
Abstract
Background
Hereditary transthyretin amyloidosis (ATTRv) is a rare, debilitating and fatal disease, mostly characterized by progressive axonal peripheral neuropathy. Diagnosis is still challenging and diagnostic delay in non-endemic area is about 3–4 years. The aim of this study was to arrange a clinical and electrophysiological score to select patients with axonal neuropathy that deserve screening for TTR mutation.
Methods
Thirty-five ATTRv patients and 55 patients with chronic idiopathic axonal polyneuropathy (CIAP) were retrospectively analyzed. Clinical and electrophysiological findings at first evaluation were collected. Based on significant results between the two groups, a compound (clinical and electrophysiological) score was arranged, and ROC analysis was performed to identify the ideal cut-off able to discriminate between the two groups.
Results
ATTRv patients presented a later age at onset, more frequent muscle weakness and carpal tunnel syndrome history. On the other hand, electrophysiological analysis showed that ATTRv patients had lower CMAP and SAP amplitude in all examined nerves. We arranged a compound score constituted by 7 total items, ranging from 0 to 12. ROC analysis showed an Area Under the Curve = 0.8655 and we set the cut-off ≥ 5 points to discriminate ATTRv patients with a sensitivity of 96.6% and a specificity of 63.6%.
Conclusion
Our study demonstrated that our compound score with cut-off ≥ 5 allows to discriminate ATTRv patients among subject affected by axonal polyneuropathy with a sensitivity > 95%. Thus, our compound score is a quick, easy and effective screening tool.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hereditary transthyretin-related amyloidosis (ATTRv; v for “variant”) is a multisystem disorder, caused by mutations in transthyretin (TTR) gene. It is an autosomal-dominant, debilitating, progressive and fatal disease. Mutated TTR tetramer is unstable, dissociates in misfolded monomers that accumulate in extracellular spaces forming oligomers and amyloid fibrils [1].
Transthyretin-mutated protein accumulates mainly in heart and peripheral nervous system (PNS), leading cardiomyopathy and progressive axonal peripheral neuropathy, respectively [2]. PNS involvement is the presenting complaint in most cases of ATTRv. Polyneuropathy is due to the accumulation of TTR fibrils in PNS causing an axonal length-dependent sensory–motor and autonomic neuropathy [3]. ATTRv is a progressive and invalidating neuropathy and over few years, patients accumulate disability, become wheelchair bound and bedridden, and ultimately die [2].
In the last decade, different treatments (tafamidis, diflunisal, patisiran, inotersen) have proved their efficacy in slowing progression of neuropathy [4,5,6] and cardiomyopathy [7]. These treatments drastically changed natural history of neuropathy, even though therapies are more efficacious if they are precociously administered [8]. Thus, early diagnosis is important to impact on natural history through the innovative disease modifying therapies.
Unfortunately, in non-endemic area, diagnosis can be delayed by 3–4 years [2] since a correct diagnosis is challenging for clinician. Patients with ATTRv amyloidosis experience multiple neurological and/or cardiovascular testing and hospitalization prior to achieve the diagnosis [9]. In 32–74% of cases, patients receive misdiagnoses [10] and undergo inadequate or inappropriate treatments. Misdiagnoses are due to the lack of family history, the heterogeneous initial clinical manifestations and nerve conduction studies (NCS) that could show some demyelinating features [11] and pathological examinations (abdominal fat and sural nerve biopsy) negative for amyloid depositions [12].
Based on disease’s red flags, suspicion index of ATTRv amyloidosis was proposed to preciously recognize ATTRv and avoid diagnostic delay [13]. Suspicion index is based on the presence of a progressive polyneuropathy in addition to at least one red flag symptom suggestive of multi-systemic involvement. However, sometimes the demonstration of a progressive neuropathy requires follow-up evaluations, risking wasting time. Moreover, some red flags (e.g., cardiomyopathy or vitreous opacities) need specialist evaluations that could be often lacking during the first neurological evaluation [14].
The aim of this study was to optimize a clinical and electrophysiological score to select patients with axonal neuropathy worthy of TTR screening.
