
Abstract
Objective
We aim to evaluate 3-year effects of ocrelizumab (humanized anti-CD20 monoclonal antibody for the treatment of multiple sclerosis (MS)) on lymphocytes, neutrophils and immunoglobulins: (1) when compared with pre-infusion assessment; (2) over the course of treatment; and (3) possible clinical correlates of the observed immunological modifications.
Methods
This real-world observational cohort study has been conducted on prospectively collected data from 78 MS patients (mean age 47.8 ± 10.5 years; females 48.7%) commencing on ocrelizumab from 2018, with mean follow-up of 36.5 ± 6.8 months. Clinical data and blood samples were collected every three months. Total lymphocyte count and subpopulations were assessed on peripheral blood using flow cytometry. Serum immunoglobulins were evaluated with nephelometry.
Results
When compared with pre-infusion values, we observed reduction of total, CD19 and CD20 lymphocyte counts; however, after the first infusion, their levels remained substantially stable. Over time we observed a progressive reduction of CD8 lymphocytes, while no changes were observed for CD4, CD27, CD3CD27, and CD19CD27. After the first infusion, we observed reduction in IgG, which further decreased during the follow-up. Higher probability of EDSS progression was associated with reduced modulation of CD8 lymphocytes.
Interpretation
Ocrelizumab affects both humoral and cellular immune responses. Disability progression over the follow-up was associated with lower CD8 cytotoxic T-lymphocyte reduction. Changes in humoral response are immediate and sustained, while modulation of cellular immunity occurs progressively through regular re-treatment, and is related to clinical stability.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Disease-modifying treatments (DMTs) for multiple sclerosis (MS) imply the chronic modulation and/or depletion of humoral and/or cellular components of immunity. In particular, a humanized anti-CD20 monoclonal antibody (ocrelizumab) is approved for relapsing–remitting MS (RRMS), active secondary progressive MS (SPMS) and primary progressive MS (PPMS), being effective on both relapses and disability progression, as also confirmed in real-world studies [1,2,3]. The effect of ocrelizumab is thought to be mediated by the depletion of B cells which plays a crucial role in the pathogenesis of MS by producing pro-inflammatory cytokines, interacting with other inflammatory cells (i.e., macrophages, natural killer cells, and cytotoxic T lymphocytes), and stimulating antibody production [4,5,6,7,8]. Further, B cells also act as antigen-presenting cells being involved in the activation of T cells in secondary lymphoid tissues, and thus contributing to chronic non-resolving inflammation [9, 10].
Over six months after the first infusion of ocrelizumab, a reduction of B and T cell subpopulations (CD19, CD20, CD4, CD8) is already seen [11, 12]. Using 7-year clinical trial data, Hauser and colleagues confirmed the action of ocrelizumab on both B and T cells, and also showed a reduction of serum IgM, IgG and IgA, when compared with pre-ocrelizumab levels [13]. However, authors only investigated changes between pre-ocrelizumab immunological status and follow-up, while any further changes occurring after the first infusion could have implications on risk of infection and on vaccine response in patients on chronic treatment with ocrelizumab [14,15,16,17]. Also, the effect of ocrelizumab on the activation of B and T cells (from naïve to active) remained unexplored.
In the present 3-years real-world observational cohort study, we aim to evaluate: (1) changes in total lymphocyte count, lymphocyte subpopulations (CD19 and CD20 B cells, CD4 T helper cells, CD8 cytotoxic T cells, CD27 activated T cells, CD3CD27 naïve and central memory T cells, CD19CD27 memory B cells), neutrophils and immunoglobulins (IgG, IgA and IgM) when compared with pre-infusion assessment; (2) any further changes after the first infusion; and (3) possible clinical correlates of the observed immunological changes.
Methods
Study design and population
This real-world observational cohort study was conducted at the MS Unit of the Federico II University Hospital of Naples, Italy, on prospectively collected data from 2018 to 2021. The study was approved by the Federico II Ethics Committee (355/19). All patients signed informed consent authorizing the use of anonymized data in line with data protection regulation (GDPR EU2016/679). The present study was performed in accordance with good clinical practice and Declaration of Helsinki.
