49]. The design matrices included demeaned age, sex, education, and total intracranial volumes as covariates [49].
Subcortical morphometry
FMRIB’s software library was used for brain extraction and tissue-type segmentation. Resulting grey-matter partial volume images were then aligned to MNI152 standard space using affine registration. A study-specific template was subsequently created, to which the grey matter images from each subject were non-linearly coregistered. Group membership and covariates were specified in study-specific design matrices and demeaned covariates included age, sex, education, and TIV. A voxelwise generalized linear model and permutation-based non-parametric testing was used to highlight density alterations in a merged subcortical grey matter mask accounting for multiple testing, age, sex, and education. [49, 50] Labels of the Harvard–Oxford subcortical probabilistic structural atlas was used to generate a merged subcortical grey mask incorporating the left and right caudate, thalamus, accumbens, hippocampus, amygdala, putamen, and pallidum. [51, 52]
Cortical grey matter analyses
A dual pipeline was implemented exploring (1) cortical thickness alterations and (2) morphometric changes using voxel-based morphometry (VBM). Following pre-processing and cortical segmentation in FreeSurfer, average cortical thickness values have been retrieved from 34 cortical regions in each hemisphere separately as per the Desikan–Killiany atlas. Cortical thickness values in the following lobes and corresponding subregions regions were appraised, Frontal lobe (13 ROIs): Superior Frontal, Rostral and Caudal Middle Frontal, Pars Opercularis, Pars Triangularis, and Pars Orbitalis, Lateral and Medial Orbitofrontal, Precentral, Paracentral, Frontal Pole, Rostral Anterior cingulate, Caudal Anterior cingulate, Parietal lobe (7 ROIs): Superior Parietal, Inferior Parietal, Supramarginal, Postcentral, Precuneus, Posterior cingulate, cingulate isthmus, Temporal lobe (9 ROIs): Superior, Middle, and Inferior Temporal, Banks of the Superior Temporal Sulcus, Fusiform, Transverse Temporal, Entorhinal, Temporal Pole, Parahippocampal, Occipital lobe (4 ROIs): lateral Occipital, Lingual, Cuneus, Pericalcarine, and the Insula (1 ROI). In addition to cortical thickness analyses, voxel-based morphometry was also performed to evaluate anatomical patterns of signal intensity reductions in mutation carriers. Cortical grey matter morphometry analyses were conducted using FSL-VBM [53, 54]. Following brain extraction, motion-correction, and tissue-type segmentation, the resulting grey-matter partial volume images were aligned to MNI152 standard space using affine registration. A study-specific template was generated to which the grey matter images from each subject were non-linearly coregistered. Permutation-based non-parametric inference and the threshold-free cluster enhancement (TFCE) approach were utilised to test for differences between study groups controlling for age, sex, TIV, and education.
Results
The main study groups were matched for age and education and differed in sex ratios (Table 1). We note that all statistical models were corrected for age, sex, and education.
While differences in overall subcortical volume differences did not reach significance (Table 2), mediodorsal-paratenial-reuniens in the left thalamus and pulvinar-limitans atrophy in the right thalamus was detected in asymptomatic C9orf72 hexanucleotide repeat expansion carriers compared to gene-negative family members (Table 3.). Furthermore, higher sensory nuclei volumes were identified in C9orf72 hexanucleotide repeat expansion carriers compared to both gene-negative controls and SOD1 mutation carriers in both thalami. Effect sizes are illustrated in Fig. 1. Differences in amygdalar nuclei and hippocampal subfield volumes did not reach significance. Relevant output statistics, univariate p value, and effect sizes are summarised in supplementary tables 1–2.
Vertex analyses identified shape deformations in C9orf72 hexanucleotide repeat expansion carriers compared to gene-negative controls in the anterior, superior, and posterior surface of both thalami as well as the lateral aspect of the left hippocampus (Fig. 2). Vertex analyses in SOD1 carriers did not identify shape deformation compared to gene-negative controls. ROI morphometric analyses revealed bi-thalamic and left pulvinar signal reductions in C9orf72 hexanucleotide repeat expansion carriers compared to gene-negative controls (Fig. 3). In our voxelwise analyses, the contrasts between SOD1 carriers and gene-negative controls did not reach significance.