Methods
Thirty-five ATTRv patients and 55 patients with chronic idiopathic axonal polyneuropathy (CIAP) were retrospectively analyzed by two Italian third-level neuromuscular centers (University of Naples “Federico II” and Fondazione Policlinico Universitario A. Gemelli IRCCS of Rome). All patients underwent clinical assessment, nerve conduction study and Sanger sequencing of TTR gene. ATTRv patients were defined as patients with axonal polyneuropathy carrying TTR pathogenic variant. CIAP patients were defined as patients with at least of 6-month history of axonal sensory–motor polyneuropathy, resulted negative for TTR variant and for other causes of neuropathy through appropriate investigations [15]. In particular, all CIAP patients had no family history nor signs of hereditary neuropathy (e.i. pes cavus), no metabolic (diabetes, liver, renal or thyroid dysfunction), deficiency (vitamin B12, thiamine or pyridoxine deficiency), toxic (no history of exposure to alcohol, neurotoxic agents, drugs or chemotherapy), immunological (rheumatological, paraneoplastic or celiac disease), haematological (paraproteinemic syndrome as AL amyloidosis or POEMS) and infective (HBV, HCV, HIV) causes were identified as etiology of neuropathy.
As clinical data, we collected gender, age of onset, disease duration (time between age of onset and first evaluation) and the presence at first evaluation of (1) family history of polyneuropathy, (2) progressive disturbance in the last 6 months as perceived by patients, (3) muscle weakness, (4) positive and negative sensory symptoms (i.e., tingling and numbness), (5) autonomic symptoms (i.e., erectile dysfunction, diarrhea/constipation, nausea and vomiting, sweating abnormalities) (6) carpal tunnel syndrome (CTS) history. Moreover, we collected data about the walking impairment (0 = no walking difficulties; 1 = walking difficulties but independent; 2 = needing support; 3 = wheelchair bound).
As electrophysiological features, we collected amplitude of compound muscular action potential (CMAP; mV), distal motor latency (DML; ms) and motor nerve conduction velocity (MNCV; m/s) of the median, ulnar, tibial and peroneal nerves. Moreover, we collected amplitude of sensory action potential (SAP; μV) and sensory nerve conduction velocity (SNCV; m/s) of median, ulnar, peroneal superficial and sural nerves. Moreover, SAP and CMAP amplitude values were categorized in normal (0), reduced (1) and absent (2) according to the normal value of each center.
Statistical analysis
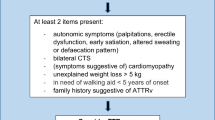
Descriptive statistics were based on mean ± standard deviation in the case of continuous variables and on frequencies (percentage) in the case of categorical variables. Statistical differences between ATTRv and CIAP groups were performed through Pearson's chi-squared test for categorical variables and Student’s T test for continuous variables. P values less than 0.05 were deemed as statistically significant. Based on the significant difference between two groups, a compound (clinical and electrophysiological) score (Fig. 1) was arranged (ranging from 0 to 12) assigning the highest scores to each variable that were more frequently abnormal in ATTRv patients as shown by the comparison analysis between the two groups. The score was constituted by 7 total items: motor symptoms (0 = none, 1 = present), CTS history (0–1), Median SAP (0 = normal, 1 = reduced; 2 = absent), Ulnar SAP (0–2), Median CMAP (0–2), Ulnar CMAP (0–2) and Tibial CMAP (0–2). The receiving operating characteristics (ROC) analyses were used to discriminate groups using the total score. To test the difference between ATTRv and CIAP patients with short disease duration, we performed a sub-analysis on the patients with disease duration ≤ 2 years through Student T test. All analyses were performed using STATA statistical software, version 13.

Composite clinical and electrophysiological score. CTS carpal tunnel syndrome, SAP sensory action potential, CMAP compound motor action potential
Results
Clinical and electrophysiological findings were summarized in Table 1.
Clinical data analysis showed that ATTRv group had more frequently motor symptoms (p = 0.002) and CTS history (p = 0.001) respect CIAP patients. Moreover, patients carrying a TTR variant had a later age of onset respect patients with idiopathic neuropathy (64.3 ± 9.9 vs 58.2 ± 11.2; p = 0.011). Conversely, no other clinical differences were found between two groups (gender, disease duration, progressive disease, sensory and autonomic symptoms, walking impairment) (Table 1).