Inclusion criteria were: (1) patients with MS at first treatment with ocrelizumab from 2018 to 2021; (2) continued ocrelizumab treatment for at least 2 years (with < 4 weeks of flexibility in 6-month infusion intervals) [18, 19]; (3) availability of clinical and laboratory data at baseline (before first ocrelizumab infusion) and over at least 2 years. Exclusion criteria were: (1) age < 18 years; (2) pregnancy; (3) concomitant diseases (i.e., immunodeficiency diseases) or treatments (i.e., chemotherapy, immunosuppressive therapy) affecting the immune system.
First, ocrelizumab infusion was split into two 300 mg infusions (300 mg at week day 0 and 14). The date of the first infusion was counted as baseline. Following infusions were performed every 6 months, with < 4 weeks of flexibility to 6-month infusion intervals, which is deemed to be within regular infusion dosing [18, 19]. Clinical evaluations and blood sample collection were performed at least every three months over the follow-up period.
Laboratory variables
An aliquot (50 μL) of anti-coagulated ethylene-diamine-tetra-acetic acid (EDTA) whole fresh blood (within 12 h) was incubated at 4 °C for 30 min in the presence of appropriate amounts of monoclonal antibodies. The mixtures were then diluted 1:20 in ammonium chloride lysing solution, incubated at room temperature for 10 min and finally washed prior to flow cytometric analysis with the FACSCanto II flow cytometer (Becton Dickinson, San Jose, CA, USA). Samples were analyzed on FACSDiva software (BD Bioscience, San Jose, CA, USA). The following antigens were analyzed: CD4 PE (from BD San Diego, CA, USA), CD8 APCcy7 (from Beckman Coulter, Marseille Cedex 9, France), CD20 FITC (from BD San Diego, CA, USA), CD19 APC (from Beckman Coulter, Marseille Cedex 9, France), CD45 PerCP (from BD San Diego, CA, USA), CD27 HV500 (from BD San Diego, CA, USA), CD3 Pacific Blu (from Beckman Coulter, Marseille Cedex 9, France). B and T lymphocytes were gated on forward scatter (FSC) and side scatter (SSC) parameters, identifying 50,000 events. Gating strategy is shown in Fig. 1. The lowest level of detection was 10–4 (as such, zero corresponds to a level below 1/10,000 cells). For lymphocyte absolute count, we coupled cytometry to complete blood count on hematological counter (double platform). Laboratory procedures were performed in accordance with UK-NEQAS quality standards (https://ukneqas.org.uk/). For quantitative testing of serum immunoglobulins, we used nephelometry with a wavelength of 840 nm (BN™ II System, Siemens Healthcare, Erlangen, Germany), in accordance with manufacturer instructions. Reference curves were generated by multi-point calibration. Serum samples were automatically diluted 1:400 (IgG), 1:20 (IgA) or 1:5 in the low concentration assay (IgAs and IgMs). Reference values were derived from Italian population [20].

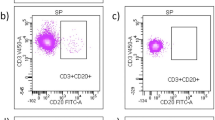
Flow cytometry gating strategy. The dot plots show the subsequent gates that were drawn to analyze the lymphocytes. SSC-Area and FSC-Area were plotted to exclude debris and to identify viable cells, among which the lymphocyte region was defined (a). Then, FSC-Area and FSC-Height were used to exclude doublets (b). From this region of lymphocyte singlets, plots were constructed to define lymphocyte subpopulations based on CD expression (e.g., CD3 + CD4 + in c, CD3 + CD8 + in d)
Demographic and clinical variables
At baseline, age, sex, disease duration (time from symptom onset to baseline visit), disease subtype (RRMS, SPMS, PPMS), and previous disease-modifying treatments (DMTs) were collected as per clinical practice. Disability was scored with the Expanded Disability Status Scale (EDSS), by certified examiners. EDSS progression at last available follow-up was defined as increase in EDSS by 1 point if baseline EDSS was 5.5 or lower, or increase in EDSS by 0.5 point if baseline EDSS was above 5.5 [21]. Relapses were recorded on the occasion of clinical consultations and infusions. Side effects were also collected on the occasion of clinical consultations and infusions; however, considering the retrospective nature of the study, we only referred to serious adverse events, which are less likely to be missed in clinical practice (defined as reaction that results in death, is life-threatening, requires hospitalization or prolongation of existing hospitalization, results in persistent or significant disability or incapacity, or is a birth defect).
Power calculation
Considering a normal distribution of variables to be analyzed in regression models, and a 30% effect size [13], a sample of 78 patients would be able to achieve 87% power with 5% α error.
Statistics
Study variables are presented as mean (standard deviation), median (range), or number (percent) as appropriate.
