
Abstract
Background
(1) Description of clinical and cranial MRI features in the original Pontine Autosomal Dominant Microangiopathy with Leukoencephalopathy (PADMAL) family and correlation with the segregation analysis of the causative collagen 4A1 gene (COL4A1) variant. (2) Sequence analysis of the COL4A1 miRNA-binding site containing the causative variant in two independent cross-sectional samples of sporadic stroke patients.
Patients and methods
Sanger sequencing of the COL4A1 miRNA-binding site in the PADMAL family and 874 sporadic stroke patients.
Results
PADMAL shows adult-onset usually between 30 and 50 years of age with initial brainstem-related symptoms most commonly dysarthria, with progression to dementia and tetraparesis. Radiologically pontine lacunes are followed by supratentorial white matter involvement. Radiological onset may precede clinical symptoms. We found no variants in the COL4A1 miRNA-binding site of sporadic stroke patients.
Conclusion
Our results allow an early diagnosis of PADMAL based on cranial MRI, clinical signs, and confirmatory sequencing of the COL4A1 miRNA-29-binding site. COL4A1 miRNA-29-binding site variants do not contribute to a sizeable proportion of sporadic stroke.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Sporadic cerebral microangiopathies are common and a leading cause of stroke and dementia [1]. Monogenic cerebral microangiopathies are comparatively rare but provide important models for the pathomechanisms underlying their sporadic counterparts [2]. Pontine Autosomal Dominant Microangiopathy with Leukoencephalopathy (PADMAL) is an autosomal dominant cerebral microangiopathy first described in a German family by Colmant and Hagel [3, 4]. PADMAL is an extremely rare disease. Only 11 families have been published and it is not possible to estimate the incidence or prevalence of the disease [5,6,7,8,9]. PADMAL belongs to the group of autosomal dominant cerebrovascular diseases caused by type IV collagen variants [10, 11]. Type IV collagens are the main constituent of basement membranes and form mesh-like structures with multiple functions including mechanic strength [12]. COL4A1/A2 proteins include a triple helix containing the classic Gly-Xaa-Yaa repeat amino acid sequence [12]. Missense variants affecting these glycine residues may lead to a failure to form proper triple helices [12]. Protein truncating variants and duplications may also cause cerebrovascular disease, presumably by reduced expression or overexpression of type IV collagens [11, 13,14,15]. Clinically, COL4A1/A2 mutations cause a wide variety of phenotypes ranging from fetal death, porencephaly, and intracerebral hemorrhages to cerebral microangiopathy, cervical artery dissection, and some individuals even remain asymptomatic [10, 16, 17]. Extra-neurological features, such as renal involvement, cardiac involvement, and ocular disease, occur in a subset of variants [18]. PADMAL is caused by variants outside the protein-coding part of COL4A1, affecting a micro-RNA (miRNA-29)-binding site in the 3’-untranslated region (3’-UTR) [5,6,7,8,9]. These variants lead to a loss of transcription repression by miRNA-29 and consequently to collagen 4A1 overexpression [8]. These findings show that both, overexpression and under-expression of type IV collagens might lead to cerebrovascular disease and suggest that the expression of type IV collagens needs to be tightly regulated to maintain vascular integrity.
Interestingly, genetic variants in COL4A1/A2 are also associated with sporadic cerebral microangiopathies [19,20,21,22,23]. However, no clear-cut relationship with COL4A1/A2 mRNA expression has yet been demonstrated [23].
Aim
Here, we (1) investigate the co-segregation of the causative COL4A1 variant in the original PADMAL family complementing published clinical and radiological [4, 24] as well as pathological data [3, 4, 25] of this family and, and describe the clinical and radiological presentation and progression of PADMAL using this family and literature data (2) report a sequence analysis of the mutated miRNA-29-binding site in two German samples of patients with sporadic cerebral microangiopathies as well as other types of ischemic stroke.
Patients and methods
Patients, standard protocol approvals, registrations, and patient consent
All individuals of the original PADMAL family were examined in the years 2007–2014. Ages in this publication are ages at examination. Typically, we performed a full history and physical examination, a cranial Magnetic Resonance Imaging (MRI) scan, and obtained an EDTA-anticoagulated blood sample. We did not perform a cranial MRI in married-in family members. Technical details and results of the MRI investigations have been previously published [24]. For some bedridden individuals, clinical cranial MRI or computed tomography (CT) scans were obtained with their or next of kin’s consent. To preserve anonymity, we include no individual patient reports and present the pedigree without age and sex. All procedures performed in studies involving human participants followed the ethical standards of the institutional and/or national research committee and the 1964 Helsinki declaration and its later amendments or comparable ethical standards. All individuals gave written informed consent and institutional review board approval was obtained from the ethical advisory boards of the Universities of Hamburg and Kiel (Hamburg M276/06, Kiel B226/08).
