
Abstract
Background
We intended to clarify the phenotypic and molecular diversities of spinocerebellar ataxia type 2 (SCA2) in Japan.
Methods
DNA was extracted from the peripheral blood of 436 patients, including 126 patients with chronic neuropathy, 108 with amyotrophic lateral sclerosis, and 202 with cerebellar ataxia. We then PCR-amplified and sequenced the ATXN2 gene. The biopsied sural nerves of mutation-positive patients were subjected to light-microscopic and electron-microscopic analyses. Transfection analyses were performed using a Schwann cell line, IMS32.
Results
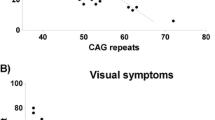
We found PCR-amplified products potentially corresponding to expanded CAG repeats in four patients. Two patients in the chronic neuropathy group had a full repeat expansion or an intermediate expansion (39 or 32 repeats), without limb ataxia. The sural nerve biopsy findings of the two patients included axonal neuropathy and mixed neuropathy (axonal changes with demyelination). Schwann cells harbored either cytoplasmic or nuclear inclusions on electron microscopic examination. Both patients recently exhibited pyramidal signs. In the third patient in the cerebellar ataxia group, we identified a novel 21-base duplication mutation near 22 CAG repeats (c.432_452dup). The transfection study revealed that the 21-base-duplication mutant Ataxin-2 proteins aggregated in IMS32 and rendered cells susceptible to oxidative stress, similar to a CAG-expanded mutant. The fourth patient, with 41 repeats, had ataxia and spasticity. The two patients with cerebellar ataxia also had peripheral neuropathy.
Conclusions
Patients with expanded CAG repeats can exhibit a neuropathy-dominant phenotype not described previously. The novel 21-base-duplication mutant seems to share the aggregation properties of polyglutamine-expanded mutants.




Similar content being viewed by others


References
Sanpei K, Takano H, Igarashi S, Sato T, Oyake M, Sasaki H, Wakisaka A, Tashiro K, Ishida Y, Ikeuchi T, Koide R, Saito M, Sato A, Tanaka T, Hanyu S, Takiyama Y, Nishizawa M, Shimizu N, Nomura Y, Segawa M, Iwabuchi K, Eguchi I, Tanaka H, Takahashi H, Tsuji S (1996) Identification of the spinocerebellar ataxia type 2 gene using a direct identification of repeat expansion and cloning technique, DIRECT. Nat Genet 14:277–284
Naruse H, Matsukawa T, Ishiura H, Mitsui J, Takahashi Y, Takano H, Goto J, Toda T, Tsuji S (2019) Association of ATXN2 intermediate-length CAG repeats with amyotrophic lateral sclerosis correlates with the distributions of normal CAG repeat alleles among individual ethnic populations. Neurogenetics 20:65–71
Elden AC, Kim HJ, Hart MP, Chen-Plotkin AS, Johnson BS, Fang X, Armakola M, Geser F, Greene R, Lu MM, Padmanabhan A, Clay-Falcone D, McCluskey L, Elman L, Juhr D, Gruber PJ, Rub U, Auburger G, Trojanowski JQ, Lee VM, Van Deerlin VM, Bonini NM, Gitler AD (2010) Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 466:1069–1075
Gwinn-Hardy K, Chen JY, Liu HC, Liu TY, Boss M, Seltzer W, Adam A, Singleton A, Koroshetz W, Waters C, Hardy J, Farrer M (2000) Spinocerebellar ataxia type 2 with parkinsonism in ethnic Chinese. Neurology 55:800–805
Baumer D, East SZ, Tseu B, Zeman A, Hilton D, Talbot K, Ansorge O (2014) FTLD-ALS of TDP-43 type and SCA2 in a family with a full ataxin-2 polyglutamine expansion. Acta Neuropathol 128:597–604
Tazen S, Figueroa K, Kwan JY, Goldman J, Hunt A, Sampson J, Gutmann L, Pulst SM, Mitsumoto H, Kuo SH (2013) Amyotrophic lateral sclerosis and spinocerebellar ataxia type 2 in a family with full CAG repeat expansions of ATXN2. JAMA Neurol 70:1302–1304
Perretti A, Santoro L, Lanzillo B, Filla A, De Michele G, Barbieri F, Martino G, Ragno M, Cocozza S, Caruso G (1996) Autosomal dominant cerebellar ataxia type I: multimodal electrophysiological study and comparison between SCA1 and SCA2 patients. J Neurol Sci 142:45–53