Changes in laboratory variables were explored using mixed-effect regression models including each laboratory variable, in turn, as dependent variable, and infusion as independent variable; covariates were age, sex, previous DMT, and follow-up duration. Patients were included in the models as random intercept to account for the hierarchical structure of the data. As unit of analysis was the infusion, in case of multiple outcome records corresponding to the same infusion, the statistical model averaged them. Our statistical references were laboratory variables at baseline (first infusion), and, then, after first infusion, to assess changes over time when compared with before ocrelizumab treatment and following its first infusion, respectively. Clinical correlates of immunological modifications (i.e., relapse occurrence, side effects and EDSS progression) were explored using logistic regression models including each clinical variable (EDSS progression, relapse occurrence, serious adverse events), in turn, as dependent variable, and each laboratory variable, in turn, as independent variable; covariates were age, sex, previous DMT, and follow-up duration.
Results are presented as coefficients (Coeff), odds ratio (OR), 95% confidence intervals (95%CI), and p-values. Statistical analyses were performed using Stata 17.0.
Results
We included 78 MS patients (age 47.8 ± 10.5 years; sex 48.7% females), with 36.5 ± 6.8 months of follow-up. 51.3% patients had 7 infusions of ocrelizumab (ranging from 5 to 8 infusions). Demographic, clinical and treatment features are reported in Table 1.
When compared with pre-infusion values, white blood cell count initially decreased after 2nd to 4th infusion, but then increased after 6th to 8th infusion; total lymphocyte count decreased after 1st and 2nd infusion; CD19 and CD20 lymphocytes decreased across all infusions; CD8 lymphocytes decreased after 3rd to 8th infusion; CD4/CD8 ratio increased after 2nd to 8th infusion; and IgG decreased after 1st to 8th infusion. No changes were observed for neutrophils, CD4 lymphocytes, CD27 lymphocytes, CD3CD27 lymphocytes, CD19CD27 lymphocytes, IgM and IgA (Table 2; Fig. 2).

Laboratory changes over time. Profile plots show changes in total lymphocyte count (a), CD19 lymphocytes (b), CD20 lymphocytes (c), CD4 lymphocytes (d), CD8 lymphocytes (e), CD27 lymphocytes (f), IgG (g), IgM (h) and IgA (i)
When compared with values after 1st infusion, white blood cell count initially decreased after 2nd to 5th infusion, but then increased after 6th to 8th infusion; neutrophils increased after 3rd to 8th infusion; CD19 and CD20 lymphocytes decreased after 2nd infusion; CD8 lymphocytes decreased after 3rd to 4th infusion; and IgG decreased after 2nd to 8th infusion. No changes were observed for total lymphocyte count, CD4 lymphocytes, CD4/CD8 ratio, CD27 lymphocytes, CD3CD27 lymphocytes, CD19CD27 lymphocytes, IgM and IgA (Table 2).
Over 36 months of follow-up, 26 patients (33.3%) had EDSS progression. The probability of EDSS progression was associated with higher CD8 (OR = 1.01; 95 CI = 1.00, 1.05; p = 0.02), and lower CD4/CD8 ratio (OR = 0.67; 95 CI = 0.46, 0.96; p = 0.03); no significant associations were found for WBC, neutrophils, total lymphocytes and other subsets (CD19, CD20, CD4, CD27, CD3CD27, and CD19CD27) (Table 3). We recorded two relapses, and eight cases of serious adverse events in seven patients; in particular, serious adverse events were infections (2 pulmonary infections, 1 herpes zoster infection, and 1 urinary tract infection), and malignancies (2 skin cancer, 1 bladder cancer, and 1 leukemia). Statistical models for relapses and side effects, separately, failed due to multicollinearity.
Discussion
Our study confirmed that ocrelizumab has wide range effects on both humoral and cellular immune response from its first infusion, and showed that most changes occurred from the first to the fourth infusions, thus pointing toward relative stability of the immunological profile afterward. Of note, cellular, but not humoral, immune effects of ocrelizumab were associated with disability progression, and occurred throughout the follow-up, suggesting that continuous treatment with ocrelizumab is needed to modulate the neurodegenerative aspects of MS.