3D representation of shape deformations in C9orf72 hexanucleotide repeat expansion carriers compared to gene-negative controls at p < 0.05 FWE (age, sex, TIV, education corr.) in the anterior, superior, and posterior surface of both thalami as well as the lateral aspect of the left hippocampus. Blue colour represents the three-dimensional study-specific mesh of the bilateral thalami, hippocampi, and amygdalae. Orange colour represents areas of surface deformations at p < 0.05 FWE corr. in C9orf72 mutation carriers. A superior view, B left lateral view, C posterior view, D anterior view, and E left lateroposterior oblique view

Morphometric alterations based on signal reductions in C9orf72 hexanucleotide repeat expansion carriers compared to gene-negative controls at p < 0.05 TFCE corrected for age, sex, education, and TIV. The aqua blue colour underlay represents the unified basal ganglia-thalamic region-pf-interest mask and the statistically significant clusters at p < 0.05 TFCE are shown in red–orange. Slice coordinates are provided with reference to the Montreal Neurosciences Institute (MNI152) space. Green arrows highlight the small cluster of signal reduction in the pulvinar region of the left thalamus
Discussion
Our computational image analyses capture thalamic and hippocampal alterations in a cohort asymptomatic C9orf72 hexanucleotide repeat expansion carriers without associated neocortical grey matter atrophy. No cortical or subcortical grey matter pathology was observed in our presymptomatic SOD1 group.
Methodologically, our study benefits from a multiparametric approach, where data were interrogated in multiple pipelines run in different image analyses suites resulting in good anatomical concordance. The results of our post-segmentation volumetric analyses and our ROI-based morphometric subcortical analyses are relatively concordant in identifying bilateral mediodorsal thalamic atrophy (Table 3, Fig. 3.). Our results indicate that as opposed to global thalamic degeneration, selective thalamic involvement characterises the asymptomatic phase of C9orf72. The shape deformation identified on vertex analyses confirms focal thalamic involvement, but the nature of these analyses is that surface-projected changes are captured instead of the intra-thalamic changes described by morphometric and post-segmentation pipelines. Resting-state fMRI studies have consistently described widespread connectivity alterations in C9orf72 mutation carriers including networks relayed through the thalamus [13, 55]. Network integrity alterations were also detected using chronnectomic approaches [56] and thalamic hypometabolism has also been consistently identified by PET studies [57, 58]. MR spectroscopy [59] identified reduced putaminal NAA/Cr, Glu/Cr and Glu/NAA ratios, and reduced thalamic Glu/NAA in asymptomatic C9orf72 mutation carriers [19]. Based on structural data, thalamic atrophy [8, 13, 16, 18, 25,26,27,28] has been previously described in presymptomatic C9orf72 hexanucleotide carriers as well as disease burden in other subcortical grey matter structures, such as the caudate [14, 60], putamen [14], and striatum, but the predilection for specific nuclei or subregions have not been comprehensively analysed. In our study, the two main thalamic regions identified by both our volumetric and morphometric analysis streams are the mediodorsal and pulvinar regions. The physiological role of cortico-basal networks relayed though specific nuclei are fairly well established [33, 35, 61]. Thalamic atrophy has been highlighted in most genetic variants of FTD [35, 62], but medial pulvinar degeneration is thought to be relatively unique to C9orf72 [62, 63]. The involvement of specific groups of nuclei at a presymptomatic phase is relativity novel as previous studies primarily focused on global thalamus pathology [18] or only described focal changes based on voxelwise analyses. In a particularly elegant recent study, spectral clustering, a graph-based partitioning technique was implemented which revealed posteromedial thalamic changes in asymptomatic C9orf72 carriers. [64] The early degeneration of mediodorsal and pulvinar regions in our C9orf72 cohort may herald future dysfunction in associated cognitive domains, such as limbic, executive, and associative processes.
It is noteworthy that the patterns of grey matter changes identified in this asymptomatic cohort, are also relatively consistent with the “post-symptomatic” signature associated of the genotype. Selective thalamic atrophy in symptomatic C9orf72 hexanucleotide expansion carriers is evidenced by a multitude of large, prospective neuroimaging studies including both ALS and FTD phenotypes [35, 65]. Grey matter involvement in C9orf72 carriers outside the neocortex has a predilection for the thalamus, [66,67,68,69,70] although hippocampal, putaminal, striatal, caudate, cerebellar, and nucleus accumbens changes are also commonly reported [14, 40, 71, 72]. Even though grey matter disease burden is thought to be more pronounced in C9orf72-associated ALS [71, 73], considerable subcortical grey matter pathology is also readily identified in C9orf72-negative ALS and PLS [40, 47, 74,75,76,77]. Basal ganglia changes have also been consistently described in sporadic patients and linked to neuropsychological and extra-pyramidal deficits [78,79,80]. Similar to our findings in this cohort of presymptomatic patients, a study of symptomatic C9orf72 patients with ALS fulfilling the EL Escorial criteria [69] also identified the selective degeneration of thalamic nuclei in C9orf72-positive ALS preferentially involving the mediodorsal–parateniual–reuniens group of nuclei, which play a central role in executive processes. Interestingly, we have detected higher sensory nuclei volumes bilaterally in C9orf72 hexanucleotide carriers compared to both “gene-negative” ALS kindreds and asymptomatic SOD1 mutation carriers. While somatosensory impairment is not classically associated with the core clinical features of ALS, in symptomatic cohorts, imaging studies have consistently confirmed the involvement of somatosensory structures [34, 70]. The marked differences between SOD1 and C9orf72 showcase the notable heterogeneity of ALS, and given the relatively young age profile of participants, the higher volumes detected hexanucleotide repeat carriers may support the role of neurodevelopmental factors [17, 81, 82]. Our vertex analyses also highlight hippocampal atrophy in hexanucleotide carriers which is also consistent with observations from symptomatic patient groups [14, 40, 71] and in line with the reports of early memory impairment in clinical subgroups of ALS [83]. Also, these subjects exhibit nucleus reuniens involvement of a key hub of thalamic afferents to the hippocampus [84]. Thus, C9orf72 carriers display concomitant thalamic and hippocampal alterations which are probably functionally interlinked. However, given that our hippocampal findings are unilateral, further validation is needed by larger studies.
