
Abstract
Background
Hereditary transthyretin amyloidosis (ATTRv amyloidosis) is a rare, but life-threatening protein misfolding disorder due to TTR gene mutations. Cardiomyopathy (ATTRv-CM) and polyneuropathy (ATTRv-PN) with early small nerve fibre involvement are the most common manifestations. Timely diagnosis and treatment initiation are key to limiting progression of disease. Corneal confocal microscopy (CCM) is a non-invasive method to quantify corneal small nerve fibres and immune cell infiltrates in vivo.
Methods
This cross-sectional study investigated the utility of CCM in 20 patients with ATTRv amyloidosis (ATTRv-CM, n = 6; ATTRv-PN, n = 14) and presymptomatic carriers (n = 5) compared to 20 age- and sex-matched healthy controls. Corneal nerve fibre density, corneal nerve fibre length, corneal nerve branch density, and cell infiltrates were assessed.
Results
Corneal nerve fibre density and nerve fibre length were significantly lower in patients with ATTRv amyloidosis compared to healthy controls regardless of the clinical phenotype (ATTRv-CM, ATTRv-PN) and corneal nerve fibre density was significantly lower in presymptomatic carriers. Immune cell infiltrates were only evident in patients with ATTRv amyloidosis, which correlated with reduced corneal nerve fibre density.
Conclusions
CCM identifies small nerve fibre damage in presymptomatic carriers and symptomatic patients with ATTRv amyloidosis and may serve as a predictive surrogate marker to identify individuals at risk of developing symptomatic amyloidosis. Furthermore, increased corneal cell infiltration suggests an immune-mediated mechanism in the pathogenesis of amyloid neuropathy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hereditary transthyretin (ATTRv) amyloidosis is a rare [1], but life-threatening autosomal-dominant systemic disorder characterised by extracellular deposition of misfolded transthyretin fibrils in various tissues [2]. Cardiomyopathy (ATTRv-CM) and progressive axonal polyneuropathy (ATTRv-PN) with early small fibre involvement and autonomic dysfunction are the most common manifestations [3]. To date, more than 120 amyloidogenic mutations of the TTR gene have been identified [4] and genetic heterogeneity is particularly high in non-endemic regions such as the USA and most of Europe except areas in Northern Portugal and Sweden [1, 5,6,7]. Phenotypic presentation as cardiac, neuropathic or mixed, partially depends on the underlying genotype [5, 8], but even in carriers of the same mutation, phenotypes may vary [9]. Age at disease onset ranges from 20 to 80 years, and penetrance is incomplete [10]. Genetic heterogeneity, clinical variability, inadequate diagnostic workup and low disease awareness frequently lead to a diagnostic delay [11], which is critical facing the fatal natural disease course. Untreated ATTRv amyloidosis leads to rapid disease progression, severe disability, and death within approximately 7–11 years [10, 12].
The approval of patisiran and vutrisiran, small interfering RNA drugs, and inotersen, an antisense oligonucleotide, which effectively supress transthyretin expression by degradation of TTR mRNA, have transformed the management of ATTRv-PN [13, 14]. Tafamidis, a stabilizer of the transthyretin tetramer is available for the treatment of isolated ATTRv-CM [15]. Given that timely treatment initiation improves outcomes in patients with ATTRv amyloidosis and amyloid deposition is considered to start before symptom onset, it remains an open question whether and when to start treatment in carriers, especially as treatments are burdensome and costly [10, 16,17,18,19,20]. A more precise assessment of underlying pathology may allow earlier intervention and assessment of disease progression.
Corneal confocal microscopy (CCM) is a rapid, reiterative, and non-invasive imaging technique to quantify small corneal nerve fibers in vivo [21]. It has proven diagnostic value in various peripheral neuropathies including diabetic [22,23,24], hereditary [25, 26] and immune-mediated neuropathies [27, 28]. It has also been used to assess immune cell infiltration in a number of neuropathies [29,30,31]. Differential origins of these cells has been discussed before. Professional antigen-presenting dendritic cells analogously to Langerhans cells of the skin [32,33,34,35] are present in the noninflamed corneal epithelium [36]. Since mature Langerhans cells feature dendrites whereas immature Langerhans cells lack dendrites, the differential cells detected can be discussed in this context [29, 37, 38].