In our study, we confirmed previous clinical trial extension results on white blood cells, which progressively decreased following the first infusion, but then came back to previous levels after year 2, and on neutrophils, which remained stable throughout the study, in the absence of cases of late-onset neutropenia [13, 22, 23]. Looking at B lymphocytes, CD19 and CD20 B lymphocytes were the first subpopulation hit by ocrelizumab, decreasing immediately after the first infusion and then remaining substantially stable. B lymphocytes play an important role in MS pathology in the context of both relapsing and progressive MS [24,25,26], promoting injury through direct activation of lymphocytes (CD8 cytotoxic T lymphocytes) [26,27,28,29], and other neuro-inflammatory cells (e.g., macrophages, microglia, astrocytes) [30, 31] or the production of pro-inflammatory cytokines [32]. In the central nervous system of MS patients, B cells are primarily located in the meninges and in the perivascular spaces, where they contribute to chronic aberrant compartmentalized inflammation [28, 33]. In animal models, treatment with anti-CD20 monoclonal antibodies eliminates CD20 B lymphocytes and disrupts B cell aggregates [34].
Antibody production is strictly dependent on B-mediated humoral response, and can directly contribute to MS inflammatory changes [35,36,37]. Accordingly, our results showed a reduction of IgG from the first infusion, alongside B lymphocytes, with progressive decrease over infusions, as already shown by clinical trial extension [13]. While the low levels of IgG have raised concerns on infectious risk and vaccine response in MS [15,16,17, 38], our study showed stable IgM levels and CD19CD27 B lymphocytes. In particular, CD19CD27 B lymphocytes in the peripheral blood are active B cells, but share the same antigens with plasma cells, located within lymphoid tissues and hereby producing immunoglobulins [39]. Taken together, these observations indirectly suggest that, while CD20 B lymphocytes are depleted, plasma cells remain unaffected, with normal possibility to mount humoral immune response against new antigens (IgM) [25], as preliminary shown in some cases [38].
Looking at T lymphocyte subpopulations, CD4 T lymphocytes remained substantially stable as previously showed in our feeder study [12], and also in clinical trial extension results [17]. On the contrary, CD8 T lymphocytes progressively decreased over infusions, as shown by regression coefficients and also confirmed by a concomitant drop of the CD4/CD8 ratio. However, we did not observe changes in CD27 activated T lymphocytes and CD3CD27 naïve cells (CD45RO−) and central memory T lymphocytes (CD45RO +), suggesting that ocrelizumab leaves unaffected the possibility to mount a cellular immune response while specifically targeting the aberrant immune response mediated by CD8 cytotoxic T lymphocytes. In keep with this, we found an association between increased probability of EDSS progression and reduced ocrelizumab-mediated modulation (i.e., higher levels) of CD8 T lymphocytes. While we need to acknowledge small effect size in this statistical model, this association is underlined by strong biological plausibility. Indeed, CD8 T lymphocytes directly contribute to non-resolving inflammation within the central nervous system, ultimately leading to demyelination and axon loss in both relapsing and progressive MS [30]. In particular, in progressive MS, inflammatory activity and demyelination is associated with infiltrates of tissue-resident CD8 cytotoxic T cells [40]. As such, the effect of ocrelizumab does not only depend on depletion of B lymphocytes, which is early event and substantially stable after the second infusion (using 6-months interval dosing), but also of CD8 cytotoxic T lymphocytes, which, on the contrary, occurs progressively during the course of treatments. Thus, while extending ocrelizumab dosing interval following B lymphocyte counts could be effective on the inflammatory aspects of MS (i.e., relapses, MRI lesions) [19], CD8 cytotoxic T lymphocytes progressively decrease during treatment and would very likely require regular infusions to be constantly modulated, with subsequent effect on disability progression [18]. In line with this, in a murine model, single administration of anti-CD20 monoclonal antibody was immediately effective on B cell aggregates in the absence of changes in other inflammatory cells [41].
Unfortunately, we were unable to provide more thoughtful insights on other clinical variables. We recorded relapses, which were too few to be analyzed statistically and correlated to laboratory variables. The low rate of relapses could be due to the strong and immediate anti-inflammatory effect of ocrelizumab [42], but also to the inclusion of progressive patients, representing about 50% of the population. We only had eight cases of serious adverse events (four infections, and four malignancies), whom statistical models failed due to the small number of observations. This could be due to the inclusion of serious adverse events only, the retrospective nature of the study, and/or the implementation of a program for infection screening and prophylaxis, which proved effective in reducing infections in patients receiving anti-CD20 agents for MS [43].