The practical relevance of multimodal presymptomatic studies stems from the prospect of developing accurate predictive models to foretell the approximate time of phenoconversion and the likely clinical phenotype. This would enable precision care planning for individual subjects and optimised timing for clinical trial inclusion. The utility of machine learning (ML) has already been demonstrated in a variety of prognostic and diagnostic applications in ALS [85,86,87]. Imaging-based ML models in ALS increasingly include subcortical measures [88,89,90,91] in addition to cortical grey matter and cerebral white matter metrics [92,93,94,95]. Feature importance analyses and cluster analyses consistently confirmed the discriminatory potential of subcortical indices and integrity metrics of networks relayed through subcortical nuclei [96, 97]. In asymptomatic C9orf72 HRE carriers, emerging spinal cord imaging techniques may be particularly useful in delineating incipient ALS from FTD [9, 98]. MRI-based predictive models have already been successfully trialled in symptomatic ALS patients [92, 99] and similar strategies could be adopted in asymptomatic cohorts to foretell likely phenoconversion.
Our results highlight the fundamental heterogeneity of genetic ALS from its earliest stages, by demonstrating the strikingly divergent disease-burden patterns between C9orf72 and SOD1 carriers. Despite the insights generated by a cross-sectional study, ultimately, large, multi-timepoint longitudinal studies are required to track mutation carriers from a very young age until fulfilling diagnostic criteria and beyond. The lack of longitudinal analyses is the biggest limitation of this cross-sectional study. While our descriptive statistics demonstrate focal grey matter changes, they only offer a mere snapshot in time. Characterising the evolution of grey matter alterations from birth to phenoconversion through several timepoints would reveal the full biological trajectory of these processes [100]. Importantly, the subjects of this study have not yet met relevant diagnostic criteria, and therefore, we cannot be sure whether individual hexanucleotide expansion carriers will develop a syndrome primarily consistent with ALS or FTD, although the index cases (affected, symptomatic family members) of the current study all had ALS. Finally, we acknowledge the limitations of the raw data and wish that additional spectroscopic or resting-state fMRI data would be at our disposal to comprehensively interrogate metabolic and connectomic alterations.
Conclusions
C9orf72 hexanucleotide repeat expansions are associated with presymptomatic thalamus and hippocampus alterations which may precede detectable neocortical involvement. The identified radiological changes may be mediated by a multitude of C9orf72-associated pathophysiological processes which take place decades before symptom manifestation.
Data availability statement
Group-level outputs, post-hoc statistics and additional information on data processing pipelines can be requested from the corresponding author. Individual-subject neuroimaging data cannot be made available due to departmental policies.
Abbreviations
- AAA:
-
Anterior amygdaloid area
- ABN:
-
Accessory basal nucleus
- ALS:
-
Amyotrophic lateral sclerosis
- AN:
-
Attention network
- ANCOVA:
-
Analysis of covariance
- AV:
-
Anteroventral nuclei
- BG:
-
Basal ganglia
- BN:
-
Basal nucleus
- C9orf72:
-
Chromosome 9 open-reading frame 72
- CA:
-
Cornu Ammonis
- CAT:
-
Cortico-amygdaloid transition
- CeM:
-
Central medial nucleus
- CL:
-
Central lateral nucleus
- CM:
-
Centromedian nucleus
- CN:
-
Cortical nucleus
- CST:
-
Corticospinal tract
- Cr:
-
Creatine‐phosphocreatine
- CT:
-
Cortical thickness
- DTI:
-
Diffusion tensor imaging
- ECAS:
-
Edinburgh Cognitive ALS Screen
- EMM:
-
Estimated marginal mean
- EPI:
-
Echo-planar imaging
- eTIV:
-
Total intracranial volume estimates
- FTD:
-
Frontotemporal dementia
- FLAIR:
-
Fluid-attenuated inversion recovery
- FOV:
-
Field-of-view
- FSL:
-
FMRIB Software Library
- FWE:
-
Familywise error
- GC-DG:
-
Granule cell layer of dentate gyrus
- GM:
-
Grey matter
- HARDI:
-
High angular resolution diffusion imaging