The Muenster stroke sample included 225 patients with sporadic microangiopathic stroke according to the TOAST criteria. The Muenster stroke sample has previously been described in detail [26, 27]. We recruited in a completely anonymized fashion consecutive consenting ischemic stroke patients from hospitals in northwest Germany through the regional Westphalian Stroke Register and from the University Hospital of Greifswald in northeast Germany. The treating physicians used standardized patient assessment forms to collect sociodemographic and disease-related data as well as blood samples. For the whole study, we included all available patients with completed ischemic stroke, proved by computed tomography (CT) or magnetic resonance imaging (MRI) and classified as: large artery atherosclerosis (TOAST 1, n = 515), cardioembolism (TOAST 2, n = 411), or small vessel occlusion (TOAST 3, n = 255). We excluded patients who had experienced a transient ischemic attack (TIA) or hemorrhagic stroke, as well as patients falling into other TOAST categories, to maximize etiologic homogeneity. For this study, we analyzed all patients with small vessel occlusion (TOAST3) for whom DNA was still available (225 out of 255). All patients gave written informed consent and institutional review board approval was obtained from the ethical advisory board of Muenster and Kiel University (Muenster 00/113stö, Kiel D552/20).
The Rostock sample was ascertained in the context of the stroke in young Fabry patients (SIFAP) study and has been described in detail [28]. The whole Rostock Stroke Sample recruited initially, 5111 patients, 88 withdrew consent, including 1 patient with Fabry disease. Thus, 5023 patients aged between 18 and 55 years and presenting with an acute cerebrovascular event (CVE) of any cause within the last 3 months before recruitment were included in the study. Seventy-eight percent of all patients had been enrolled in the study within 10 days after the qualifying stroke event. The diagnosis of stroke had to be verified by brain MRI analysis (82%), or in case of negative or missing MRI, the clinical diagnosis of CVE had to be confirmed by a qualified stroke neurologist, who had at least 5 years of experience in treating stroke (18%). Patients not meeting these criteria had to be excluded from the study. All patients underwent thorough evaluation including brain and vascular imaging, and extensive laboratory testing according to the local laboratory routine (hemogram, blood fat, glucose, HbA1C, liver transaminases, creatinine, electrolytes, total albumin in serum, C reactive protein, antinuclear antibodies, anti-neutrophil cytoplasmic antibodies, rheumatoid factor, Factor V mutation, prothrombin mutation, antiphospholipid antibodies), Fabry disease diagnostics, cardiac ultrasound examination, and ECG. Data on comorbidities and vascular risk factors were collected in a standardized pre-specified case report form from medical records and self-reported by the patient.14 In addition, centers provided vascular and cardiac imaging, as well as stroke-associated comorbidities like depression, pain, or headache. For this study, we selected the samples of patients fulfilling the following criteria: (1) only patients who tested negative for Fabry disease and CADASIL, (2) only patients who consented to further genetic studies and the sharing of samples as well as data with collaborators, (3) from this subsample, patients with MRI signs of cerebral microangiopathy, either lacunar stroke or leukoaraiosis Fazekas grad 2/3 and/or vertebrobasilar involvement/stroke. All patients gave written informed consent and institutional review board approval was obtained from the ethical advisory board of Rostock and Kiel University (Rostock II PV 03/2006, Kiel D552/20).
Sequence analysis of the COL4A1 miRNA-29-binding site
Genomic DNA was isolated from EDTA-anticoagulated blood samples using standard procedures in all but deceased patients from the PADMAL family in whom DNA was obtained from paraffin-embedded brain sections. The miRNA-binding site and surrounding DNA sequence were Polymerase Chain Reaction (PCR) amplified for 35 cycles using the oligonucleotide primers COL4A1_UF1 (5′-TACGCCGTCCACCTTGAA-3′) and COL4A1_UR1 (5′-AGGTCAATGAAGCAGGGTGT-3′) employing standard procedures. Sanger sequencing was performed using BigDye Terminator chemistry (Applied Biosystems, MA, USA) on an Applied Biosystems 3730xl DNA analyzer (Applied Biosystems, MA, USA). Sequence electropherograms were assembled and compared to the human genome reference sequence (build GRCh38/hg38) using the SeqMan module of the Lasergene software (DNAstar, WI, USA).
Power analysis
We tested for the statistical power to detect at least one individual with a variant in the analyzed region of the COL4A1 gene versus none at a p value threshold of p < 0.05. We used RStudio (Version 2022.07.1 Build 554) with the package “pwr” and the function “pwr.p.test” with mutation frequencies between 0 and 0.5 percent and a sample size of 874 individuals.