However, data on CCM in patients with ATTRv amyloidosis are scarce, despite small fibre involvement being a frequent [39], early [40, 41] and prominent manifestation of ATTRv amyloidosis. This cross-sectional study assessed the utility of CCM as a tool for early diagnosis in a genetically heterogeneous and phenotypically variable cohort of patients with ATTRv amyloidosis and presymptomatic carriers.
Patients and methods
Patients
Investigations were performed in the Department of Neurology, University Hospital Essen between January 2020 and December 2021. Data from 20 patients with confirmed amyloidogenic TTR mutation and either isolated ATTRv-CM or ATTRv-PN with or without cardiac involvement, and 5 presymptomatic carriers (relatives of the aforementioned patients) were collected. All patients and carriers underwent transthoracic echocardiography, nerve conduction studies (NCS), detailed medical history and laboratory evaluation to exclude other causes of neuropathy e.g. exposure to alcohol or chemotherapy, or predisposing conditions such as diabetes, thyroid disorders or vitamin deficiencies. Carriers were considered presymptomatic if NCS, echocardiography, and clinical examination were normal. ATTRv amyloidosis was classsified as ATTRv-CM if there was an abnormality on transthoracic echocardiography, consistent with amyloid cardiomyopathy, but NCS and neurological examination were normal. Patients with neurological and NCS abnormality were classified as ATTRv-PN, with or without cardiomyopathy.
A reference group of 20 age- and sex-matched healthy individuals who had undergone clinical, neurological and neurophysiological testing and laboratory testing to exclude neuropathy was recruited from the University of Manchester, United Kingdom.
No participant had a known ophthalmologic disease or ocular symptoms at the time of the investigation, as these were exclusion criteria.
Corneal confocal microscopy
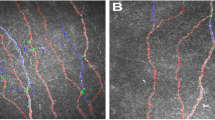
CCM was performed using a Heidelberg Retina Tomograph (HRT III, Rostock Cornea Module, Heidelberg Engineering, Heidelberg, Germany). A local anaesthetic (0.4% benoxinate hydrochloride) was administered immediately before the examination. Viscotears liquid gel was applied to the eye for lubrication and to establish a thin gel bridge between the corneal surface and a sterile, single-use lens cap. Several scan cycles of the entire depth of the cornea were carried out focusing on the sub-basal nerve plexus at the center of the cornea. The integrity of the corneal surface was visually confirmed and at least six images per patient meeting accepted quality criteria were analyzed [42]. Well-established software (ACCMetrics Image Analysis tool v1.1, University of Manchester, UK) was used for automated quantification of corneal nerve fibre density (CNFD, major nerves/mm2), corneal nerve fibre length (CNFL, mm/mm2), and corneal nerve branch density (CNBD, major branches/mm2, see Fig. 1). Corneal cells were counted manually by an independent investigator in a blinded fashion using ImageJ software (version 1.41, National Institutes of Health, Bethesda, Maryland, USA) and were classified according to their morphology, i.e. presence or absence of dendritic cell extensions, and their location relative to the corneal nerve fibres, as previously described [27]. Four subtypes, dendritic cells with fibre contact (DCF), dendritic cells in the periphery without fibre contact (DCP), non-dendritic cells with fibre contact (NCF), and non-dendritic cells in the periphery without fibre contact (NCP) were identified (see Fig. 2).

Representative image of corneal nerve fibres (left panel) and illustration of automated analysis of nerve fibre parameters (right panel) using ACCMetrics Image Analysis tool v1.1, University of Manchester, UK. Major nerves (red lines), axon collaterals (blue lines), and nerve branches (green dots)

Representative images of the corneal cell subtypes identified according to their morphology and localisation. Dendritic cell with fibre contact (DCF, upper left panel), dendritic cell in the periphery without fibre contact (DCP, lower left panel), non-dendritic cell with fibre contact (NCF, upper right panel), and non-dendritic cell in the periphery without fibre contact (NCP, lower right panel). Scale bars = 100 µm
Statistical analysis
Statistical analyses were performed using GraphPad Prism (version 9.1.0 for Windows, GraphPad Software, San Diego, California, USA). All data are presented as the mean, standard error of the mean, and p values. Differences in cell and nerve fibre parameters between patients with ATTRv-CM, ATTRv-PN and healthy controls and between ATTRv amyloidosis patients, presymptomatic carriers and healthy controls were assessed using a Kruskal–Wallis test and Dunn’s test as post hoc analysis. P values < 0.05 were considered to be statistically significant. Correlations were calculated using Spearman’s rank correlation coefficient.