- HATA:
-
Hippocampus-amygdala transition area
- HC:
-
Healthy control
- HRE:
-
Hexanucleotide repeat expansions
- IR-SPGR:
-
Inversion Recovery prepared Spoiled Gradient Recalled echo
- LD:
-
Laterodorsal nuclei
- LN:
-
Lateral nucleus
- LGN:
-
Lateral geniculate
- LMN:
-
Lower motor neuron
- LP:
-
Lateral posterior nuclei
- L-SG:
-
Limitans/suprageniculate nuclei
- Lt:
-
Left
- M:
-
Mean
- MANCOVA:
-
Multivariate analysis of covariance
- MDI:
-
Mediodorsal lateral parvocellular nuclei
- MDm:
-
Mediodorsal medial magnocellular nuclei
- MGN:
-
Medial geniculate nuclei
- ML:
-
Machine learning
- MN:
-
Medial nucleus
- MND:
-
Motor neuron disease
- MNI152:
-
Montreal Neurological Institute 152 standard space
- MR:
-
Magnetic resonance
- MRS:
-
Magnetic resonance spectroscopy
- MT:
-
Magnetisation transfer
- MV-re:
-
Reuniens/medial ventral nuclei
- Myo:
-
Myoinositol
- NAA:
-
N-Acetylaspartate
- NODDI:
-
Neurite orientation dispersion and density imaging
- PBA:
-
Pseudobulbar affect
- Pc:
-
Paracentral nuclei
- PCL:
-
Pathological crying and laughing
- Pf:
-
Parafascicular nuclei
- PLS:
-
Primary lateral sclerosis
- PMA:
-
Progressive muscular atrophy
- PMC:
-
Primary motor cortex
- PN:
-
Paralaminar nucleus
- PPZ:
-
Perforant pathway zone
- Pt:
-
Paratenial nuclei
- pTDP-43:
-
Phosphorylated 43 kDa TAR DNA-binding protein
- PuA:
-
Pulvinar anterior nuclei
- PuI:
-
Pulvinar inferior nucleus
- PuL:
-
Pulvinar lateral nucleus
- pTDP-43:
-
Phosphorylated 43 kDa TAR DNA- binding protein
- PuM:
-
Pulvinar medial nucleus
- QSM:
-
Quantitative susceptibility mapping
- RE:
-
Repeat expansion
- ROI:
-
Region-of-interest
- Rt:
-
Right
- SBMA:
-
Spinal and bulbar muscular atrophy / Kennedy's disease
- SD:
-
Standard deviation
- SE:
-
Standard error
- SOD1:
-
Superoxide dismutase 1
- SN:
-
Salience network
- SPSS:
-
Statistical product and service solutions
- T1W:
-
T1-weighted imaging
- TE:
-
Echo time
- TFCE:
-
Threshold-free cluster enhancement
- TI:
-
Inversion time
- TIV:
-
Total intracranial volume
- TR:
-
Repetition time
- UMN:
-
Upper motor neuron
- VA:
-
Ventral anterior nuclei
- VAmc:
-
Ventral anterior magnocellular nuclei
- VBM:
-
Voxel-based morphometry
- VLa:
-
Ventral lateral anterior nuclei
- VLp:
-
Ventral lateral posterior nuclei
- VM:
-
Ventromedial nuclei
- VPL:
-
Ventral posterolateral nuclei
- WM:
-
White matter
References
Miller T, Cudkowicz M, Shaw PJ et al (2020) Phase 1–2 trial of antisense oligonucleotide tofersen for SOD1 ALS. N Engl J Med 383:109–119
Mueller C, Berry JD, McKenna-Yasek DM et al (2020) SOD1 suppression with adeno-associated virus and microRNA in familial ALS. N Engl J Med 383:151–158
Omer T, Finegan E, Hutchinson S et al (2017) Neuroimaging patterns along the ALS-FTD spectrum: a multiparametric imaging study. Amyotroph Lateral Scler Frontotemporal Degener 18:611–623
Burke T, Pinto-Grau M, Lonergan K et al (2017) A Cross-sectional population-based investigation into behavioral change in amyotrophic lateral sclerosis: subphenotypes, staging, cognitive predictors, and survival. Ann Clin Transl Neurol 4:305–317
Finegan E, Chipika RH, Shing SLH, Hardiman O, Bede P (2019) Primary lateral sclerosis: a distinct entity or part of the ALS spectrum? Amyotroph Lateral Scler Frontotemporal Degener 20:133–145
Burke T, Elamin M, Bede P et al (2016) Discordant performance on the “reading the mind in the eyes” test, based on disease onset in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener 17:467–472
Kiernan MC, Ziemann U, Eisen A (2019) Amyotrophic lateral sclerosis: origins traced to impaired balance between neural excitation and inhibition in the neonatal period. Muscle Nerve 60:232–235
Bertrand A, Wen J, Rinaldi D et al (2018) Early cognitive, structural, and microstructural changes in presymptomatic C9orf72 carriers younger than 40 years. JAMA Neurol 75:236–245
Querin G, Bede P, El Mendili MM et al (2019) Presymptomatic spinal cord pathology in c9orf72 mutation carriers: a longitudinal neuroimaging study. Ann Neurol 86:158–167
Cirak S, Arechavala-Gomeza V, Guglieri M et al (2011) Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study. Lancet 378:595–605