Results
Segregation analysis in the PADMAL family
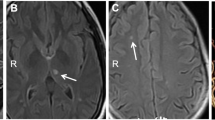
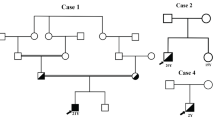
We defined radiological affection status based on the presence of at least one lacune in the pons because pontine lacunes were always present in MRIs demonstrating supratentorial white matter lesions (WML) while isolated pontine involvement—even in the absence of clinical symptoms and signs—was present in some individuals (Fig. 1). Age at radiological onset was defined as the age at the first MRI showing evidence of PADMAL. We based clinical affection status on a history of stroke, a history of symptoms suggesting a stroke, and clinical signs suggesting a past stroke in radiologically affected individuals. The anonymized pedigree (Fig. 2) shows that genotypes were available for 17 family members excluding married-in individuals of whom 6 were clinically and radiologically affected, and 3 were only radiologically affected. Three individuals were clinically and radiologically unaffected but carried the causative COL4A1 variant and were therefore defined as at risk. Figure 3 shows the causative COL4A1 variant in the sequence context of the miRNA-29-binding site with the cognate miRNAs. Table 1 summarizes the main clinical and radiological characteristics of the affected and the at-risk individuals at the time of examination. Despite the small number of patients, it becomes apparent that the radiological onset is around a decade earlier than the clinical onset. However, the radiological onset can not be estimated reliably because it is determined by the time of the first MRI. Note that the radiological onset in the individuals who are also affected clinically seems to be later than in individuals with radiological signs but no clinical symptoms. This is an artifact caused by the fact that the first MRI in the clinically affected individuals was performed at the clinical onset. In all but one clinically affected individual, the first symptom (dysarthria, vertigo, ataxia) was most likely related to a brainstem stroke. The death occurred in the 6th to 8th decade.

MRI features of PADMAL. T2-weighted cranial MRI scans of an early and a later stage PADMAL patient demonstrating pontine and supratentorial involvement

Anonymized pedigree of the family with PADMAL. Empty symbols show clinically and radiologically unaffected individuals. Half-filled symbols show radiologically affected but clinically unaffected individuals. Filled symbols show radiologically and clinically affected individuals. Slashed symbols show deceased individuals. Red “v” stands for heterozygous COL4A1, NM_001845.6:c.*31G > T carriers and wt for individuals who are homozygous wildtype. Sex is omitted from the pedigree

COL4A1, NM_001845.6:c.*31G > T variant causing PADMAL in this family. The upper panel shows miRNA-29 variants within the binding site. The middle panel shows the COL4A1 wildtype sequence and the lower panel the COL4A1, NM_001845.6:c.*31G > T variant in a patient from the family with PADMAL
Sequence analysis of the COL4A1 miRNA-29-binding site in two German sporadic stroke samples
In the Muenster stroke sample, genomic DNA was still available for 225 of 255 patients with microangiopathic stroke described in a previous publication [26]. Table 2 summarizes the demographic and clinical data for the Muenster and Rostock sporadic stroke samples. The Muenster sample represents a typical cross-section of patients with a microangiopathic stroke. The Rostock sample (649 patients) consists of young-onset stroke patients with preferential brainstem and cerebellar localization but with diverse etiologies [28]. Therefore, the age at stroke is higher (mean 68 vs. 44 years) and more variable (standard deviation of 13 vs. 8 years) in the Muenster sample compared to the Rostock sample. The Muenster sample is sex balanced while the Rostock sample shows a male preponderance (50% vs. 66% male). Detailed information on the lesion distribution is present for the Rostock but not for the Muenster sample. Power analysis (Supplemental Fig. 1) showed a power of 80% to detect a variant in the COL4A1 miRNA-29-binding site at a frequency of 0.18% for our sample of 874 patients.
Sequence analysis in two independent German sporadic stroke samples did not identify a single variant in the core COL4A1 miRNA-29-binding site (5′-GGTGCT-3′) in any of the 874 sequenced patients with sporadic stroke.