Results
Twenty patients with ATTRv amyloidosis (15 male, 5 female, mean age 64.1 ± 2.9 years) were compared to 20 healthy controls (15 male, 5 female, mean age 62.7 ± 1.7 years) and 5 presymptomatic carriers (2 male, 3 female, mean age 40.4 ± 2.8 years; for individual CCM data see Online Resource 1). Six patients were classified as ATTRv-CM patients (3 male, 3 female, mean age 67.7 ± 5.8 years), and 14 as ATTRv-PN patients (12 male, 2 female, mean age 62.9 ± 3.1 years). Patients and presymptomatic carriers were genetically heterogeneous with a wide variety of TTR mutations. Small fibre-mediated symptoms such as neuropathic pain and autonomic dysfunction were frequent in patients with symptomatic ATTRv amyloidosis. For detailed demographic and clinical characteristics see Table 1 and for NCS of the ATTRv-PN patients, see Online Resource 2.
CNFD (21.05 ± 1.59 vs. 30.75 ± 1.58 fibres/mm2, p = 0.0003) and CNFL (12.95 ± 0.79 vs. 17.40 ± 0.93 mm/mm2, p = 0.0032) were significantly lower (Fig. 1), whilst CNBD did not differ significantly (29.05 ± 4.03 vs. 39.02 ± 4.55 branches/mm2, p = 0.119) in patients with ATTRv amyloidosis compared to healthy controls. In case of a CNFD below a threshold of 24 fibres/mm2 (two standard deviations below the mean of the reference group) the relative risk of symptomatic amyloidosis instead of being presymptomatic/healthy was 3,75 (15 out of 20 participants with CNFD < 24 fibres/mm2 and only 5 out of 25 participants with CNFD ≥ 24 fibres/mm2 were patients with symptomatic ATTRv amyloidosis). In presymptomatic carriers, CNFD was significantly lower compared to healthy controls (19.00 ± 4.11 vs. 30.75 ± 1.58 fibres/mm2, p = 0.025) and was comparable to patients with symptomatic amyloidosis (Fig. 3).

Corneal nerve fibre parameters in patients with ATTRv amyloidosis (ATTRv, black), presymptomatic carriers (PC, light grey), and healthy controls (HC, dark grey). Corneal nerve fibre density (CNFD), corneal nerve fibre length (CNFL), and corneal nerve branch density (CNBD) are displayed as Mean ± SEM, *p < 0.05, **p < 0.01. ***p < 0.001, ns, not significant
In patients with ATTRv-PN, CNFD (20.36 ± 1.77 vs. 30.75 ± 1.58 fibres/mm2, p < 0.001) and CNFL (12.71 ± 0.87 vs. 17.40 ± 0.93 mm/mm2, p = 0.008, Fig. 4) were significantly lower compared to controls. In patients with ATTRv-CM, CNFD (22.67 ± 3.51 vs. 30.75 ± 1.58 fibres/mm2, p = 0.038, Fig. 4), but not CNFL was significantly reduced compared to healthy controls. CNBD did not differ from healthy controls in both ATTRv amyloidosis phenotypes.

Subgroup analysis of corneal nerve fibre parameters in ATTRv patients with peripheral neuropathy (ATTRv-PN, black), cardiomyopathy (ATTRv-CM, light grey), and healthy controls (HC). Corneal nerve fibre density (CNFD), corneal nerve fibre length (CNFL), and corneal nerve branch density (CNBD) are displayed as mean ± SEM, *p < 0.05, **p < 0.01. ***p < 0.001, ns, not significant
In relation to corneal immune cell infiltration, NCF was significantly higher in ATTRv patients compared to healthy controls (11.80 ± 3.98 vs. 2.30 ± 0.68 cells/mm2, p = 0.021), but not in presymptomatic carriers (4.20 ± 2.43 vs. 2.30 ± 0.68 cells/mm2, p > 0.999). Total cell count (TC), DCF, DCP, and NCP did not differ between ATTRv patients and healthy controls (Fig. 5).