Mendell JR, Rodino-Klapac LR, Sahenk Z et al (2013) Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann Neurol 74:637–647
Mendell JR, Goemans N, Lowes LP et al (2016) Longitudinal effect of eteplirsen versus historical control on ambulation in Duchenne muscular dystrophy. Ann Neurol 79:257–271
Lee SE, Sias AC, Mandelli ML et al (2017) Network degeneration and dysfunction in presymptomatic C9ORF72 expansion carriers. Neuroimage Clin 14:286–297
Walhout R, Schmidt R, Westeneng HJ et al (2015) Brain morphologic changes in asymptomatic C9orf72 repeat expansion carriers. Neurology 85:1780–1788
Le Blanc G, Jetté Pomerleau V, McCarthy J et al (2020) Faster cortical thinning and surface area loss in presymptomatic and symptomatic C9orf72 repeat expansion adult carriers. Ann Neurol 2:2
Papma JM, Jiskoot LC, Panman JL et al (2017) Cognition and gray and white matter characteristics of presymptomatic C9orf72 repeat expansion. Neurology 89:1256–1264
Lulé DE, Müller HP, Finsel J et al (2020) Deficits in verbal fluency in presymptomatic C9orf72 mutation gene carriers-a developmental disorder. J Neurol Neurosurg Psychiatry 91:1195–1200
Wen J, Zhang H, Alexander DC et al (2019) Neurite density is reduced in the presymptomatic phase of C9orf72 disease. J Neurol Neurosurg Psychiatry 90:387–394
De Vocht J, Blommaert J, Devrome M et al (2020) Use of multimodal imaging and clinical biomarkers in presymptomatic carriers of C9orf72 repeat expansion. JAMA Neurol 77:1–10
Babić Leko M, Župunski V, Kirincich J et al (2019) Molecular mechanisms of neurodegeneration related to C9orf72 hexanucleotide repeat expansion. Behav Neurol 2019:2909168
Balendra R, Isaacs AM (2018) C9orf72-mediated ALS and FTD: multiple pathways to disease. Nat Rev Neurol 14:544–558
Ng MC, Ho JT, Ho SL et al (2008) Abnormal diffusion tensor in nonsymptomatic familial amyotrophic lateral sclerosis with a causative superoxide dismutase 1 mutation. J Magn Reson Imaging 27:8–13
Carew JD, Nair G, Andersen PM et al (2011) Presymptomatic spinal cord neurometabolic findings in SOD1-positive people at risk for familial ALS. Neurology 77:1370–1375
Turner MR, Hammers A, Al-Chalabi A et al (2005) Distinct cerebral lesions in sporadic and “D90A” SOD1 ALS: studies with [11C]flumazenil PET. Brain 128:1323–1329
Panman JL, Jiskoot LC, Bouts M et al (2019) Gray and white matter changes in presymptomatic genetic frontotemporal dementia: a longitudinal MRI study. Neurobiol Aging 76:115–124
Rohrer JD, Nicholas JM, Cash DM et al (2015) Presymptomatic cognitive and neuroanatomical changes in genetic frontotemporal dementia in the Genetic Frontotemporal dementia Initiative (GENFI) study: a cross-sectional analysis. Lancet Neurol 14:253–262
Olney NT, Ong E, Goh SM et al (2020) Clinical and volumetric changes with increasing functional impairment in familial frontotemporal lobar degeneration. Alzheimers Dement 16:49–59
Cash DM, Bocchetta M, Thomas DL et al (2018) Patterns of gray matter atrophy in genetic frontotemporal dementia: results from the GENFI study. Neurobiol Aging 62:191–196
Diehl-Schmid J, Licata A, Goldhardt O et al (2019) FDG-PET underscores the key role of the thalamus in frontotemporal lobar degeneration caused by C9ORF72 mutations. Transl Psychiatry 9:54
Malpetti M, Holland N, Jones PS et al (2021) Synaptic density in carriers of C9orf72 mutations: a [(11) C]UCB-J PET study. Ann Clin Transl Neurol 8:1515–1523
van Veenhuijzen K, Westeneng HJ, Tan HHG et al (2022) Longitudinal effects of asymptomatic C9orf72 carriership on brain morphology. Ann Neurol 2:2
Shoukry RS, Waugh R, Bartlett D, Raitcheva D, Floeter MK (2020) Longitudinal changes in resting state networks in early presymptomatic carriers of C9orf72 expansions. Neuroimage Clin 28:102354
Bonelli RM, Cummings JL (2007) Frontal-subcortical circuitry and behavior. Dialog Clin Neurosci 9:141–151
Lule D, Diekmann V, Muller HP, Kassubek J, Ludolph AC, Birbaumer N (2010) Neuroimaging of multimodal sensory stimulation in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 81:899–906
McKenna MC, Lope J, Bede P, Tan EL (2023) Thalamic pathology in frontotemporal dementia: predilection for specific nuclei, phenotype-specific signatures, clinical correlates, and practical relevance. Brain Behav. 2:e2881
