
Abstract
Background
Congenital myasthenic syndromes (CMS) are a heterogeneous group of disorders caused by genetic defects resulting in impaired neuromuscular transmission. Although effective treatments are available, CMS is probably underdiagnosed, and systematic clinico-genetic investigations are warranted.
Methods
We used a nationwide approach to collect Austrian patients with genetically confirmed CMS. We provide a clinical and molecular characterization of this cohort and aimed to ascertain the current frequency of CMS in Austria.
Results
Twenty-eight cases with genetically confirmed CMS were identified, corresponding to an overall prevalence of 3.1 per million (95% CI 2.0–4.3) in Austria. The most frequent genetic etiology was CHRNE (n = 13), accounting for 46.4% of the cohort. Within this subgroup, the variant c.1327del, p.(Glu443Lysfs*64) was detected in nine individuals. Moreover, causative variants were found in DOK7 (n = 4), RAPSN (n = 3), COLQ (n = 2), GMPPB (n = 2), CHAT (n = 1), COL13A1 (n = 1), MUSK (n = 1) and AGRN (n = 1). Clinical onset within the first year of life was reported in one half of the patients. Across all subtypes, the most common symptoms were ptosis (85.7%), lower limb (67.9%), upper limb (60.7%) and facial weakness (60.7%). The majority of patients (96.4%) received specific treatment, including acetylcholinesterase inhibitors in 20, adrenergic agonists in 11 and 3,4-diaminopyridine in nine patients.
Conclusions
Our study presents the first systematic characterization of individuals with CMS in Austria, providing prevalence estimates and genotype–phenotype correlations that may help to improve the diagnostic approach and patient management.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Congenital myasthenic syndromes (CMS) encompass a heterogeneous group of inherited disorders caused by genetic defects that result in impaired signal transmission at the neuromuscular junction (NMJ) [1]. While early-onset fatigable muscle weakness is considered the clinical hallmark of CMS, the phenotypic spectrum may also include other organ manifestations, and symptom severity as well as age of onset are highly variable between genetic subtypes. With the rapid advancements of next-generation sequencing (NGS) technology over the past decade, the number of known molecular defects has constantly grown, and more than 30 underlying monogenic disease genes encoding presynaptic, synaptic and postsynaptic components of the NMJ have been identified to date [2].
CMS are rare with only few prevalence estimates in the literature, ranging between 1.8 per million total population and 22.2 per million children [3, 4]. Epidemiological studies, however, probably underestimate the real prevalence of CMS due to the lack of large disease registries, limited access to NGS-based genetic testing in some countries and the misclassification of CMS as (seronegative) autoimmune myasthenia gravis [5]. Nonetheless, the early identification of underlying genetic defects is crucial in the light of various effective treatment options that are already available for most CMS forms [6].
In our study, we aimed to systematically describe the clinical and molecular spectrum of CMS in Austria. We used a nationwide approach to recruit patients from both pediatric and adult neuromuscular centers and, for the first time, provide prevalence estimates for CMS in Austria.
Patients and methods
Patient ascertainment and ethics approval
This retrospective, nationwide cohort study included all patients with genetically confirmed CMS (i.e., related to genes previously associated with CMS) who were treated in one of the Austrian pediatric or adult neuromuscular centers between 01/01/2000 and 31/12/2021. The ethics committee of the Medical University of Vienna approved this study prior to the nationwide acquisition of clinical and genetic patient data (EK: 2065/2018). The study was performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments.
Data collection
Collaborating centers were contacted via e-mail and invited to complete a pseudonymized case report form including demographic, clinical and genetic details. All genetic variants were re-analyzed, and only pathogenic and likely pathogenic variants according to the standards of the American College of Medical Genetics and Genomics (ACMG) were eligible for inclusion in the study, if the variants were compatible with the expected mode of inheritance [7]. Reported variants were submitted to the Global Variome shared Leiden Open-source Variation Database (LOVD, https://databases.lovd.nl) [8]. Given the multicentric nature of this study and the application of genetic tests in different laboratories, applied gene panels, exome and genome analyses may have encompassed different sets of genes with variable coverage. These different NGS approaches were pooled and analyzed together in this study.
Data analysis
The prevalence of CMS in Austria was estimated based on population data (2020) extracted from “Statistik Austria” (https://www.statistik.at), with 95% confidence intervals (CI) calculated assuming a Poisson distribution. Data were analyzed by SPSS version 28 (IBM Corp., Armonk, NY, USA) using descriptive measures. Figures were created using Prism, version 9.1.0 (GraphPad Software Inc., San Diego, CA, USA).