Discussion
This manuscript serves two purposes. First, it shows the co-segregation of a COL4A1 3’UTR variant with the disease in the family for which the name Pontine Autosomal Dominant Microangiopathy with Leukoencephalopathy (PADMAL) was coined. This completes our previous reports on clinical features, MRI findings, and pathology and underpins the causality of the detected variant [3, 4, 8, 24]. Taken together and adding the results of other publications, a delineation of the sequence of clinical and radiological progression of the disease is possible. The first radiological signs are pontine lacunes most likely appearing in the 4th to 5th decade in some individuals while others present with normal MRIs in this age range. Whether the latter individuals remain asymptomatic due to reduced penetrance can not be answered yet. There is uncertainty in the age at radiological onset because detection depends on a cranial MRI performed in a clinically healthy individual which is usually not performed outside a study context. Therefore, radiological onset might be somewhat earlier than in this study. The clinical onset, mostly in the form of brainstem-related neurological symptoms is in the 4th to 6th decade. The disease then progresses fairly rapidly and leads to death in the 6th to 8th decade. The first MRI signs are brainstem lacunes, followed by supratentorial white matter lesions and atrophy of pons and medulla oblongata. Several reports describing families or single cases suffering from PADMAL have been published [5, 7,8,9, 29, 30]. For none of these families, extensive MRI studies are available which could be compared in detail to our study [24]. The causative variants vary slightly between studies but all affect the COL4A1 miRNA-29-binding site. Nevertheless, data in these studies allow the conclusion that the disease started in most cases with brainstem symptoms, namely with dysarthria. There is also some variation in the age of onset and the age at death between studies but adult-onset (mostly 4th to 5th decade) and disease duration between 10 and 20 years were found in all patients. Compared to disease caused by variants in the coding regions of COL4A1 and COL4A2, PADMAL has a later age at onset and does not lead to cerebral hemorrhages, or porencephaly. The second purpose of this manuscript is to answer the question of whether some sporadic stroke patients also show a variant in the COL4A1 miRNA-29-binding site, we sequenced 874 German stroke patients, of whom 291 suffered from a microangiopathic stroke but did not find a single variant in the COL4A1 miRNA-29-binding site. Our power analysis shows that it is unlikely that variants in the COL4A1 miRNA-29-binding site are present at a frequency above ~ 0.2%. Of note, the COL4A1 miRNA-29-binding site is extremely conserved across species, none of the mammals, birds, and bony fish available for analysis in the UCSC human genome browser (Supplemental Fig. 2 and https://genome.ucsc.edu) show a single base change. Further, no variant in the 6-base-binding motif is found in the approximately 117.007 human sequences covering this motif in the gnomAD database (https://gnomad.broadinstitute.org, GRCh38/hg38, NC_000013.11:g.110.150.327–110.150.332) and no single-nucleotide polymorphism has been reported up to dbSNP version 153 (https://www.ncbi.nlm.gov/snp/). Targeted sequencing of genes causing monogenic disease has in some cases revealed a higher frequency of pathogenic variants in sporadic patients or a contribution of less pathogenic variants to sporadic diseases. An example of the former is Fabry disease which is a moderately common cause of young-onset stroke [28]. An example of the latter is the contribution of common heterozygous variants in the gene causing Gaucher disease in the pathogenesis of Parkinson’s disease [31, 32].
Our study has several limitations. The first is the small number of family members available for clinical and radiological analysis in the PADMAL family. More family members would allow a more detailed description of the phenotype. However, compared to other studies of PADMAL, we have analyzed one of the largest families. The second is the size and composition of the samples with sporadic stroke. All patients in the Muenster sample had a diagnosis of cerebral microangiopathy but the lesion distribution is unknown. Therefore, we do not know in whom the lesion pattern was similar to PADMAL. For the Rostock sample, this data was available. The much younger age at stroke onset increases the likelihood of a monogenic cause. We can not exclude that variants in the COL4A1 miRNA-29-binding site contribute to the pathogenesis of sporadic stroke at low frequencies below ~ 0.2% or to specific stroke subtypes not sufficiently represented in our samples. A third limitation might be the lack of functional data. However, these data have already been provided by other publications [6, 8, 25].