Corneal cell counts in patients with ATTRv amyloidosis (ATTRv, black), presymptomatic carriers (PC, light grey), and healthy controls (HC, dark grey)
The total cell count (TC), cell counts of dendritic cells with fibre contact (DCF), dendritic cells in the periphery (DCP), non-dendritic cells with fibre contact (NCF), non-dendritic cells in the periphery (NCP), and all cells with fibre contact regardless of their morphology (CF) are displayed as mean ± SEM, *p < 0.05, **p < 0.01. ns, not significant.
There were no significant differences between ATTRv-CM and ATTRv-PN (data not shown). NCF (r = − 0.50, p = 0.026), but not DCF, correlated inversely with CNFD (Fig. 6) in patients with ATTRv amyloidosis. The proportion of corneal cells with fibre contact was higher in patients with ATTRv amyloidosis (44%), but only marginally in presymptomatic carriers (28%) compared to healthy controls (26%) (Fig. 7).

Correlation of non-dendritic cells with fibre contact (NCF) with corneal nerve fibre density (CNFD) in patients with ATTRv amyloidosis

Proportion of cells with (black) and without (grey) fibre contact in patients with ATTRv amyloidosis (left), presymptomatic carriers (middle), and healthy controls (right)
Discussion and conclusions
This study has utilized CCM to identify a significant reduction of corneal nerve fibres in patients with ATTRv amyloidosis regardless of clinical phenotype or TTR mutation and in presymptomatic carriers decades before reaching the age of disease onset in index patients.
Early diagnosis and treatment are key to the management of patients with ATTRv amyloidosis. However, defining disease onset in oligosymptomatic patients can be challenging and monitoring algorithms in presymptomatic carriers remain a matter of debate [43]. Current expert consensus recommends annual follow-up of carriers commencing ten years prior to the predicted age of disease onset (PADO), depending on the TTR mutation, age of onset in the individual and family members with amyloidosis [44]. However, cases of early- and late-onset ATTRv amyloidosis can be found within the same family or genotype [45] and amyloid deposition is thought to precede symptom onset by several years similar to other protein misfolding disorders [2]. There is an urgent need for reliable, reproducible and non-invasive biomarkers of disease onset and progression.
Our findings suggest that CCM may identify the onset of subclinical amyloid neuropathy in presymptomatic carriers, as there was a striking loss of nerve fibres in carriers who were approximately twenty years younger than their relatives with apparent amyloidosis at disease onset. CCM may therefore qualify as a predictive surrogate marker to identify presymptomatic carriers at risk of developing ATTR related neuropathy. The fact that CNBD was not altered in ATTRv amyloidosis patients, whilst CNFD and CNFL were reduced, might be explained by a compensatory axonal regeneration at early stages of the disease.
Additionally, corneal immune cell infiltration could be used to differentiate between presymptomatic carriers and symptomatic patients, as significant corneal immune cell infiltration, namely an increase in NCF, only occurred in the latter. The corneal immune cells we classified as non-dendritic cells according to morphological criteria have been discussed before to be resident immature Langerhans cells. Previously, it has been shown that the number, proportion and distribution of corneal Langerhans cell subtypes depends on various factors such as the ocular microenvironment [46], the corneal region since mature Langerhans cells are predominantly present in the periphery [34, 37], and even diurnal variations [47] in healthy individuals. Nevertheless, the increase in cells with fibre contact in this study, albeit non-dendritic ones, and their correlation with reduced nerve fibre density suggest an immune mediated [27, 28] mechanism of amyloid neuropathy or at least secondary inflammatory processes. Although an upregulation of pro-inflammatory cytokines [48] and an induction of inflammatory responses through macrophage activation in the sural nerve [49] have been shown previously in patients with amyloidosis, current research into the disease mechanisms has focused on mechanical compression of nerve fibres, ischemia due to perivascular amyloid deposition, and toxic effects of non-fibrillar aggregates [50,51,52]. Our data are supported by a recent study showing a clustering of immature Langerhans cells at the inferior whorl in ATTRv amyloidosis patients [53]. Finally, it remains to be determined, whether CCM can help to identify secondary inflammatory processes as a therapeutic target in patients with ATTRv amyloidosis.