Results
Demographic and clinical characteristics
Twenty-eight individuals from 25 different families with genetically confirmed CMS were identified through our nationwide patient recruitment, corresponding to an overall prevalence of 3.1 per million (95% CI 2.0–4.3) in Austria. The calculated prevalence in the pediatric population (< 19 years) was 10.5 per million (95% CI 5.6–15.3). In our study cohort, females accounted for 64.3%. One half of the patients experienced first symptoms just after birth or during the first year of life (50%). Disease onset in adolescence (13–18 years) and adulthood (> 18 years), by contrast, was very rare, with only one case represented in each age group (both associated with biallelic variants in GMPPB) (Table 1).
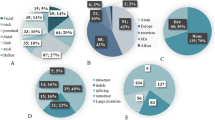
The most commonly observed clinical features across all genetic subtypes were ptosis (85.7%), lower limb (67.9%), upper limb (60.7%) and facial weakness (60.7%). Ophthalmoparesis/ophthalmoplegia was present in 50.0% of the cohort and the dominant symptom in patients with causative CHNRE variants (Fig. 1A). Apart from neuromuscular symptoms, further manifestations included scoliosis (25%), facial dysmorphism (14.3%) and gastrointestinal symptoms (10.7%). One patient (with MUSK-related CMS) was very severely affected requiring percutaneous endoscopic gastrostomy and invasive ventilation. Another patient with AGRN-related CMS required non-invasive ventilation. Lung function tests were available for 10 individuals, showing a mean forced vital capacity (FVC) of 71.4% (total range: 34–94%).

Clinical and genetic spectrum of patients with CMS in Austria. A Frequency of phenotypic features compared between patients with CHRNE-associated CMS and the remaining molecular etiologies. The CHRNE subgroup is predominantly characterized by ocular features (ophthalmoparesis/ophthalmoplegia and ptosis) and the absence of respiratory symptoms, while limb and bulbar weakness was more commonly observed in patients with other CMS forms. B The identification of nine different molecular etiologies within the cohort reflects the remarkable genetic heterogeneity of CMS. CHRNE and DOK7 are the two most commonly mutated genes together accounting for more than 60% of the whole cohort
A pathological decrement on repetitive nerve stimulation was reported in five patients and elevated serum creatine kinase (CK) activity in two patients (both with pathogenic GMPPB variants).
Treatment-related aspects
The vast majority of the cohort (96.4%) had received CMS-specific medical treatment at any time point, with acetylcholinesterase inhibitors (pyridostigmine, dosage range: 1.25–7.5 mg/kg/d) used in 20 patients (71.4%), and adrenergic agonists (salbutamol, dosage range: 0.1–1.39 mg/kg/d) and 3,4-diaminopyridine (3,4-DAP, dosage range: 0.1–1.25 mg/kg/d) used in 11 (39.3%) and nine patients (32.1%), respectively. A significant proportion of patients (39.3%) had multiple treatments, either used sequentially or in combination. General clinical improvement attributable to medication was reported in two thirds (66.7%) and specific improvement of ocular symptoms in 37% of treated patients.
All 13 patients with CHRNE-associated CMS received targeted medical treatment. Pyridostigmine was used in all patients, while 3,4-DAP was used in four and salbutamol only in one. In addition to medication, ptosis surgery (blepharoplasty) was performed in two individuals with CHRNE-related CMS.
The second most common genetic etiology in our series was DOK7, which shows an excellent response to treatment with salbutamol [9, 10]. All four DOK7 cases reported in this study received adrenergic agonists, which resulted in marked clinical improvement in three of them.
Summarized case report files of the 28 individuals with CMS including clinical and demographic details are provided as supplementary information.
Genetic findings
Twenty-four different pathogenic or likely pathogenic (i.e., diagnostic) variants according to ACMG standards were identified in nine different CMS-related genes (Fig. 1B and Table 2), with variants in the most commonly affected genes CHRNE and DOK7 accounting for 46.4% and 14.3% of the cohort, respectively. All genetically solved cases displayed an autosomal recessive inheritance pattern with homozygous or compound heterozygous variants in known disease genes. The spectrum of detected variants included 14 different missense variants, five frameshift variants, two canonical splice-site variants, one nonsense variant, one near-splice variant and one copy number variant (microdeletion). Overall, more molecular diagnoses could be established by NGS-based approaches than by single gene sequencing (16 vs. six cases, data missing in six cases).
CHRNE-associated CMS
The majority of CHRNE-associated CMS cases (9/13 patients, 69.2%) were caused by the truncating founder variant c.1327del, p.(Glu443Lysfs*64), either in a homozygous or a compound heterozygous carrier state.