Electrophysiological findings analysis showed that ATTRv patients had a more reduced amplitude of SAP and CMAP in all examined nerves (p < 0.05) (Table 2). In detail, ATTRv group presented more frequently absent CMAP in tibial nerve (47% vs 29%), SAP in median (75% vs 24%) and ulnar (54% vs 26%) nerves, and more frequently reduced CMAP in median (75% vs 26%) and ulnar (82% vs 26%) CMAP (Table 1). Moreover, significant differences between two groups were MNCV across the elbow in the ulnar nerve and DML of peroneal nerve (p < 0.05) (Table 2). Using ROC analysis, we established that the total score that best separated ATTRv patients from CIAP was a value ≥ 5 (AUC = 0.86, Fig. 2) with a sensitivity of 96.6% and a specificity of 63.6%. In particular, in our cohort, a total score ≥ 5 points was present in 96.6% ATTRv patients and in 36.4% CIAP patients. Lastly, the difference between the two groups with disease duration ≤ 2 years showed that the ATTRv patients had a greater score (11 patients; 7.4 + 1.2) respect to the CIAP patients (24 patients; 4 + 3.1) (p < 0.001).

ROC analysis of composite score. ROC analysis of composite score in patients with ATTRv and CIAP patients showing an area under the curve (AUC) of 0.8655
Discussion
The neuropathy in ATTRv patients represents one of the most disabling and progressive conditions and sometimes electrophysiological findings can misinterpreted by clinician although the neuropathy is due to a primary axonal degeneration [11]. Our study aimed to mark peculiar clinical and electrophysiological characteristics which can help clinicians to suspect ATTRv among patients with axonal polyneuropathy.
Clinical findings showed that ATTRv patients referred more frequently motor symptoms (86% vs 54%) and CTS history (57% vs 24%) respect patients with CIAP. These results confirmed that ATTRv patients have a precocious involvement of motor system respect CIAP patients which complain especially sensory symptoms [16]. In fact, although statistical analysis missed to reach significance, only 50% (vs 70%) of our ATTRv patients can walk independently.
Conversely, in our cohort, autonomic symptoms did not represent a discriminative feature. In fact, our population was constituted by late-onset ATTRv patients [17] in which autonomic involvement at the disease onset is often subtle and undetected if not adequately investigated and become clinically prominent in advanced stage [1]. Another possible explanation for this lacking significant was that the autonomic dysfunction was detect through the reported symptoms during patient’s interview and not by appropriate questionnaire or specific instrumental test (e.g., tilt test, skin sympathetic response).
Lastly, disease progressivity unexpectedly did not differ between ATTRv and CIAP patients. The reason of this result could be due to the retrospectively design of the study. In fact, the disease progression was considered as perceived by patients at first evaluation and not by follow-up evaluations. As occurs in other conditions, in which the patient perception of health status does not parallel functional and disability measures [18], CIAP patients could perceive their neuropathy as progressive disease as well.
Electrophysiological findings showed that ATTRv patients, although they had the same disease duration of CIAP patients, had a greater reduction of amplitude of potentials in all nerves with a more frequently absence of potential at lower limbs and reduction at upper limbs. Our data confirm that axonal degeneration is the primary patho-mechanism in ATTRv disease and suggests early involvement of upper limbs nerves respect to CIAP patients in which simultaneous development of upper and lower extremity rarely occurs [19]. Although ATTRv neuropathy is defined as length-dependent, the early involvement of upper limb nerves could be the expression of a ganglionopathic pattern damage [20]. In ATTRv amyloid, accumulation starts in dorsal root ganglia and nerve roots and afterward amyloid deposits spread through a proximo-distal gradient over time [11].