Conclusion
PADMAL is an extremely rare autosomal dominant cerebral microangiopathy caused by variants in the COL4A1 miRNA-29-binding site. Our clinical description in conjunction with other publications delineates an autosomal dominant inheritance pattern, adult-onset (mostly 4th to 5th decade), initial brainstem-related symptoms (most commonly dysarthria), progression to tetraparesis and dementia and pontine lacunes and atrophy followed by supratentorial white matter lesions as the main clinical and radiological characteristics which should suffice to raise the suspicion of PADMAL. Variants in the COL4A1 miRNA-29-binding site are not involved in the pathogenesis of a sizeable proportion of sporadic stroke.
Data availability
All data pertaining to COL4A1 miRNA-29-binding site sequencing are contained in the article itself.
References
Cannistraro RJ, Badi M, Eidelman BH, Dickson DW, Middlebrooks EH, Meschia JF (2019) CNS small vessel disease: a clinical review. Neurology 92:1146–1156. https://doi.org/10.1212/wnl.0000000000007654
Marini S, Anderson CD, Rosand J (2020) Genetics of cerebral small vessel disease. Stroke 51:12–20. https://doi.org/10.1161/strokeaha.119.024151
Colmant HJ, Hagel C, Makrigeorgi-Butera M, Stavrou D (2000) Neuropathology of hereditary subcortical angiopathic encephalopathy. Clin Neuropathol 19:254–255
Hagel C, Groden C, Niemeyer R, Stavrou D, Colmant HJ (2004) Subcortical angiopathic encephalopathy in a German kindred suggests an autosomal dominant disorder distinct from CADASIL. Acta Neuropathol (Berl) 108:231–240
Li Q, Wang C, Li W, Zhang Z, Wang S, Wupuer A, Hu X, Wumaier K, Zhu Y, Li H, Yu W (2022) A novel mutation in COL4A1 gene in a Chinese family with pontine autosomal dominant microangiopathy and leukoencephalopathy. Transl Stroke Res 13:238–244. https://doi.org/10.1007/s12975-021-00926-0
Siitonen M, Borjesson-Hanson A, Poyhonen M, Ora A, Pasanen P, Bras J, Kern S, Kern J, Andersen O, Stanescu H, Kleta R, Baumann M, Kalaria R, Kalimo H, Singleton A, Hardy J, Viitanen M, Myllykangas L, Guerreiro R (2017) Multi-infarct dementia of Swedish type is caused by a 3’UTR mutation of COL4A1. Brain 140:e29. https://doi.org/10.1093/brain/awx062
Grobe-Einsler M, Urbach H, Paus S (2020) Recurrent pontine strokes in a young male. J Stroke Cerebrovasc Dis 29:105386. https://doi.org/10.1016/j.jstrokecerebrovasdis.2020.105386
Verdura E, Herve D, Bergametti F, Jacquet C, Morvan T, Prieto-Morin C, Mackowiak A, Manchon E, Hosseini H, Cordonnier C, Girard-Buttaz I, Rosenstingl S, Hagel C, Kuhlenbaumer G, Leca-Radu E, Goux D, Fleming L, Van Agtmael T, Chabriat H, Chapon F, Tournier-Lasserve E (2016) Disruption of a miR-29 binding site leading to COL4A1 upregulation causes pontine autosomal dominant microangiopathy with leukoencephalopathy. Ann Neurol 80:741–753. https://doi.org/10.1002/ana.24782
Zhao YY, Duan RN, Ji L, Liu QJ, Yan CZ (2019) Cervical spinal involvement in a Chinese pedigree with pontine autosomal dominant microangiopathy and leukoencephalopathy caused by a 3’ untranslated region mutation of COL4A1 Gene. Stroke 50:2307–2313. https://doi.org/10.1161/STROKEAHA.119.024875
Zagaglia S, Selch C, Nisevic JR, Mei D, Michalak Z, Hernandez-Hernandez L, Krithika S, Vezyroglou K, Varadkar SM, Pepler A, Biskup S, Leão M, Gärtner J, Merkenschlager A, Jaksch M, Møller RS, Gardella E, Kristiansen BS, Hansen LK, Vari MS, Helbig KL, Desai S, Smith-Hicks CL, Hino-Fukuyo N, Talvik T, Laugesaar R, Ilves P, Õunap K, Körber I, Hartlieb T, Kudernatsch M, Winkler P, Schimmel M, Hasse A, Knuf M, Heinemeyer J, Makowski C, Ghedia S, Subramanian GM, Striano P, Thomas RH, Micallef C, Thom M, Werring DJ, Kluger GJ, Cross JH, Guerrini R, Balestrini S, Sisodiya SM (2018) Neurologic phenotypes associated with COL4A1/2 mutations: expanding the spectrum of disease. Neurology 91:e2078–e2088. https://doi.org/10.1212/wnl.0000000000006567