Similar to our findings in the total cohort, around one half of the patients with CHRNE-associated CMS (53.8%) experienced symptom onset during the first year of life, and all patients before adolescence. Disease severity was generally milder in this subgroup with predominantly ocular symptoms including ophthalmoparesis in 12/13 patients and ptosis in 13/13 patients. By contrast, 12/13 patients (92.3%) in this subgroup were still able to walk independently at the last follow-up visit (as compared to 73.3% in the remainder of the cohort), and respiratory weakness was not observed at all in patients with CHRNE-CMS. Only one patient carrying the abovementioned founder variant was wheelchair-dependent.
Discussion
In this study, we present the first clinical and molecular characterization of pediatric and adult patients with genetically confirmed CMS in Austria who were ascertained through a systematic, nationwide approach. Such collaborative efforts are generally important, as they have the potential to raise awareness for orphan diseases and may therefore improve diagnostic and therapeutic patient services in the long run.
Patient recruitment covered all specialized pediatric and adult neuromuscular centers in Austria and thus enabled us to perform population-based calculations on CMS epidemiology, yielding a prevalence of 3.1 per million in the total population and 10.5 per million in the pediatric population. These figures were comparable to the CMS prevalence of 9.2 per million children in the United Kingdom [11].
However, it is likely that existing epidemiological data rather underestimate the real number of affected patients. First, this may partly be explained by the fact that disease gene discovery is an ongoing process with novel molecular etiologies yet to be identified. Second, there is emerging evidence that hereditary myasthenic syndromes are prone to misdiagnoses and may falsely be classified as (seronegative) autoimmune myasthenia gravis and even be treated with immunosuppressant drugs [5, 12]. All the more so, a precise and early molecular diagnosis is of paramount importance to avoid long diagnostic delays and unnecessary treatments with potential side effects. Third, the identification of CMS cases may also depend on specific conventions in different countries and may be limited by a restricted access to NGS applications. It has already been demonstrated that NGS yields higher diagnostic rates in heterogeneous neuromuscular disorders than sequential gene-by-gene testing [13,14,15].
Several nationwide studies have already been performed to delineate the genetic spectrum of CMS, and the findings indicate a variable distribution of molecular etiologies and specific mutations between ethnicities and different geographic regions [3, 4, 11, 16,17,18,19,20]. In accordance with data from other European countries, CHRNE also represented the most frequently mutated gene in our cohort [3, 4, 11]. Among patients with CHRNE-related CMS, the largest part could be attributed to the founder variant c.1327del, p.(Glu443Lysfs*64), which has formerly been referred to as epsilon1267delG. Originating from the Romani (Gypsy) people, this specific variant is particularly frequent in Europe [21]. In line with previous reports, the clinical presentation associated with CHRNE variants is heterogeneous and comparably mild, mainly characterized by ophthalmoparesis, which can be helpful to guide genetic testing [22]. Specific physiological features of extraocular muscles such as exceptionally high motor neuron firing rates together with the accumulation of acetylcholine receptors in desensitized states have been suggested to underlie ophthalmoparesis in these patients [23]. In our cohort, over 90% of patients with variants in CHRNE had marked limitation of ocular movements but were still able to walk unaided. Patients with variants in CHRNE tend to respond favorably to adrenergic agonists [24]. Nonetheless, only one patient in our study was treated with salbutamol, suggesting that available and well-tolerated therapies are still underutilized in these patients.
It is also worthy of note that our study includes a previously unreported individual with MUSK-related CMS, a very rare genetic subtype with only a few patients reported to date. It has already been suggested that the phenotypic spectrum associated with MUSK variants is very broad, ranging from a mild, fatigable limb-girdle weakness to a severe neonatal syndrome with refractory respiratory failure [25, 26]. Our presented case with MUSK-related CMS had a severe phenotype with an onset just after birth, requiring invasive ventilation. However, salbutamol treatment was effective, remarkably improving motor function and reducing the effort of mechanical ventilation.
Only two of the 28 patients in our cohort had their first symptoms reported in adolescence or adulthood. Although clinically heterogeneous, it is well established that the majority of CMS patients have an early onset of disease (in infancy or childhood) [27]. Interestingly, the only two patients in our cohort with an onset beyond childhood carried causative variants in GMPPB, a gene that has already been associated with late-onset CMS and limb-girdle muscular dystrophy [28, 29]. In accordance with previous reports, these were also the only patients with significantly elevated CK activity levels [30]. This genetic etiology (together with other genetic defects of glycosylation) should therefore be taken into particular consideration in adult patients presenting with a limb-girdle pattern of myasthenic weakness and significant hyperCKemia [31].