Additional limitations include the lack of more detailed clinical (e.g., walking and hand dexterity tests), and MRI (e.g., lesion load, atrophy) outcome measures, which would have been helpful for improved population characterization and treatment effect estimation [44, 45], but unfortunately not routinely collected across the follow-up. Finally, we did not include CD45RO in the panel, and thus were unable to differentiate CD3 + CD27 + cells into naïve and central memory T lymphocytes.
In conclusion, we provided real-world evidence on the mechanisms of action of ocrelizumab, and confirmed its effect on both cellular and humoral immunity. While the effect on B lymphocytes and, subsequently, on immunoglobulins is well known and expected, the mechanisms by which ocrelizumab reduces CD8 cytotoxic T lymphocytes deserves to be further explored also in light of its association with disability progression.
References
Ellwardt E, Rolfes L, Klein J et al (2020) Ocrelizumab initiation in patients with MS: a multicenter observational study. Neurol Neuroimmunol Neuroinflamm 7:1–9. https://doi.org/10.1212/NXI.0000000000000719
Montalban X, Hauser SL, Kappos L et al (2017) Ocrelizumab versus placebo in primary progressive multiple sclerosis. N Engl J Med 376:209–220. https://doi.org/10.1056/NEJMoa1606468
Hauser SL, Bar-Or A, Comi G et al (2016) Ocrelizumab versus interferon β-1a in relapsing multiple sclerosis. N Engl J Med 376:221–234. https://doi.org/10.1056/nejmoa1601277
Archelos JJ, Storch MK, Hartung MH-P (2000) The role of B cells and autoantibodies in multiple sclerosis. Ann Neurol 47:694–706. https://doi.org/10.1016/j.autrev.2016.07.009
Bar-Or A, Fawaz L, Fan B et al (2010) Abnormal B-cell cytokine responses a trigger of T-cell-mediated disease in MS? Ann Neurol 67:452–461. https://doi.org/10.1002/ana.21939
Anderson DR, Grillo-Löpez A, Varns C et al (1997) Targeted anti-cancer therapy using rituximab, a chimaeric anti-CD20 antibody (IDEC-C2B8) in the treatment of non-Hodgkin’s B-cell lymphoma. Biochem Soc Trans 25:705–708. https://doi.org/10.1042/bst0250705
Clynes RA, Towers TL, Presta LG, Ravetch JV (2000) Inhibitory Fc receptors modulate in vivo cytoxicity against tumor targets. Nat Med 6:443–446. https://doi.org/10.1038/74704
Disanto G, Morahan JM, Barnett MH et al (2012) The evidence for a role of B cells in multiple sclerosis. Neurology 78:823–832. https://doi.org/10.1212/WNL.0b013e318249f6f0
Christensen JR, Börnsen L, Ratzer R et al (2013) Systemic inflammation in progressive multiple sclerosis involves follicular T-Helper, Th17- and activated B-cells and correlates with progression. PLoS One 8:1–11. https://doi.org/10.1371/journal.pone.0057820
Tangye SG, Ma CS, Brink R, Deenick EK (2013) The good, the bad and the ugly-T FH cells in human health and disease. Nat Rev Immunol 13:412–426. https://doi.org/10.1038/nri3447
Fernández-Velasco JI, Kuhle J, Monreal E et al (2021) Effect of ocrelizumab in blood leukocytes of patients with primary progressive MS. Neurol Neuroimmunol Neuroinflamm 8:1–10. https://doi.org/10.1212/NXI.0000000000000940
Capasso N, Nozzolillo A, Scalia G et al (2021) Ocrelizumab depletes T-lymphocytes more than rituximab in multiple sclerosis. Mult Scler Relat Disord. https://doi.org/10.1016/j.msard.2021.102802
Hauser SL, Kappos L, Montalban X et al (2021) Safety of ocrelizumab in patients with relapsing and primary progressive multiple sclerosis. Neurology 97(16):e1546–e1559
Zabalza A, Arrambide G, Tagliani P et al (2022) Humoral and cellular responses to SARS-CoV-2 in convalescent COVID-19 patients with multiple sclerosis. Neurol Neuroimmunol Neuroinflamm 9:e1143. https://doi.org/10.1212/nxi.0000000000001143
Baker D, MacDougall A, Kang AS, Schmierer K, Gavin Giovannoni RD (2021) Seroconversion following COVID-19 vaccination: Can we optimize protective response in CD20-treated individuals? Clin Exp Immunol. https://doi.org/10.1093/cei/uxab015