Two small cohort studies have previously utilized CCM in patients with ATTRv amyloidosis. Rousseau et al. [54] found reduced CNFL in 15 patients with ATTRv amyloidosis (10 patients with p.Val50Met) which is consistent with our results and was associated with more severe autonomic dysfunction and reduced intraepidermal nerve fibre density. There was no change in CNFL in 2 presymptomatic carriers included, also consistent with our data. However, this study did not examine other nerve fibre parameters than CNFL. A recent study from China [44] showed a reduction in corneal nerve fibre length at the inferior whorl in patients with ATTRv amyloidosis, which correlated with disease severity defined by clinical disease stages. However, they included 5 presymptomatic carriers and only 10 patients with ATTRv amyloidosis with the same mutation (p.Ala117Ser) and the healthy controls were considerably younger, potentially affecting CNFD which was indeed much higher than in our controls, and they did not investigate clinically defined subgroups as we did.
A relevant limitation of this study and the mentioned studies is the small cohort of investigated patients and especially carriers. This is a result of ATTRv amyloidosis being a very rare disease outside endemic areas. Furthermore, management of presymptomatic carriers requires careful genetic counseling and many individuals at risk avoid molecular genetic testing expecting socioeconomic disadvantages associated with a positive result.
In conclusion, CCM, a non-invasive ophthalmic technique enables the identification of small fibre involvement across diverse genotypes and clinical phenotypes, especially early in the course of ATTRv amyloidosis. It also sheds new light on possible immune-mediated mechanisms of small fibre neuropathy in ATTRv amyloidosis. CCM could be a sensitive diagnostic tool to enable early diagnosis and to monitor mutation carriers, even earlier than currently recommended following the PADO concept. Longitudinal studies are warranted to assess the predictive value of CCM in the development and progression of different ATTRv phenotypes.
Data availability
Data are available from the corresponding author upon reasonable request by any qualified investigator.
References
Schmidt H, Waddington-Cruz M, Botterman M et al (2017) Estimating the global prevalence of transthyretin familial amyloid polyneuropathy. Muscle Nerve 57:829–837. https://doi.org/10.1002/mus.26034
Adams D, Koike H, Slama M, Coelho T (2019) Hereditary transthyretin amyloidosis: a model of medical progress for a fatal disease. Nat Rev Neurol 15:387–404. https://doi.org/10.1038/s41582-019-0210-4
Planté-Bordeneuve V, Said G (2011) Familial amyloid polyneuropathy. Lancet Neurol 10:1086–1097. https://doi.org/10.1016/S1474-4422(11)70246-0
Rowczenio D, Noor I, Gillmore J et al (2014) Online registry for mutations in hereditary amyloidosis including nomenclature recommendations. Hum Mutat 35:E2403-2412. https://doi.org/10.1002/humu.22619
Castano A, Drachman B, Judge D, Maurer M (2015) Natural history and therapy of TTR-cardiac amyloidosis: emerging disease-modifying therapies from organ transplantation to stabilizer and silencer drugs. Heart Fail Rev 20:163–178. https://doi.org/10.1007/s10741-014-9462-7
Ikeda S, Nakazato M, Ando Y, Sobue G (2002) Familial transthyretin-type amyloid polyneuropathy in Japan: clinical and genetic heterogeneity. Neurology 58:1001–1007. https://doi.org/10.1212/wnl.58.7.1001
Parman Y, Adams D, Obici L et al (2016) Sixty years of transthyretin familial amyloid polyneuropathy (TTR-FAP) in Europe: where are we now? A European network approach to defining the epidemiology and management patterns for TTR-FAP. Curr Opin Neurol. https://doi.org/10.1097/WCO.0000000000000288
Rapezzi C, Quarta C, Obici L et al (2013) Disease profile and differential diagnosis of hereditary transthyretin-related amyloidosis with exclusively cardiac phenotype: an Italian perspective. Eur Heart J 34:520–528. https://doi.org/10.1093/eurheartj/ehs123