Moreover, our study confirmed the role of CTS history as red flag of ATTRv as it could precede by several years the onset of polyneuropathy [21]. Of interest, our electrophysiological data findings in symptomatic ATTRv patients did not show a greater SNCV slowing and DML prolongation in median nerve respect to CIAP patients as expected. Our findings suggest that in a patient with polyneuropathy, the clinical history of CTS is important in the suspicion of ATTR rather than electrophysiological findings of CTS. In fact, the CTS physiopathology in ATTRv patient seems to have a peculiar behavior respect the idiopathic CTS. The ultrasound results showed that CTS in ATTRv is characterized by a peculiar mismatch between electrophysiological and ultrasound abnormalities of the median nerve at wrist, differentiating from idiopathic CTS, in which ultrasound findings mirror electrophysiology severity [22]. Altogether, we can suppose that the entrapment injury of the median nerve can occur in pre-symptomatic stage through the deposition of amyloid in the carpal ligament [23], but contextually, there is already a systemic damage of nerves that starts proximally [24].
Based on this peculiar characteristic of ATTRv patients, we arranged a compound clinical and electrophysiological score. A total score ≥ 5 allows to identify with a sensitivity over 95% ATTRv patients among subject with chronic axonal polyneuropathy. We have decided to set a cut-off with higher sensitivity respect the specificity, since the score was arranged as a screening tool. We opted to have more false positive respect to lose the possibility to detect an ATTRv patient, given that the disease is debilitating but curable especially in the early stage [25]. Moreover, the score is able to discriminate ATTRv also in patients with short disease duration (≤ 2 years), strengthening that the ATTRv leads to a severe neuropathy since the first year of disease.
The score emphasizes the predominant motor involvement in ATTRv disease, respective to CIAP, and this difference might cause an imbalance between the two populations. However, in front of a single patient with neuropathy, the severity of motor involvement can be difficult to valorize. In fact, CIAP still represents a common misdiagnosis for ATTRv patients [26]. Our study identified the principal differences between the two groups and valorized them in a compound score which can help clinician through a specific cut-off to recognize patients deserving TTR genetic analysis. Moreover, the score is easy to perform during clinical and/or electrophysiological examination and it does not require other special exam (e.g., cardiac imaging or ophthalmological examination to detect cardiomyopathy and vitreous corpus respectively).
If the score can influence marginally the choice to perform genetic analysis in a third-level center, where TTR genetic test is easily accessible, conversely it can help physician in primary centers, where the patients are evaluated for the first time and genetic test can be difficult to perform. Especially in this context, the application of the compound score in patients with sensory–motor neuropathy may have a major role, representing a first screening tool to drive the choice of referring patients in an amyloidosis center, avoiding wasting time and therefore shortening the time to reach a correct diagnosis.
In conclusion, although the small number of patients and the needing to confirm the result through a prospective study, our study suggests that some clinical and electrophysiological features in sensory–motor neuropathy patients, such as CTS history, relevant motor impairment and a greater axonal loss with a precocious upper limb involvement, should alert the clinicians [27]. Applying our score to patients with sensory–motor neuropathy, clinicians could easily establish if patients deserve TTR genetic analysis. This score can be easily performed also during electrophysiological evaluation and can be extremely useful in those area where genetic analysis is not easily accessible.