McKenna MC, Li Hi Shing S, Murad A et al (2022) Focal thalamus pathology in frontotemporal dementia: phenotype-associated thalamic profiles. J Neurol Sci 436:120221
Chipika RH, Christidi F, Finegan E et al (2020) Amygdala pathology in amyotrophic lateral sclerosis and primary lateral sclerosis. J Neurol Sci 417:117039
Pinkhardt EH, van Elst LT, Ludolph AC, Kassubek J (2006) Amygdala size in amyotrophic lateral sclerosis without dementia: an in vivo study using MRI volumetry. BMC Neurol 6:48
Chipika RH, Siah WF, Shing SLH et al (2020) MRI data confirm the selective involvement of thalamic and amygdalar nuclei in amyotrophic lateral sclerosis and primary lateral sclerosis. Data Brief 2:106246
Christidi F, Karavasilis E, Rentzos M et al (2019) Hippocampal pathology in amyotrophic lateral sclerosis: selective vulnerability of subfields and their associated projections. Neurobiol Aging 84:178–188
Christidi F, Karavasilis E, Velonakis G et al (2018) The clinical and radiological spectrum of hippocampal pathology in amyotrophic lateral sclerosis. Front Neurol 9:523
Kassubek J, Müller HP, Del Tredici K et al (2018) Imaging the pathoanatomy of amyotrophic lateral sclerosis in vivo: targeting a propagation-based biological marker. J Neurol Neurosurg Psychiatry 89:374–381
Fischl B (2012) FreeSurfer. Neuroimage 62:774–781
Iglesias JE, Insausti R, Lerma-Usabiaga G et al (2018) A probabilistic atlas of the human thalamic nuclei combining ex vivo MRI and histology. Neuroimage 183:314–326
Saygin ZM, Kliemann D, Iglesias JE et al (2017) High-resolution magnetic resonance imaging reveals nuclei of the human amygdala: manual segmentation to automatic atlas. Neuroimage 155:370–382
Iglesias JE, Augustinack JC, Nguyen K et al (2015) A computational atlas of the hippocampal formation using ex vivo, ultra-high resolution MRI: application to adaptive segmentation of in vivo MRI. Neuroimage 115:117–137
Machts J, Loewe K, Kaufmann J et al (2015) Basal ganglia pathology in ALS is associated with neuropsychological deficits. Neurology 85:1301–1309
Patenaude B, Smith SM, Kennedy DN, Jenkinson M (2011) A Bayesian model of shape and appearance for subcortical brain segmentation. Neuroimage 56:907–922
Winkler AM, Ridgway GR, Webster MA, Smith SM, Nichols TE (2014) Permutation inference for the general linear model. Neuroimage 92:381–397
Nichols TE, Holmes AP (2002) Nonparametric permutation tests for functional neuroimaging: a primer with examples. Hum Brain Mapp 15:1–25
Frazier JA, Chiu S, Breeze JL et al (2005) Structural brain magnetic resonance imaging of limbic and thalamic volumes in pediatric bipolar disorder. Am J Psychiatry 162:1256–1265
Desikan RS, Segonne F, Fischl B et al (2006) An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. Neuroimage 31:968–980
Douaud G, Smith S, Jenkinson M et al (2007) Anatomically related grey and white matter abnormalities in adolescent-onset schizophrenia. Brain 130:2375–2386
Good CD, Johnsrude IS, Ashburner J, Henson RN, Friston KJ, Frackowiak RS (2001) A voxel-based morphometric study of ageing in 465 normal adult human brains. Neuroimage 14:21–36
Proudfoot M, Bede P, Turner MR (2018) Imaging cerebral activity in amyotrophic lateral sclerosis. Front Neurol 9:1148
Premi E, Calhoun VD, Diano M et al (2019) The inner fluctuations of the brain in presymptomatic frontotemporal dementia: the chronnectome fingerprint. Neuroimage 189:645–654
Van Laere K, Vanhee A, Verschueren J et al (2014) Value of 18fluorodeoxyglucose-positron-emission tomography in amyotrophic lateral sclerosis: a prospective study. JAMA Neurol 71:553–561
Cistaro A, Pagani M, Montuschi A et al (2014) The metabolic signature of C9ORF72-related ALS: FDG PET comparison with nonmutated patients. Eur J Nucl Med Mol Imaging. 2:2
Christidi F, Karavasilis E, Argyropoulos GD et al (2022) Neurometabolic alterations in motor neuron disease: insights from magnetic resonance spectroscopy. J Integr Neurosci 21:87
Popuri K, Dowds E, Beg MF et al (2018) Gray matter changes in asymptomatic C9orf72 and GRN mutation carriers. Neuroimage Clin 18:591–598
O’Callaghan C, Bertoux M, Hornberger M (2013) Beyond and below the cortex: the contribution of striatal dysfunction to cognition and behaviour in neurodegeneration. J Neurol Neurosurg Psychiatry. 2:2