In conclusion, our study provides the first comprehensive clinical and genetic characterization of patients with CMS in Austria. CHRNE was the most commonly mutated gene and associated with severe ophthalmoparesis in most patients, which can help to guide diagnosis. In addition, the presented data may be useful to improve diagnostic and therapeutic services for patients and foster collaborative research on this rare and treatable condition.
Data availability
Anonymized data not published in this article will be made available by request from the corresponding author.
References
Vanhaesebrouck AE, Beeson D (2019) The congenital myasthenic syndromes: expanding genetic and phenotypic spectrums and refining treatment strategies. Curr Opin Neurol 32:696–703
Ramdas S, Beeson D (2021) Congenital myasthenic syndromes: where do we go from here? Neuromuscul Disord 31:943–954
Natera-de Benito D, Topf A, Vilchez JJ, Gonzalez-Quereda L, Dominguez-Carral J, Diaz-Manera J, Ortez C, Bestue M, Gallano P, Dusl M et al (2017) Molecular characterization of congenital myasthenic syndromes in Spain. Neuromuscul Disord 27:1087–1098
Troha Gergeli A, Neubauer D, Golli T, Butenko T, Loboda T, Maver A, Osredkar D (2020) Prevalence and genetic subtypes of congenital myasthenic syndromes in the pediatric population of Slovenia. Eur J Paediatr Neurol 26:34–38
Kao JC, Milone M, Selcen D, Shen XM, Engel AG, Liewluck T (2018) Congenital myasthenic syndromes in adult neurology clinic: a long road to diagnosis and therapy. Neurology 91:e1770–e1777
Lee M, Beeson D, Palace J (2018) Therapeutic strategies for congenital myasthenic syndromes. Ann N Y Acad Sci 1412:129–136
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E et al (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17:405–424
Fokkema IF, Taschner PE, Schaafsma GC, Celli J, Laros JF, den Dunnen JT (2011) LOVD v2.0: the next generation in gene variant databases. Hum Mutat 32:557–563
Burke G, Hiscock A, Klein A, Niks EH, Main M, Manzur AY, Ng J, de Vile C, Muntoni F, Beeson D, Robb S (2013) Salbutamol benefits children with congenital myasthenic syndrome due to DOK7 mutations. Neuromuscul Disord 23:170–175
Lorenzoni PJ, Scola RH, Kay CS, Filla L, Miranda AP, Pinheiro JM, Chaouch A, Lochmuller H, Werneck LC (2013) Salbutamol therapy in congenital myasthenic syndrome due to DOK7 mutation. J Neurol Sci 331:155–157
Parr JR, Andrew MJ, Finnis M, Beeson D, Vincent A, Jayawant S (2014) How common is childhood myasthenia? The UK incidence and prevalence of autoimmune and congenital myasthenia. Arch Dis Child 99:539–542
Lorenzoni PJ, Ducci RD, Arndt RC, Hrysay NMC, Fustes OJH, Topf A, Lochmuller H, Werneck LC, Kay CSK, Scola RH (2022) Congenital myasthenic syndrome in a cohort of patients with “double” seronegative myasthenia gravis. Arq Neuropsiquiatr 80:69–74
Gorokhova S, Cerino M, Mathieu Y, Courrier S, Desvignes JP, Salgado D, Beroud C, Krahn M, Bartoli M (2015) Comparing targeted exome and whole exome approaches for genetic diagnosis of neuromuscular disorders. Appl Transl Genom 7:26–31
Krenn M, Tomschik M, Rath J, Cetin H, Grisold A, Zulehner G, Milenkovic I, Stogmann E, Zimprich A, Strom TM et al (2020) Genotype-guided diagnostic reassessment after exome sequencing in neuromuscular disorders: experiences with a two-step approach. Eur J Neurol 27:51–61
Krenn M, Tomschik M, Wagner M, Zulehner G, Weng R, Rath J, Klotz S, Gelpi E, Bsteh G, Keritam O et al (2022) Clinico-genetic spectrum of limb-girdle muscular weakness in Austria: A multicentre cohort study. Eur J Neurol 29:1815–1824
Aharoni S, Sadeh M, Shapira Y, Edvardson S, Daana M, Dor-Wollman T, Mimouni-Bloch A, Halevy A, Cohen R, Sagie L et al (2017) Congenital myasthenic syndrome in Israel: genetic and clinical characterization. Neuromuscul Disord 27:136–140