Tortorella C, Aiello A, Gasperini C et al (2022) Humoral- and T-cell–specific immune responses to SARS-CoV-2 mRNA vaccination in patients with MS using different disease-modifying therapies. Neurology 98:e541–e554. https://doi.org/10.1212/wnl.0000000000013108
Baker D, MacDougall A, Kang AS et al (2022) CD19 B cell repopulation after ocrelizumab, alemtuzumab and cladribine: implications for SARS-CoV-2 vaccinations in multiple sclerosis. Mult Scler Relat Disord 57:103448. https://doi.org/10.1016/j.msard.2021.103448
van Lierop ZYGJ, Toorop AA, van Ballegoij WJC et al (2021) Personalized B-cell tailored dosing of ocrelizumab in patients with multiple sclerosis during the COVID-19 pandemic. Mult Scler J. https://doi.org/10.1177/13524585211028833
Rolfes L, Pawlitzki M, Pfeuffer S et al (2021) Ocrelizumab extended interval dosing in multiple sclerosis in times of COVID-19. Neurol Neuroimmunol Neuroinflamm. https://doi.org/10.1212/NXI.0000000000001035
Santagostino A, Garbaccio G, Pistorio A, Bolis V, Camisasca G, Pagliaro PGM (1999) An Italian national multicenter study for the definition of reference ranges for normal values of peripheral blood lymphocyte subsets in healthy adults. Haematologica 84:499–504
Kalincik T, Cutter G, Spelman T et al (2015) Defining reliable disability outcomes in multiple sclerosis. Brain 138:3287–3298. https://doi.org/10.1093/brain/awv258
Cohen BA (2019) Late-onset neutropenia following ocrelizumab therapy for multiple sclerosis. Neurology 92:435–436. https://doi.org/10.1212/WNL.0000000000006924
Marrodan M, Laviano J, Oneto S et al (2021) Rituximab- and ocrelizumab-induced early- and late-onset neutropenia in a multiple sclerosis patient. Neurol Sci 42:3893–3895. https://doi.org/10.1007/s10072-021-05357-1
Graf J, Mares J, Barnett M et al (2021) Targeting B cells to modify MS, NMOSD, and MOGAD: part 1. Neurol Neuroimmunol Neuroinflamm 8:1–12. https://doi.org/10.1212/NXI.0000000000000918
Baker D, Jacobs BM, Gnanapavan S et al (2019) Plasma cell and B cell-targeted treatments for use in advanced multiple sclerosis. Mult Scler Relat Disord 35:19–25. https://doi.org/10.1016/j.msard.2019.06.030
Jelcic I, Al Nimer F, Wang J et al (2018) Memory B cells activate brain-homing, autoreactive CD4+ T cells in multiple sclerosis. Cell 175:85-100.e23. https://doi.org/10.1016/j.cell.2018.08.011
Machado-Santos J, Saji E, Tröscher AR et al (2018) The compartmentalized inflammatory response in the multiple sclerosis brain is composed of tissue-resident CD8+ T lymphocytes and B cells. Brain 141:2066–2082. https://doi.org/10.1093/brain/awy151
Reali C, Magliozzi R, Roncaroli F et al (2020) B cell rich meningeal inflammation associates with increased spinal cord pathology in multiple sclerosis. Brain Pathol 30:779–793. https://doi.org/10.1111/bpa.12841
Monaco S, Nicholas R, Reynolds R, Magliozzi R (2020) Intrathecal inflammation in progressive multiple sclerosis. Int J Mol Sci 21:1–11. https://doi.org/10.3390/ijms21218217
Moccia M, Haider L, Eshaghi A et al (2022) B cells in the CNS at postmortem are associated with worse outcome and cell types in multiple sclerosis. Neurol Neuroimmunol Neuroinflamm 9:1–12. https://doi.org/10.1212/NXI.0000000000001108
Jain RW, Yong VW (2021) B cells in central nervous system disease: diversity, locations and pathophysiology. Nat Rev Immunol. https://doi.org/10.1038/s41577-021-00652-6
Göbel K, Ruck T, Meuth SG (2018) Cytokine signaling in multiple sclerosis: lost in translation. Mult Scler J 24:432–439. https://doi.org/10.1177/1352458518763094
Choi SR, Howell OW, Carassiti D et al (2012) Meningeal inflammation plays a role in the pathology of primary progressive multiple sclerosis. Brain 135:2925–2937. https://doi.org/10.1093/brain/aws189