Bocchetta M, Iglesias JE, Neason M, Cash DM, Warren JD, Rohrer JD (2020) Thalamic nuclei in frontotemporal dementia: Mediodorsal nucleus involvement is universal but pulvinar atrophy is unique to C9orf72. Hum Brain Mapp 41:1006–1016
Lee SE, Khazenzon AM, Trujillo AJ et al (2014) Altered network connectivity in frontotemporal dementia with C9orf72 hexanucleotide repeat expansion. Brain 137:3047–3060
Bruffaerts R, Gors D, Bárcenas Gallardo A et al (2022) Hierarchical spectral clustering reveals brain size and shape changes in asymptomatic carriers of C9orf72. Brain Commun. 4:182
Li Hi Shing S, McKenna MC, Siah WF, Chipika RH, Hardiman O, Bede P (2021) The imaging signature of C9orf72 hexanucleotide repeat expansions: implications for clinical trials and therapy development. Brain Imaging Behav 2:2
Floeter MK, Bageac D, Danielian LE, Braun LE, Traynor BJ, Kwan JY (2016) Longitudinal imaging in C9orf72 mutation carriers: Relationship to phenotype. Neuroimage Clin 12:1035–1043
Agosta F, Ferraro PM, Riva N et al (2017) Structural and functional brain signatures of C9orf72 in motor neuron disease. Neurobiol Aging 57:206–219
McMillan CT, Russ J, Wood EM et al (2015) C9orf72 promoter hypermethylation is neuroprotective: neuroimaging and neuropathologic evidence. Neurology 84:1622–1630
Chipika RH, Finegan E, Li Hi Shing S et al (2020) Switchboard malfunction in motor neuron diseases: Selective pathology of thalamic nuclei in amyotrophic lateral sclerosis and primary lateral sclerosis. Neuroimage Clin. 27:102300
Chipika RH, Mulkerrin G, Murad A, Lope J, Hardiman O, Bede P (2022) Alterations in somatosensory, visual and auditory pathways in amyotrophic lateral sclerosis: an under-recognised facet of ALS. J Integr Neurosci 21:88
Bede P, Elamin M, Byrne S et al (2013) Basal ganglia involvement in amyotrophic lateral sclerosis. Neurology 81:2107–2115
Bede P, Chipika RH, Christidi F et al (2021) Genotype-associated cerebellar profiles in ALS: focal cerebellar pathology and cerebro-cerebellar connectivity alterations. J Neurol Neurosurg Psychiatry 92:1197–1205
Westeneng HJ, Walhout R, Straathof M et al (2016) Widespread structural brain involvement in ALS is not limited to the C9orf72 repeat expansion. J Neurol Neurosurg Psychiatry 87:1354–1360
van der Burgh HK, Westeneng HJ, Walhout R et al (2020) Multimodal longitudinal study of structural brain involvement in amyotrophic lateral sclerosis. Neurology 94:e2592–e2604
Finegan E, Li Hi Shing S, Chipika RH et al (2019) Widespread subcortical grey matter degeneration in primary lateral sclerosis: a multimodal imaging study with genetic profiling. Neuroimage Clin. 24:102089
Trojsi F, Di Nardo F, Caiazzo G et al (2020) Hippocampal connectivity in amyotrophic lateral sclerosis (ALS): more than Papez circuit impairment. Brain Imaging Behav. 2:2
Agosta F, Ferraro PM, Riva N et al (2016) Structural brain correlates of cognitive and behavioral impairment in MND. Hum Brain Mapp 37:1614–1626
Feron M, Couillandre A, Mseddi E et al (2018) Extrapyramidal deficits in ALS: a combined biomechanical and neuroimaging study. J Neurol 265:2125–2136
Abidi M, de Marco G, Grami F et al (2021) Neural correlates of motor imagery of gait in amyotrophic lateral sclerosis. J Magn Reson Imaging 53:223–233
Abidi M, Pradat PF, Termoz N, Couillandre A, Bede P, de Marco G (2022) Motor imagery in amyotrophic lateral Sclerosis: An fMRI study of postural control. Neuroimage Clin 35:103051
Lule D, Ludolph AC, Ludolph AG (2008) Neurodevelopmental and neurodegenerative diseases—is there a pathophysiological link? Attention-deficit/hyperactivity disorder and amyotrophic lateral sclerosis as examples. Med Hypotheses 70:1133–1138
Bede P, Siah WF, McKenna MC, Shing LH, S. (2020) Consideration of C9orf72-associated ALS-FTD as a neurodevelopmental disorder: insights from neuroimaging. J Neurol Neurosurg Psychiatry. 2:2
Finsel J, Uttner I, Vázquez Medrano CR, Ludolph AC, Lulé D (2022) Cognition in the course of ALS-a meta-analysis. Amyotroph Lateral Scler Frontotemporal Degener. 2:1–12