Guey S, Herve D (2022) Main features of COL4A1-COL4A2 related cerebral microangiopathies. Cereb Circ Cogn Behav 3:100140. https://doi.org/10.1016/j.cccb.2022.100140
Volonghi I, Pezzini A, Del Zotto E, Giossi A, Costa P, Ferrari D, Padovani A (2010) Role of COL4A1 in basement-membrane integrity and cerebral small-vessel disease. The COL4A1 stroke syndrome. Curr Med Chem 17:1317–1324. https://doi.org/10.2174/092986710790936293
Kuuluvainen L, Mönkäre S, Kokkonen H, Zhao F, Verkkoniemi-Ahola A, Schleutker J, Hakonen AH, Hartikainen P, Pöyhönen M, Myllykangas L (2021) COL4A1 and COL4A2 duplication causes cerebral small vessel disease with recurrent early onset ischemic strokes. Stroke 52:e624–e625. https://doi.org/10.1161/strokeaha.120.033864
Meuwissen ME, Halley DJ, Smit LS, Lequin MH, Cobben JM, de Coo R, van Harssel J, Sallevelt S, Woldringh G, van der Knaap MS, de Vries LS, Mancini GM (2015) The expanding phenotype of COL4A1 and COL4A2 mutations: clinical data on 13 newly identified families and a review of the literature. Genet Med 17:843–853. https://doi.org/10.1038/gim.2014.210
Jeanne M, Gould DB (2017) Genotype-phenotype correlations in pathology caused by collagen type IV alpha 1 and 2 mutations. Matrix Biol 57–58:29–44. https://doi.org/10.1016/j.matbio.2016.10.003
Lanfranconi S, Markus HS (2010) COL4A1 mutations as a monogenic cause of cerebral small vessel disease: a systematic review. Stroke 41:e513-518. https://doi.org/10.1161/strokeaha.110.581918
Traenka C, Kloss M, Strom T, Lyrer P, Brandt T, Bonati LH, Grond-Ginsbach C, Engelter S (2019) Rare genetic variants in patients with cervical artery dissection. Eur Stroke J 4:355–362. https://doi.org/10.1177/2396987319861869
Rannikmäe K, Henshall DE, Thrippleton S, Ginj Kong Q, Chong M, Grami N, Kuan I, Wilkinson T, Wilson B, Wilson K, Paré G, Sudlow C (2020) Beyond the brain: systematic review of extracerebral phenotypes associated with monogenic cerebral small vessel disease. Stroke 51:3007–3017. https://doi.org/10.1161/strokeaha.120.029517
Traylor M, Persyn E, Tomppo L, Klasson S, Abedi V, Bakker MK, Torres N, Li L, Bell S, Rutten-Jacobs L, Tozer DJ, Griessenauer CJ, Zhang Y, Pedersen A, Sharma P, Jimenez-Conde J, Rundek T, Grewal RP, Lindgren A, Meschia JF, Salomaa V, Havulinna A, Kourkoulis C, Crawford K, Marini S, Mitchell BD, Kittner SJ, Rosand J, Dichgans M, Jern C, Strbian D, Fernandez-Cadenas I, Zand R, Ruigrok Y, Rost N, Lemmens R, Rothwell PM, Anderson CD, Wardlaw J, Lewis CM, Markus HS (2021) Genetic basis of lacunar stroke: a pooled analysis of individual patient data and genome-wide association studies. Lancet Neurol 20:351–361. https://doi.org/10.1016/s1474-4422(21)00031-4
Mishra A, Malik R, Hachiya T, Jurgenson T, Namba S, Posner DC, Kamanu FK, Koido M, Le Grand Q, Shi M, He Y, Georgakis MK, Caro I, Krebs K, Liaw YC, Vaura FC, Lin K, Winsvold BS, Srinivasasainagendra V, Parodi L, Bae HJ, Chauhan G, Chong MR, Tomppo L, Akinyemi R, Roshchupkin GV, Habib N, Jee YH, Thomassen JQ, Abedi V, Carcel-Marquez J, Nygaard M, Leonard HL, Yang C, Yonova-Doing E, Knol MJ, Lewis AJ, Judy RL, Ago T, Amouyel P, Armstrong ND, Bakker MK, Bartz TM, Bennett DA, Bis JC, Bordes C, Borte S, Cain A, Ridker PM, Cho K, Chen Z, Cruchaga C, Cole JW, de Jager PL, de Cid R, Endres M, Ferreira LE, Geerlings MI, Gasca NC, Gudnason V, Hata J, He J, Heath AK, Ho YL, Havulinna AS, Hopewell JC, Hyacinth HI, Inouye M, Jacob MA, Jeon CE, Jern C, Kamouchi M, Keene KL, Kitazono T, Kittner SJ, Konuma T, Kumar A, Lacaze P, Launer LJ, Lee KJ, Lepik K, Li J, Li L, Manichaikul A, Markus HS, Marston NA, Meitinger T, Mitchell BD, Montellano FA, Morisaki T, Mosley TH, Nalls MA, Nordestgaard BG, O'Donnell MJ, Okada Y, Onland-Moret NC, Ovbiagele B, Peters A, Psaty BM, Rich SS, Rosand J, Sabatine MS, Sacco RL, Saleheen D, Sandset EC, Salomaa V, Sargurupremraj M, Sasaki M, Satizabal CL, Schmidt CO, Shimizu A, Smith NL, Sloane KL, Sutoh Y, Sun YV, Tanno K, Tiedt S, Tatlisumak T, Torres-Aguila NP, Tiwari HK, Tregouet DA, Trompet S, Tuladhar AM, Tybjaerg-Hansen A, van Vugt M, Vibo R, Verma SS, Wiggins KL, Wennberg P, Woo D, Wilson PWF, Xu H, Yang Q, Yoon K, Consortium C, Consortium I, Dutch Parelsnoer Initiative Cerebrovascular Disease Study G, Estonian B, Consortium PQ, FinnGen C, Network NSG, Consortium M, Consortium S, China Kadoorie Biobank Collaborative G, Program VAMV, International Stroke Genetics C, Biobank