Roodselaar J, Zhou Y, Leppert D et al (2021) Anti-CD20 disrupts meningeal B-cell aggregates in a model of secondary progressive multiple sclerosis. Neurol Neuroimmunol Neuroinflamm. https://doi.org/10.1212/NXI.0000000000000975
Muñoz U, Sebal C, Escudero E et al (2021) Main role of antibodies in demyelination and axonal damage in multiple sclerosis. Cell Mol Neurobiol. https://doi.org/10.1007/s10571-021-01059-6
Sádaba MC, Tzartos J, Paíno C et al (2012) Axonal and oligodendrocyte-localized IgM and IgG deposits in MS lesions. J Neuroimmunol 247:86–94. https://doi.org/10.1016/j.jneuroim.2012.03.020
Genain CP, Cannella B, SLH & CSR (1999) Identification of autoantibodies associated with myelin damage in multiple sclerosis. Nat Med 5:1–6
Novi G, Ivaldi F, Sbragia E et al (2020) Ocrelizumab does not impair B- and T-cell responses to primary VZV infection in a patient with MS. Neurol Neuroimmunol Neuroinflamm. https://doi.org/10.1212/NXI.0000000000000695
Fecteau JF, Roy A, Néron S (2009) Peripheral blood CD27+ IgG+ B cells rapidly proliferate and differentiate into immunoglobulin-secreting cells after exposure to low CD154 interaction. Immunology. https://doi.org/10.1111/j.1365-2567.2008.02976.x
Fransen NL, Hsiao CC, Van Der Poel M et al (2020) Tissue-resident memory T cells invade the brain parenchyma in multiple sclerosis white matter lesions. Brain 143:1714–1730. https://doi.org/10.1093/brain/awaa117
Brand RM, Friedrich V, Diddens J, Pfaller M (2021) Anti-CD20 depletes meningeal B cells but does not halt the formation of meningeal ectopic lymphoid tissue. Neurol Neuroimmunol Neuroinflamm. https://doi.org/10.1212/NXI.0000000000001012
Barkhof F, Kappos L, Wolinsky JS et al (2019) Onset of clinical and MRI efficacy of ocrelizumab in relapsing multiple sclerosis. Neurology 93:e1778–e1786. https://doi.org/10.1212/WNL.0000000000008189
Zappulo E, Buonomo AR, Saccà F et al (2019) Incidence and predictive risk factors of infective events in patients with multiple sclerosis treated with agents targeting CD20 and CD52 surface antigens. Open Forum Infect Dis 6:1–9. https://doi.org/10.1093/ofid/ofz445
Moccia M, de Stefano N, Barkhof F (2017) Imaging outcome measures for progressive multiple sclerosis trials. Mult Scler 23:1614–1626. https://doi.org/10.1177/1352458517729456
Tur C, Moccia M, Barkhof F et al (2018) Assessing treatment outcomes in multiple sclerosis trials and in the clinical setting. Nat Rev Neurol 14:75–93. https://doi.org/10.1038/nrneurol.2017.171
Funding
Open access funding provided by Università degli Studi di Napoli Federico II within the CRUI-CARE Agreement. The present study received no specific funding; laboratory and clinical variables were acquired with standard procedures within the clinical practice.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
RL has received honoraria from Biogen, Merck, Novartis, Roche, and Teva. MP discloses travel/meeting expenses from Novartis, Roche and Merck, speaking honoraria from HEALTH&LIFE S.r.l. and honoraria for consulting services from Biogen. VBM has received research grants from the Italian MS Society, and Roche, and honoraria from Bayer, Biogen, Merck, Mylan, Novartis, Roche, Sanofi-Genzyme, and Teva. MM has received research grants from the ECTRIMS-MAGNIMS, the UK MS Society, and Merck, and honoraria from Biogen, Merck, Novartis and Roche. Other authors have nothing to disclose.
Additional information
Authors who completed the statistical analysis: Marcello Moccia and Raffaele Palladino.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Capasso, N., Palladino, R., Cerbone, V. et al. Ocrelizumab effect on humoral and cellular immunity in multiple sclerosis and its clinical correlates: a 3-year observational study. J Neurol 270, 272–282 (2023). https://doi.org/10.1007/s00415-022-11350-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-022-11350-1