Change history
21 July 2022
Missing Open Access funding information has been added in the Funding Note
References
Manganelli F, Fabrizi GM, Luigetti M, Mandich P, Mazzeo A, Pareyson D (2020) Hereditary transthyretin amyloidosis overview. Neurol Sci. https://doi.org/10.1007/s10072-020-04889-2 ((Epub ahead of print. PMID: 33188616))
Adams D, Koike H, Slama M, Coelho T (2019) Hereditary transthyretin amyloidosis: a model of medical progress for a fatal disease. Nat Rev Neurol 15(7):387–404. https://doi.org/10.1038/s41582-019-0210-4 (Epub 2019 Jun 17 PMID: 31209302)
Luigetti M, Romozzi M, Bisogni G, Cardellini D, Cavallaro T, Di Paolantonio A, Fabrizi GM, Fenu S, Gentile L, Grandis M, Marucci G, Massucco S, Mazzeo A, Pareyson D, Romano A, Russo M, Schenone A, Tagliapietra M, Tozza S, Vita G, Sabatelli M (2020) hATTR pathology: nerve biopsy results from italian referral centers. Brain Sci 10(11):780. https://doi.org/10.3390/brainsci10110780 ((PMID:33114611; PMCID:PMC7692609))
Coelho T, Maia LF, Martins da Silva A, Waddington Cruz M, Planté-Bordeneuve V, Lozeron P, Suhr OB, Campistol JM, Conceição IM, Schmidt HH, Trigo P, Kelly JW, Labaudinière R, Chan J, Packman J, Wilson A, Grogan DR (2012) Tafamidis for transthyretin familial amyloid polyneuropathy: a randomized, controlled trial. Neurology 79(8):785–792. https://doi.org/10.1212/WNL.0b013e3182661eb1 (PMID: 22843282; PMCID: PMC4098875)
Adams D, Gonzalez-Duarte A, O’Riordan WD, Yang CC, Ueda M, Kristen AV, Tournev I, Schmidt HH, Coelho T, Berk JL, Lin KP, Vita G, Attarian S, Planté-Bordeneuve V, Mezei MM, Campistol JM, Buades J, Brannagan TH 3rd, Kim BJ, Oh J, Parman Y, Sekijima Y, Hawkins PN, Solomon SD, Polydefkis M, Dyck PJ, Gandhi PJ, Goyal S, Chen J, Strahs AL, Nochur SV, Sweetser MT, Garg PP, Vaishnaw AK, Gollob JA, Suhr OB (2018) Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N Engl J Med 379(1):11–21. https://doi.org/10.1056/NEJMoa1716153 (PMID: 29972753)
Benson MD, Waddington-Cruz M, Berk JL, Polydefkis M, Dyck PJ, Wang AK, Planté-Bordeneuve V, Barroso FA, Merlini G, Obici L, Scheinberg M, Brannagan TH 3rd, Litchy WJ, Whelan C, Drachman BM, Adams D, Heitner SB, Conceição I, Schmidt HH, Vita G, Campistol JM, Gamez J, Gorevic PD, Gane E, Shah AM, Solomon SD, Monia BP, Hughes SG, Kwoh TJ, McEvoy BW, Jung SW, Baker BF, Ackermann EJ, Gertz MA, Coelho T (2018) Inotersen treatment for patients with hereditary transthyretin amyloidosis. N Engl J Med 379(1):22–31. https://doi.org/10.1056/NEJMoa1716793 (PMID: 29972757)
Maurer MS, Schwartz JH, Gundapaneni B, Elliott PM, Merlini G, Waddington-Cruz M, Kristen AV, Grogan M, Witteles R, Damy T, Drachman BM, Shah SJ, Hanna M, Judge DP, Barsdorf AI, Huber P, Patterson TA, Riley S, Schumacher J, Stewart M, Sultan MB, Rapezzi C, ATTR-ACT Study Investigators (2018) ATTR-ACT study investigators tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med 379(11):1007–1016. https://doi.org/10.1056/NEJMoa1805689 (Epub 2018 Aug 27 PMID: 30145929)
Sekijima Y, Ueda M, Koike H, Misawa S, Ishii T, Ando Y (2018) Diagnosis and management of transthyretin familial amyloid polyneuropathy in Japan: red-flag symptom clusters and treatment algorithm. Orphanet J Rare Dis 13(1):6. https://doi.org/10.1186/s13023-017-0726-x (Erratum. In: Orphanet J Rare Dis. 2019 May 21; 14(1): 111. PMID:29343286; PMCID:PMC5773042)
Vera-Llonch M, Reddy SR, Chang E, Tarbox MH, Pollock M (2021) The patient journey toward a diagnosis of hereditary transthyretin (ATTRv) amyloidosis. Orphanet J Rare Dis 16(1):25. https://doi.org/10.1186/s13023-020-01623-1.PMID:33430941;PMCID:PMC7798313