Durmus H, Shen XM, Serdaroglu-Oflazer P, Kara B, Parman-Gulsen Y, Ozdemir C, Brengman J, Deymeer F, Engel AG (2018) Congenital myasthenic syndromes in Turkey: clinical clues and prognosis with long term follow-up. Neuromuscul Disord 28:315–322
Yis U, Becker K, Kurul SH, Uyanik G, Bayram E, Haliloglu G, Polat AI, Ayanoglu M, Okur D, Tosun AF et al (2017) Genetic landscape of congenital myasthenic syndromes from turkey: novel mutations and clinical insights. J Child Neurol 32:759–765
Zhao Y, Li Y, Bian Y, Yao S, Liu P, Yu M, Zhang W, Wang Z, Yuan Y (2021) Congenital myasthenic syndrome in China: genetic and myopathological characterization. Ann Clin Transl Neurol 8:898–907
Pattrakornkul N, Ittiwut C, Boonsimma P, Boonyapisit K, Khongkhatithum C, Sanmaneechai O, Suphapeetiporn K, Shotelersuk V (2020) Congenital myasthenic syndromes in the Thai population: clinical findings and novel mutations. Neuromuscul Disord 30:851–858
Abicht A, Stucka R, Karcagi V, Herczegfalvi A, Horvath R, Mortier W, Schara U, Ramaekers V, Jost W, Brunner J et al (1999) A common mutation (epsilon1267delG) in congenital myasthenic patients of Gypsy ethnic origin. Neurology 53:1564–1569
Natera-de Benito D, Dominguez-Carral J, Muelas N, Nascimento A, Ortez C, Jaijo T, Arteaga R, Colomer J, Vilchez JJ (2016) Phenotypic heterogeneity in two large Roma families with a congenital myasthenic syndrome due to CHRNE 1267delG mutation. A long-term follow-up. Neuromuscul Disord 26:789–795
Cetin H, Liu W, Cheung J, Cossins J, Vanhaesebrouck A, Maxwell S, Vincent A, Beeson D, Webster R (2019) Rapsyn facilitates recovery from desensitization in fetal and adult acetylcholine receptors expressed in a muscle cell line. J Physiol 597:3713–3725
Rodriguez Cruz PM, Palace J, Ramjattan H, Jayawant S, Robb SA, Beeson D (2015) Salbutamol and ephedrine in the treatment of severe AChR deficiency syndromes. Neurology 85:1043–1047
Shen Y, Wang B, Zheng X, Zhang W, Wu H, Hei M (2020) A neonate with musk congenital myasthenic syndrome presenting with refractory respiratory failure. Front Pediatr 8:166
Owen D, Topf A, Preethish-Kumar V, Lorenzoni PJ, Vroling B, Scola RH, Dias-Tosta E, Geraldo A, Polavarapu K, Nashi S et al (2018) Recessive variants of MuSK are associated with late onset CMS and predominant limb girdle weakness. Am J Med Genet A 176:1594–1601
Engel AG, Shen XM, Selcen D, Sine SM (2015) Congenital myasthenic syndromes: pathogenesis, diagnosis, and treatment. Lancet Neurol 14:420–434
Topf A, Johnson K, Bates A, Phillips L, Chao KR, England EM, Laricchia KM, Mullen T, Valkanas E, Xu L et al (2020) Sequential targeted exome sequencing of 1001 patients affected by unexplained limb-girdle weakness. Genet Med 22:1478–1488
Rodriguez Cruz PM, Belaya K, Basiri K, Sedghi M, Farrugia ME, Holton JL, Liu WW, Maxwell S, Petty R, Walls TJ et al (2016) Clinical features of the myasthenic syndrome arising from mutations in GMPPB. J Neurol Neurosurg Psychiatry 87:802–809
Belaya K, Rodriguez Cruz PM, Liu WW, Maxwell S, McGowan S, Farrugia ME, Petty R, Walls TJ, Sedghi M, Basiri K et al (2015) Mutations in GMPPB cause congenital myasthenic syndrome and bridge myasthenic disorders with dystroglycanopathies. Brain 138:2493–2504
Rodriguez Cruz PM, Palace J, Beeson D (2018) The neuromuscular junction and wide heterogeneity of congenital myasthenic syndromes. Int J Mol Sci 19:1677
Funding
Open access funding provided by Medical University of Vienna. No targeted funding.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare that they have no conflict of interest.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Krenn, M., Sener, M., Rath, J. et al. The clinical and molecular landscape of congenital myasthenic syndromes in Austria: a nationwide study. J Neurol 270, 909–916 (2023). https://doi.org/10.1007/s00415-022-11440-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-022-11440-0