Vertes RP, Hoover WB, Szigeti-Buck K, Leranth C (2007) Nucleus reuniens of the midline thalamus: link between the medial prefrontal cortex and the hippocampus. Brain Res Bull 71:601–609
Grollemund V, Pradat PF, Querin G et al (2019) Machine learning in amyotrophic lateral sclerosis: achievements, pitfalls, and future directions. Front Neurosci 13:135
Grollemund V, Le Chat G, Secchi-Buhour MS et al (2021) Manifold learning for amyotrophic lateral sclerosis functional loss assessment: development and validation of a prognosis model. J Neurol 268:825–850
Westeneng HJ, Debray TPA, Visser AE et al (2018) Prognosis for patients with amyotrophic lateral sclerosis: development and validation of a personalised prediction model. Lancet Neurol 17:423–433
Bede P, Iyer PM, Finegan E, Omer T, Hardiman O (2017) Virtual brain biopsies in amyotrophic lateral sclerosis: diagnostic classification based on in vivo pathological patterns. Neuroimage Clin 15:653–658
Bede P, Chang KM, Tan EL (2022) Machine-learning in motor neuron diseases: prospects and pitfalls. Eur J Neurol. 2:2
Bede P, Murad A, Lope J et al (2021) Phenotypic categorisation of individual subjects with motor neuron disease based on radiological disease burden patterns: a machine-learning approach. J Neurol Sci 432:120079
Bede P, Murad A, Hardiman O (2021) Pathological neural networks and artificial neural networks in ALS: diagnostic classification based on pathognomonic neuroimaging features. J Neurol 2:2
Schuster C, Hardiman O, Bede P (2016) Development of an automated MRI-based diagnostic protocol for amyotrophic lateral sclerosis using disease-specific pathognomonic features: a quantitative disease-state classification study. PLoS ONE 11:e0167331
Behler A, Müller HP, Ludolph AC, Lulé D, Kassubek J (2022) A multivariate Bayesian classification algorithm for cerebral stage prediction by diffusion tensor imaging in amyotrophic lateral sclerosis. Neuroimage Clin 35:103094
Schuster C, Hardiman O, Bede P (2017) Survival prediction in Amyotrophic lateral sclerosis based on MRI measures and clinical characteristics. BMC Neurol 17:73
Behler A, Müller HP, Del Tredici K et al (2022) Multimodal in vivo staging in amyotrophic lateral sclerosis using artificial intelligence. Ann Clin Transl Neurol 9:1069–1079
Tan HHG, Westeneng HJ, Nitert AD et al (2022) MRI clustering reveals three ALS subtypes with unique neurodegeneration patterns. Ann Neurol 2:2
Bede P, Murad A, Lope J, Hardiman O, Chang KM (2022) Clusters of anatomical disease-burden patterns in ALS: a data-driven approach confirms radiological subtypes. J Neurol 45:23
El Mendili MM, Querin G, Bede P, Pradat PF (2019) Spinal cord imaging in amyotrophic lateral sclerosis: historical concepts-novel techniques. Front Neurol 10:350
van der Burgh HK, Schmidt R, Westeneng HJ, de Reus MA, van den Berg LH, van den Heuvel MP (2017) Deep learning predictions of survival based on MRI in amyotrophic lateral sclerosis. Neuroimage Clin 13:361–369
Chipika RH, Siah WF, McKenna MC, Li Hi Shing S, Hardiman O, Bede P (2021) The presymptomatic phase of amyotrophic lateral sclerosis: are we merely scratching the surface? J Neurol 268:4607–4629
Acknowledgements
We wholeheartedly acknowledge all study participants for contributing to this research study.
Funding
Open Access funding provided by the IReL Consortium. This study was supported by BMBF (01GM1103A, 01ED1405), the Deutsches Zentrum für Neurodegenerative Erkrankungen (DZNE), and the Kompetenznetzwerk Präventivmedizin Baden-Württemberg (K.N.K.B.008.04). Professor Bede is supported by the Health Research Board (HRB EIA-2017-019 & JPND-Cofund-2-2019-1), the Irish Institute of Clinical Neuroscience (IICN), the EU Joint Programme—Neurodegenerative Disease Research (JPND), the Andrew Lydon scholarship, The Hayes Family Charitable Fund and the Iris O'Brien Foundation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no conflict of interest.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bede, P., Lulé, D., Müller, HP. et al. Presymptomatic grey matter alterations in ALS kindreds: a computational neuroimaging study of asymptomatic C9orf72 and SOD1 mutation carriers. J Neurol 270, 4235–4247 (2023). https://doi.org/10.1007/s00415-023-11764-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-023-11764-5