Ando Y, Coelho T, Berk J et al (2013) Guideline of transthyretin-related hereditary amyloidosis for clinicians. Orphanet J Rare Dis 8:1–18. https://doi.org/10.1186/1750-1172-8-31
Adams D, Suhr O, Hund E et al (2016) First European consensus for diagnosis, management, and treatment of transthyretin familial amyloid polyneuropathy. Curr Opin Neurol 29:14–26. https://doi.org/10.1097/WCO.0000000000000289
Coelho T, Ines M, Conceicao I, Soares M, Carvalho MD, Costa J (2018) Natural history and survival in stage 1 Val30Met transthyretin familial amyloid polyneuropathy. Neurology 91:e1999–e2009. https://doi.org/10.1212/WNL.0000000000006543
Adams D, Coelho T, Obici L et al (2015) Rapid progression of familial amyloidotic polyneuropathy: a multinational natural history study. Neurology 85:675–682. https://doi.org/10.1212/WNL.0000000000001870
Adams D, Polydefkis M, González-Duarte A et al (2021) Long-term safety and efficacy of patisiran for hereditary transthyretin-mediated amyloidosis with polyneuropathy: 12-month results of an open-label extension study. Lancet Neurol 20:49–59. https://doi.org/10.1016/S1474-4422(20)30368-9
Brannagan T, Wang A, Coelho T et al (2020) Early data on long-term efficacy and safety of inotersen in patients with hereditary transthyretin-amyloidosis: a 2-year update from the open-label extension of the NEURO-TTR trial. Eur J Neurol 27:1374–1381. https://doi.org/10.1111/ene.14285
Yamamoto H, Yokochi T (2019) Transthyretin cardiac amyloidosis: an update on diagnosis and treatment. ESC Heart Fail 6:1128–1139. https://doi.org/10.1002/ehf2.12518
Planté-Bordeneuve V (2018) Transthyretin familial amyloid polyneuropathy: an update. J Neurol 265:976–983. https://doi.org/10.1007/s00415-017-8708-4
Adams D, Ando Y, Beirao J et al (2021) Expert consensus recommendations to improve diagnosis of ATTR amyloidosis with polyneuropathy. J Neurol 268:2109–2122. https://doi.org/10.1007/s00415-019-09688-0
Adams D, Beaudonnet G, Adam C et al (2016) Familial amyloid polyneuropathy: when does it stop to be asymptomatic and need a treatment? Rev Neurol (Paris) 172:645–652. https://doi.org/10.1016/j.neurol.2016.08.007
Jiang X, Labaudinière R, Buxbaum J et al (2021) A circulating, disease-specific, mechanism-linked biomarker for ATTR polyneuropathy diagnosis and response to therapy prediction. Proc Natl Acad Sci USA 118:e2016072118. https://doi.org/10.1073/pnas.2016072118
Adams D, Algalarrondo V, Polydefkis M, Sarswat N, Slama M, Nativi-Nicolau J (2021) Expert opinion on monitoring symptomatic hereditary transthyretin-mediated amyloidosis and assessment of disease progression. Orphanet J Rare Dis 16:411. https://doi.org/10.1186/s13023-021-01960-9
Petropoulos I, Ponirakis G, Khan A et al (2020) Corneal confocal microscopy: ready for prime time. Clin Exp Optom 103:265–277. https://doi.org/10.1111/cxo.12887
Hossain P, Sachdev A, Malik R (2005) Early detection of diabetic peripheral neuropathy with corneal confocal microscopy. Lancet 366:1340–1343. https://doi.org/10.1016/S0140-6736(05)67546-0
Quattrini C, Tavakoli M, Jeziorska M et al (2007) Surrogate markers of small fiber damage in human diabetic neuropathy. Diabetes 56:2148–2154. https://doi.org/10.2337/db07-0285
Ziegler D, Papanas N, Zhivov A et al (2014) Early detection of nerve fiber loss by corneal confocal microscopy and skin biopsy in recently diagnosed type 2 diabetes. Diabetes 63:2454–2463. https://doi.org/10.2337/db13-1819
Mimura T, Amano S, Fukuoka S et al (2008) In vivo confocal microscopy of hereditary sensory and autonomic neuropathy. Curr Eye Res 33:940–945. https://doi.org/10.1080/02713680802450992
Tavakoli M, Marshall A, Banka S et al (2012) Corneal confocal microscopy detects small-fiber neuropathy in Charcot–Marie–Tooth disease type 1A patients. Muscle Nerve 46:698–704. https://doi.org/10.1002/mus.23377
Stettner M, Hinrichs L, Guthoff R et al (2016) Corneal confocal microscopy in chronic inflammatory demyelinating polyneuropathy. Ann Clin Transl Neurol 3:88–100. https://doi.org/10.1002/acn3.275