J, Consortium C, Consortium G, Millwood IY, Gieger C, Ninomiya T, Grabe HJ, Jukema JW, Rissanen IL, Strbian D, Kim YJ, Chen PH, Mayerhofer E, Howson JMM, Irvin MR, Adams H, Wassertheil-Smoller S, Christensen K, Ikram MA, Rundek T, Worrall BB, Lathrop GM, Riaz M, Simonsick EM, Korv J, Franca PHC, Zand R, Prasad K, Frikke-Schmidt R, de Leeuw FE, Liman T, Haeusler KG, Ruigrok YM, Heuschmann PU, Longstreth WT, Jung KJ, Bastarache L, Pare G, Damrauer SM, Chasman DI, Rotter JI, Anderson CD, Zwart JA, Niiranen TJ, Fornage M, Liaw YP, Seshadri S, Fernandez-Cadenas I, Walters RG, Ruff CT, Owolabi MO, Huffman JE, Milani L, Kamatani Y, Dichgans M, Debette S (2022) Stroke genetics informs drug discovery and risk prediction across ancestries. Nature 611:115–123. https://doi.org/10.1038/s41586-022-05165-3
Malik R, Chauhan G, Traylor M, Sargurupremraj M, Okada Y, Mishra A, Rutten-Jacobs L, Giese AK, van der Laan SW, Gretarsdottir S, Anderson CD, Chong M, Adams HHH, Ago T, Almgren P, Amouyel P, Ay H, Bartz TM, Benavente OR, Bevan S, Boncoraglio GB, Brown RD, Jr., Butterworth AS, Carrera C, Carty CL, Chasman DI, Chen WM, Cole JW, Correa A, Cotlarciuc I, Cruchaga C, Danesh J, de Bakker PIW, DeStefano AL, den Hoed M, Duan Q, Engelter ST, Falcone GJ, Gottesman RF, Grewal RP, Gudnason V, Gustafsson S, Haessler J, Harris TB, Hassan A, Havulinna AS, Heckbert SR, Holliday EG, Howard G, Hsu FC, Hyacinth HI, Ikram MA, Ingelsson E, Irvin MR, Jian X, Jimenez-Conde J, Johnson JA, Jukema JW, Kanai M, Keene KL, Kissela BM, Kleindorfer DO, Kooperberg C, Kubo M, Lange LA, Langefeld CD, Langenberg C, Launer LJ, Lee JM, Lemmens R, Leys D, Lewis CM, Lin WY, Lindgren AG, Lorentzen E, Magnusson PK, Maguire J, Manichaikul A, McArdle PF, Meschia JF, Mitchell BD, Mosley TH, Nalls MA, Ninomiya T, O'Donnell MJ, Psaty BM, Pulit SL, Rannikmae K, Reiner AP, Rexrode KM, Rice K, Rich SS, Ridker PM, Rost NS, Rothwell PM, Rotter JI, Rundek T, Sacco RL, Sakaue S, Sale MM, Salomaa V, Sapkota BR, Schmidt R, Schmidt CO, Schminke U, Sharma P, Slowik A, Sudlow CLM, Tanislav C, Tatlisumak T, Taylor KD, Thijs VNS, Thorleifsson G, Thorsteinsdottir U, Tiedt S, Trompet S, Tzourio C, van Duijn CM, Walters M, Wareham NJ, Wassertheil-Smoller S, Wilson JG, Wiggins KL, Yang Q, Yusuf S, Consortium AF, Cohorts for H, Aging Research in Genomic Epidemiology C, International Genomics of Blood Pressure C, Consortium I, Starnet, Bis JC, Pastinen T, Ruusalepp A, Schadt EE, Koplev S, Bjorkegren JLM, Codoni V, Civelek M, Smith NL, Tregouet DA, Christophersen IE, Roselli C, Lubitz SA, Ellinor PT, Tai ES, Kooner JS, Kato N, He J, van der Harst P, Elliott P, Chambers JC, Takeuchi F, Johnson AD, BioBank Japan Cooperative Hospital G, Consortium C, Consortium E-C, Consortium EP-I, International Stroke Genetics C, Consortium M, Neurology Working Group of the CC, Network NSG, Study UKYLD, Consortium M, Sanghera DK, Melander O, Jern C, Strbian D, Fernandez-Cadenas I, Longstreth WT, Jr., Rolfs A, Hata J, Woo D, Rosand J, Pare G, Hopewell JC, Saleheen D, Stefansson K, Worrall BB, Kittner SJ, Seshadri S, Fornage M, Markus HS, Howson JMM, Kamatani Y, Debette S, Dichgans M (2018) Multiancestry genome-wide association study of 520,000 subjects identifies 32 loci associated with stroke and stroke subtypes. Nat Genet 50:524–537.https://doi.org/10.1038/s41588-018-0058-3
Rannikmäe K, Davies G, Thomson PA, Bevan S, Devan WJ, Falcone GJ, Traylor M, Anderson CD, Battey TW, Radmanesh F, Deka R, Woo JG, Martin LJ, Jimenez-Conde J, Selim M, Brown DL, Silliman SL, Kidwell CS, Montaner J, Langefeld CD, Slowik A, Hansen BM, Lindgren AG, Meschia JF, Fornage M, Bis JC, Debette S, Ikram MA, Longstreth WT, Schmidt R, Zhang CR, Yang Q, Sharma P, Kittner SJ, Mitchell BD, Holliday EG, Levi CR, Attia J, Rothwell PM, Poole DL, Boncoraglio GB, Psaty BM, Malik R, Rost N, Worrall BB, Dichgans M, Van Agtmael T, Woo D, Markus HS, Seshadri S, Rosand J, Sudlow CL (2015) Common variation in COL4A1/COL4A2 is associated with sporadic cerebral small vessel disease. Neurology 84:918–926. https://doi.org/10.1212/wnl.0000000000001309
Chung J, Marini S, Pera J, Norrving B, Jimenez-Conde J, Roquer J, Fernandez-Cadenas I, Tirschwell DL, Selim M, Brown DL, Silliman SL, Worrall BB, Meschia JF, Demel S, Greenberg SM, Slowik A, Lindgren A, Schmidt R, Traylor M, Sargurupremraj M, Tiedt S, Malik R, Debette S, Dichgans M, Langefeld CD, Woo D, Rosand J, Anderson CD (2019) Genome-wide association study of cerebral small vessel disease reveals established and novel loci. Brain 142:3176–3189. https://doi.org/10.1093/brain/awz233