Cortese A, Vegezzi E, Lozza A, Alfonsi E, Montini A, Moglia A, Merlini G, Obici L (2017) Diagnostic challenges in hereditary transthyretin amyloidosis with polyneuropathy: avoiding misdiagnosis of a treatable hereditary neuropathy. J Neurol Neurosurg Psychiatry 88(5):457–458. https://doi.org/10.1136/jnnp-2016-315262 (Epub 2017 Feb 10. PMID: 28188196; PMCID: PMC5529976)
Tozza S, Severi D, Spina E, Iovino A, Aruta F, Ruggiero L, Dubbioso R, Iodice R, Nolano M, Manganelli F (2021) The neuropathy in hereditary transthyretin amyloidosis: a narrative review. J Peripher Nerv Syst 26(2):155–159. https://doi.org/10.1111/jns.12451 (Epub 2021 May 11 PMID: 33960565)
Luigetti M, Conte A, Del Grande A, Bisogni G, Madia F, Lo Monaco M, Laurenti L, Obici L, Merlini G, Sabatelli M (2013) TTR-related amyloid neuropathy: clinical, electrophysiological and pathological findings in 15 unrelated patients. Neurol Sci 34(7):1057–1063. https://doi.org/10.1007/s10072-012-1105-y (Epub 2012 May 17 PMID: 22592564)
Adams D, Ando Y, Beirão JM, Coelho T, Gertz MA, Gillmore JD, Hawkins PN, Lousada I, Suhr OB, Merlini G (2021) Expert consensus recommendations to improve diagnosis of ATTR amyloidosis with polyneuropathy. J Neurol. 268(6):2109–2122. https://doi.org/10.1007/s00415-019-09688-0Epub 2020 Jan 6. PMID: 31907599; PMCID: PMC8179912
Luigetti M, Romano A, Di Paolantonio A, Bisogni G, Sabatelli M (2020) Diagnosis and treatment of hereditary transthyretin amyloidosis (hATTR) polyneuropathy: current perspectives on improving patient care. Ther Clin Risk Manag 21(16):109–123. https://doi.org/10.2147/TCRM.S219979.PMID:32110029;PMCID:PMC7041433
Zis P, Sarrigiannis PG, Rao DG, Hewamadduma C, Hadjivassiliou M (2016) Chronic idiopathic axonal polyneuropathy: a systematic review. J Neurol 263(10):1903–1910. https://doi.org/10.1007/s00415-016-8082-7 (Epub 2016 Mar 9 PMID: 26961897)
Singer MA, Vernino SA, Wolfe GI (2012) Idiopathic neuropathy: new paradigms, new promise. J Peripher Nerv Syst 17(Suppl 2):43–49. https://doi.org/10.1111/j.1529-8027.2012.00395.x (PMID: 22548623)
Russo M, Obici L, Bartolomei I, Cappelli F, Luigetti M, Fenu S, Cavallaro T, Chiappini MG, Gemelli C, Pradotto LG, Manganelli F, Leonardi L, My F, Sampaolo S, Briani C, Gentile L, Stancanelli C, Di Buduo E, Pacciolla P, Salvi F, Casagrande S, Bisogni G, Calabrese D, Vanoli F, Di Iorio G, Antonini G, Santoro L, Mauro A, Grandis M, Di Girolamo M, Fabrizi GM, Pareyson D, Sabatelli M, Perfetto F, Rapezzi C, Merlini G, Mazzeo A, Vita G (2020) ATTRv amyloidosis Italian registry: clinical and epidemiological data. Amyloid 27(4):259–265. https://doi.org/10.1080/13506129.2020.1794807 (Epub 2020 Jul 22 PMID: 32696671)
Tozza S, Bruzzese D, Pisciotta C, Iodice R, Esposito M, Dubbioso R, Ruggiero L, Topa A, Spina E, Santoro L, Manganelli F (2018) Motor performance deterioration accelerates after 50 years of age in Charcot-Marie-Tooth type 1A patients. Eur J Neurol 25(2):301–306. https://doi.org/10.1111/ene.13494 (Epub 2017 Dec 14 PMID: 29053907)
Wolfe GI, Baker NS, Amato AA, Jackson CE, Nations SP, Saperstein DS, Cha CH, Katz JS, Bryan WW, Barohn RJ (1999) Chronic cryptogenic sensory polyneuropathy: clinical and laboratory characteristics. Arch Neurol 56(5):540-7. https://doi.org/10.1001/archneur.56.5.540PMID: 10328248
Théaudin M, Lozeron P, Algalarrondo V, Lacroix C, Cauquil C, Labeyrie C, Slama MS, Adam C, Guiochon-Mantel A, Adams D; French FAP Network (CORNAMYL) Study Group (2019) Upper limb onset of hereditary transthyretin amyloidosis is common in non-endemic areas. Eur J Neurol 26(3):497-e36. https://doi.org/10.1111/ene.13845. Epub 2018 Dec 11. PMID: 30350904.