Fleischer M, Lee I, Erdlenbruch F et al (2021) Corneal confocal microscopy differentiates inflammatory from diabetic neuropathy. J Neuroinflamm 18:89. https://doi.org/10.1186/s12974-021-02130-1
Zhivov A, Stave J, Vollmar B, Guthoff R (2005) In vivo confocal microscopic evaluation of Langerhans cell density and distribution in the normal human corneal epithelium. Graefes Arch Exp Clin Ophthalmol 243:1056–1061. https://doi.org/10.1007/s00417-004-1075-8
Chen W, Hara K, Tian Q, Zhao K, Yoshitomi T (2007) Existence of small slow-cycling Langerhans cells in the limbal basal epithelium that express ABCG2. Exp Eye Res 84:626–634. https://doi.org/10.1016/j.exer.2006.11.006
Mayer W, Irschick U, Moser P et al (2007) Characterization of antigen-presenting cells in fresh and cultured human corneas using novel dendritic cell markers. Investig Ophthalmol Vis Sci 48:4459–4467. https://doi.org/10.1167/iovs.06-1184
Hamrah P, Huq SO, Liu Y et al (2003) Corneal immunity is mediated by heterogeneous population of antigen-presenting cells. J Leukoc Biol 74(2):172–178. https://doi.org/10.1189/jlb.1102544
Hamrah P, Zhang Q, Liu Y et al (2002) Novel characterization of MHC class II-negative populatio of resident corneal Langerhans cell-type dendritic cells. Investig Ophthalmol Vis Sci 43(3):639–646
Mayer WJ, Mackert MJ, Kranebitter N et al (2012) Distribution of antigen presenting cells in the human cornea: correlation of in vivo confocal microscopy and immunohistochemistry in different pathologic entities. Curr Eye Res 37(11):1012–1018. https://doi.org/10.3109/02713683.2012.696172
Alhatem A, Cavalcanti B, Hamrah P (2012) In vivo confocal microscopy in dry eye disease and related conditions. Semin Ophthalmol 27(5–6):138–148. https://doi.org/10.3109/08820538.2012.711416
Colorado LH, Markoulli M, Edwards K (2019) The relationship between corneal dendritic cells, corneal nerve morphology and tear inflammatory mediators and neuropeptides in healthy individuals. Curr Eye Res 44(8):840–848. https://doi.org/10.1080/027113683.2019.1600196
Mastropasqua L, Nubile M, Lanzini M et al (2006) Epithelial dendritic cells distribution in normal and inflamed human cornea: in vivo confocal microscopy study. Am J Ophhtalmol 142(5):736–744. https://doi.org/10.1016/j.ajo.2006.06.057
Chiang JCB, Goldstein D, Tavakoli A et al (2021) Corneal dendritic cells and the subbasal nerve plexus following neurotoxic treatment with oxaliplatin or paclitaxel. Sci Rep 11(1):22884. https://doi.org/10.1038/s41598-021-02439-0
Conceicao I, Gonzalez-Duarte A, Obici L et al (2016) “Red-flag” symptom clusters in transthyretin familial amyloid polyneuropathy. J Peripher Nerv Syst 21:5–9. https://doi.org/10.1111/jns.12153
González-Duarte A, Cárdenas-Soto K, Fueyo O, Banuelos C, Gibbons C, Freeman R (2019) Small fibre neuropathy assessments in early stages of hATTR amyloidosis. Amyloid 26:55–56. https://doi.org/10.1080/13506129.2019.1600852
Chiang M, Yeh T, Sung J et al (2021) Early changes of nerve integrity in preclinical carriers of hereditary transthyretin Ala117Ser amyloidosis with polyneuropathy. Eur J Neurol 28:982–991. https://doi.org/10.1111/ene.14698
Smith A, Kim G, Porzio M et al (2013) Corneal confocal microscopy is efficient, well-tolerated, and reproducible. J Peripher Nerv Syst 18:54–58. https://doi.org/10.1111/jns5.12008
Dohrn M, Auer-Grumbach M, Baron R et al (2021) Chance or challenge, spoilt for choice? New recommendations on diagnostic and therapeutic considerations in hereditary transthyretin amyloidosis with polyneuropathy: the German/Austrian position and review of the literature. J Neurol 268:3610–3625. https://doi.org/10.1007/s00415-020-09962-6
Conceicao I, Damy T, Romero M et al (2019) Early diagnosis of ATTR amyloidosis through targeted follow-up of identified carriers of TTR gene mutations. Amyloid 26:3–9. https://doi.org/10.1080/13506129.2018
Obici L, Kuks J, Bades J et al (2016) Recommendations for presymptomatic genetic testing and management of individuals at risk for hereditary transthyretin amyloidosis. Curr Opin Neurol 29:S27-35. https://doi.org/10.1097/WCO.0000000000000290