Ding XQ, Hagel C, Ringelstein B, Buchheit S, Zeumer H, Kuhlenbaumer G, Appenzeller S, Fiehler J (2009) MRI features of pontine autosomal dominant microangiopathy and leukoencephalopathy (PADMAL). J Neuroimaging 20:134–140. https://doi.org/10.1111/j.1552-6569.2008.00336.x. (JON336 [pii])
Craggs LJ, Hagel C, Kuhlenbaeumer G, Borjesson-Hanson A, Andersen O, Viitanen M, Kalimo H, McLean CA, Slade JY, Hall RA, Oakley AE, Yamamoto Y, Deramecourt V, Kalaria RN (2013) Quantitative vascular pathology and phenotyping familial and sporadic cerebral small vessel diseases. Brain Pathol 23:547–557. https://doi.org/10.1111/bpa.12041
Kuhlenbaumer G, Berger K, Huge A, Lange E, Kessler C, John U, Funke H, Nabavi DG, Stogbauer F, Ringelstein EB, Stoll M (2006) Evaluation of single nucleotide polymorphisms in the phosphodiesterase 4D gene (PDE4D) and their association with ischaemic stroke in a large German cohort. J Neurol Neurosurg Psychiatry 77:521–524
Adams HP Jr, Woolson RF, Biller J, Clarke W (1992) Studies of Org 10172 in patients with acute ischemic stroke. TOAST Study Group. Haemostasis 22:99–103
Rolfs A, Fazekas F, Grittner U, Dichgans M, Martus P, Holzhausen M, Bottcher T, Heuschmann PU, Tatlisumak T, Tanislav C, Jungehulsing GJ, Giese AK, Putaala J, Huber R, Bodechtel U, Lichy C, Enzinger C, Schmidt R, Hennerici MG, Kaps M, Kessler C, Lackner K, Paschke E, Meyer W, Mascher H, Riess O, Kolodny E, Norrving B, Stroke in Young Fabry Patients I (2013) Acute cerebrovascular disease in the young: the Stroke in Young Fabry Patients study. Stroke 44:340–349. https://doi.org/10.1161/STROKEAHA.112.663708
Sourander P, Walinder J (1977) Hereditary multi-infarct dementia. Morphological and clinical studies of a new disease. Acta Neuropathol 39:247–254
Low WC, Junna M, Borjesson-Hanson A, Morris CM, Moss TH, Stevens DL, St Clair D, Mizuno T, Zhang WW, Mykkanen K, Wahlstrom J, Andersen O, Kalimo H, Viitanen M, Kalaria RN (2007) Hereditary multi-infarct dementia of the Swedish type is a novel disorder different from NOTCH3 causing CADASIL. Brain 130:357–367
Hopfner F, Mueller SH, Szymczak S, Junge O, Tittmann L, May S, Lohmann K, Grallert H, Lieb W, Strauch K, Muller-Nurasyid M, Berger K, Schormair B, Winkelmann J, Mollenhauer B, Trenkwalder C, Maetzler W, Berg D, Kasten M, Klein C, Hoglinger GU, Gasser T, Deuschl G, Franke A, Krawczak M, Dempfle A, Kuhlenbaumer G (2020) Rare variants in specific lysosomal genes are associated with Parkinson’s disease. Mov Disord. https://doi.org/10.1002/mds.28037
Creese B, Bell E, Johar I, Francis P, Ballard C, Aarsland D (2018) Glucocerebrosidase mutations and neuropsychiatric phenotypes in Parkinson’s disease and Lewy body dementias: review and meta-analyses. Am J Med Genet B Neuropsychiatr Genet 177:232–241. https://doi.org/10.1002/ajmg.b.32549
Acknowledgements
We thank all family members and stroke patients who have made this research possible. This research was partially sponsored by a grant from the medical faculty of Kiel University to GK.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare that there is no conflict of interest.
Ethical standard
Integrity of research and reporting: Family data: To preserve anonymity, we include no individual patient reports and present the pedigree without age and sex. All individuals gave written informed consent and institutional review board approval was obtained from the ethical advisory boards of the Universities of Hamburg and Kiel (Hamburg M276/06, Kiel B226/08). Muenster sample: All patients gave written informed consent and institutional review board approval was obtained from the ethical advisory board of Muenster and Kiel University (Muenster 00/113stö, Kiel D552/20). All patients gave written informed consent and institutional review board approval was obtained from the ethical advisory board of Rostock and Kiel University (Rostock II PV 03/2006, Kiel D552/20).
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Roos, J., Müller, S., Giese, A. et al. Pontine autosomal dominant microangiopathy with leukoencephalopathy: Col4A1 gene variants in the original family and sporadic stroke. J Neurol 270, 2631–2639 (2023). https://doi.org/10.1007/s00415-023-11590-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-023-11590-9