Karam C, Dimitrova D, Christ M, Heitner SB (2019) Carpal tunnel syndrome and associated symptoms as first manifestation of hATTR amyloidosis. Neurol Clin Pract 9(4):309–313. https://doi.org/10.1212/CPJ.0000000000000640.PMID:31583185;PMCID:PMC6745748
Salvalaggio A, Coraci D, Cacciavillani M, Obici L, Mazzeo A, Luigetti M, Pastorelli F, Grandis M, Cavallaro T, Bisogni G, Lozza A, Gemelli C, Gentile L, Ermani M, Fabrizi GM, Plasmati R, Campagnolo M, Castellani F, Gasparotti R, Martinoli C, Padua L, Briani C (2021) Nerve ultrasound in hereditary transthyretin amyloidosis: red flags and possible progression biomarkers. J Neurol 268(1):189–198. https://doi.org/10.1007/s00415-020-10127-8. Epub 2020 Aug 4. PMID: 32749600; PMCID: PMC7815618.
Samões R, Taipa R, Valdrez K, Gonçalves I, Melo Pires M, Martins da Silva A, Coelho T (2017) Amyloid detection in the transverse carpal ligament of patients with hereditary ATTR V30M amyloidosis and carpal tunnel syndrome. Amyloid. 24(2):73–77. https://doi.org/10.1080/13506129.2017.1313222. Epub 2017 Apr 16. PMID: 28413892
Koike H, Morozumi S, Kawagashira Y, Iijima M, Yamamoto M, Hattori N, Tanaka F, Nakamura T, Hirayama M, Ando Y, Ikeda S, Sobue G (2009) The significance of carpal tunnel syndrome in transthyretin Val30Met familial amyloid polyneuropathy. Amyloid 16(3):142–148. https://doi.org/10.1080/13506120903094074 (PMID: 19626479)
Herman C (2006) What makes a screening exam “good”? Virtual Mentor 8(1):34–37. https://doi.org/10.1001/virtualmentor.2006.8.1.cprl1-0601 (PMID: 23232314)
Gertz M, Adams D, Ando Y, Beirão JM, Bokhari S, Coelho T, Comenzo RL, Damy T, Dorbala S, Drachman BM, Fontana M, Gillmore JD, Grogan M, Hawkins PN, Lousada I, Kristen AV, Ruberg FL, Suhr OB, Maurer MS, Nativi-Nicolau J, Quarta CC, Rapezzi C, Witteles R, Merlini G (2020) Avoiding misdiagnosis: expert consensus recommendations for the suspicion and diagnosis of transthyretin amyloidosis for the general practitioner. BMC Fam Pract 21(1):198. https://doi.org/10.1186/s12875-020-01252-4 (PMID:32967612; PMCID: PMC7513485)
Ayrignac X, Viala K, Koutlidis RM, Taïeb G, Stojkovic T, Musset L, Léger JM, Fournier E, Maisonobe T, Bouche P (2013) Sensory chronic inflammatory demyelinating polyneuropathy: an under-recognized entity? Muscle Nerve 48(5):727–732. https://doi.org/10.1002/mus.23821 (Epub 2013 Aug 30 PMID: 23424105)
Acknowledgements
ST would like to thank Rachele Delogu and Vincenzo Pierri for collecting data.
Funding
Open access funding provided by Università degli Studi di Napoli Federico II within the CRUI-CARE Agreement.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare that they have no competing interests.
Ethics approval and informed consent
Ethical approval was waived by the local Ethics Committee of University of Naples “Federico II” in view of the retrospective nature of the study and all the procedures being performed were part of the routine care.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tozza, S., Severi, D., Spina, E. et al. A compound score to screen patients with hereditary transthyretin amyloidosis. J Neurol 269, 4281–4287 (2022). https://doi.org/10.1007/s00415-022-11056-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-022-11056-4