Shen L, Barabino S, Taylor AW et al (2007) Effect of the ocular microenvironment in regulating corneal dendritic cell maturation. Arch Ophthalmol 125(7):908–915. https://doi.org/10.1001/archopht.125.7.908
Alotaibi S, Ozkan J, Papas E et al (2022) Diurnal variation of corneal dendritic cell density. Curr Eye Res 47(9):1239–1245. https://doi.org/10.1080/02713683.2022.2088799
Azevedo E, Guimaraes-Costa A, Bandeira-Melo C et al (2019) Inflammatory profiling of patients with familial amyloid polyneuropathy. BMC Neurol 19:146. https://doi.org/10.1186/s12883-019-1369-4
Sommer C, Schröder J (1989) Amyloid neuropathy: immunocytochemical localization of intra- and extracellular immunoglobulin light chains. Acta Neuropathol 79:190–199. https://doi.org/10.1007/BF00294378
Andersson K, Olofsson A, Nielsen E, Svehag S, Lundgren E (2002) Only amyloidogenic intemediates of transthyretin induce apoptosis. Biochem Biophys Res Commun 294:309–314. https://doi.org/10.1016/S0006-291X(02)00465-5
Sousa M, Saraiva M (2003) Neurodegeneration in familial amyloid polyneuropathy: from pathology to molecular signaling. Prog Neurobiol 71:385–400. https://doi.org/10.1016/j.pneurobio.2003.11.002
Koike H, Ikeda S, Takahashi M et al (2016) Schwann cell and endothelial cell damage in transthyretin familial amyloid polyneuropathy. Neurology 87:2220–2229. https://doi.org/10.1212/WNL.0000000000003362
Zhang Y, Liu Z, Zhang Y et al (2021) Corneal sub-basal whorl-like nerve plexus: a landmark for early and follow-up evaluation in transthyretin familial amyloid polyneuropathy. Eur J Neurol 28:630–638. https://doi.org/10.1111/ene.14563
Rousseau A, Cauquil C, Dupas B et al (2016) Potential role of in vivo confocal microscopy for imaging corneal nerves in transthyretin familial amyloid polyneuropathy. JAMA Opthalmol 134:983–989. https://doi.org/10.1001/jamaophthalmol.2016.1889
Funding
Open Access funding enabled and organized by Projekt DEAL. This study was supported by the Universitätsmedizin Essen Clinician Scientist Academy (UMEA, Grant number FU 356/12-2 to A.T.).
Author information
Authors and Affiliations
Contributions
AT designed the study, collected and analysed the data, and wrote the manuscript. AC, SO, MP, LK, CR contributed substantially to data acquisition and revised the manuscript for intellectual content. RAM collected data of the control group and revised the manuscript for important intellectual content. HCR, TR, KH, and CK revised the manuscript for important intellectual content. MS and TH contributed equally to data acquisition and interpretation, conceptualization of the study and revised the manuscript for important intellectual content. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflicts of interest
A.T. received speaker honoraria from Alnylam and Sobi. H.C.R. received consulting and lecture fees from Abbvie, AstraZeneca, Vertex, Roche, Novartis and Merck. H.C.R. received research funding from Gilead Pharmaceuticals. H.C.R. is a co-founder of CDL Therapeutics GmbH. The other authors report no competing interests.
Ethics approval
The study has been performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments and was approved by the ethics committee of the University Duisburg-Essen (approval number 20-9583-BO) and the North Manchester Ethics committee (control group).
Consent to participate
Informed consent was obtained from all individual participants included in the study.
Consent to publish
All participants gave their written informed consent to publishing their data.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Thimm, A., Carpinteiro, A., Oubari, S. et al. Corneal confocal microscopy identifies corneal nerve loss and increased Langerhans cells in presymptomatic carriers and patients with hereditary transthyretin amyloidosis. J Neurol 270, 3483–3491 (2023). https://doi.org/10.1007/s00415-023-11689-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-023-11689-z