Filla A, De Michele G, Campanella G, Perretti A, Santoro L, Serlenga L, Ragno M, Calabrese O, Castaldo I, De Joanna G, Cocozza S (1996) Autosomal dominant cerebellar ataxia type I. Clinical and molecular study in 36 Italian families including a comparison between SCA1 and SCA2 phenotypes. J Neurol Sci 142:140–147
Malandrini A, Galli L, Villanova M, Palmeri S, Parrotta E, DeFalco D, Cappelli M, Grieco GS, Renieri A, Guazzi G (1998) CAG repeat expansion in an italian family with spinocerebellar ataxia type 2 (SCA2): a clinical and genetic study. Eur Neurol 40:164–168
Stezin A, Venkatesh SD, Thennarasu K, Purushottam M, Jain S, Yadav R, Pal PK (2018) Non-ataxic manifestations of Spinocerebellar ataxia-2, their determinants and predictors. J Neurol Sci 394:14–18
Higuchi Y, Hashiguchi A, Yuan J, Yoshimura A, Mitsui J, Ishiura H, Tanaka M, Ishihara S, Tanabe H, Nozuma S, Okamoto Y, Matsuura E, Ohkubo R, Inamizu S, Shiraishi W, Yamasaki R, Ohyagi Y, Kira J, Oya Y, Yabe H, Nishikawa N, Tobisawa S, Matsuda N, Masuda M, Kugimoto C, Fukushima K, Yano S, Yoshimura J, Doi K, Nakagawa M, Morishita S, Tsuji S, Takashima H (2016) Mutations in MME cause an autosomal-recessive Charcot-Marie-Tooth disease type 2. Ann Neurol 79:659–672
Hirano M, Nakamura Y, Saigoh K, Sakamoto H, Ueno S, Isono C, Mitsui Y, Kusunoki S (2015) VCP gene analyses in Japanese patients with sporadic amyotrophic lateral sclerosis identify a new mutation. Neurobiol Aging 36(1604):e1601-1606
Hirano M, Nakamura Y, Saigoh K, Sakamoto H, Ueno S, Isono C, Miyamoto K, Akamatsu M, Mitsui Y, Kusunoki S (2013) Mutations in the gene encoding p62 in Japanese patients with amyotrophic lateral sclerosis. Neurology 80:458–463
Hirano M, Samukawa M, Isono C, Saigoh K, Nakamura Y, Kusunoki S (2018) Noncoding repeat expansions for ALS in Japan are associated with the ATXN8OS gene. Neurol Genet 4:e252
Okazaki M, Suzuki H, Takahashi Y, Ishiura H, Goto J, Hirano M, Saigoh K, Nakamura Y, Naruse H, Mitsui J, Tsuji S, Kusunoki S (2017) Novel mutation in the SOD1 gene in a patient with early-onset, rapidly progressive amyotrophic lateral sclerosis. Neurol Clin Neurosci 5:189–191
Hirano M, Matsumura R, Nakamura Y, Saigoh K, Sakamoto H, Ueno S, Inoue H, Kusunoki S (2017) Unexpectedly mild phenotype in an ataxic family with a two-base deletion in the APTX gene. J Neurol Sci 378:75–79
Zhao Z, Hashiguchi A, Hu J, Sakiyama Y, Okamoto Y, Tokunaga S, Zhu L, Shen H, Takashima H (2012) Alanyl-tRNA synthetase mutation in a family with dominant distal hereditary motor neuropathy. Neurology 78:1644–1649
Oka N, Kawasaki T, Unuma T, Shigematsu K, Sugiyama H (2013) Different profiles of onion bulb in CIDP and CMT1A in relation to extracellular matrix. Clin Neuropathol 32:406–412
Watabe K, Fukuda T, Tanaka J, Honda H, Toyohara K, Sakai O (1995) Spontaneously immortalized adult mouse Schwann cells secrete autocrine and paracrine growth-promoting activities. J Neurosci Res 41:279–290
Linnemann C, Tezenas du Montcel S, Rakowicz M, Schmitz-Hubsch T, Szymanski S, Berciano J, van de Warrenburg BP, Pedersen K, Depondt C, Rola R, Klockgether T, Garcia A, Mutlu G, Schols L (2016) Peripheral Neuropathy in Spinocerebellar ataxia Type 1, 2, 3, and 6. Cerebellum 15:165–173
Schroder JM, Sommer C (1991) Mitochondrial abnormalities in human sural nerves: fine structural evaluation of cases with mitochondrial myopathy, hereditary and non-hereditary neuropathies, and review of the literature. Acta Neuropathol 82:471–482
Yasaki S, Tukamoto Y, Yuasa N, Ohnuki T, Yoshii F (2016) Possible coexisting autoimmunity in a variant case of familial amyotrophic lateral sclerosis associated with anti-GM2 immunoglobulin M antibody. Neurol Clin Neurosci 4:118–120
Berciano J, Ferrer I (2005) Glial cell cytoplasmic inclusions in SCA2 do not express alpha-synuclein. J Neurol 252:742–744
Ettle B, Schlachetzki JCM, Winkler J (2016) Oligodendroglia and myelin in neurodegenerative diseases: more than just bystanders? Mol Neurobiol 53:3046–3062
Adachi H, Katsuno M, Minamiyama M, Waza M, Sang C, Nakagomi Y, Kobayashi Y, Tanaka F, Doyu M, Inukai A, Yoshida M, Hashizume Y, Sobue G (2005) Widespread nuclear and cytoplasmic accumulation of mutant androgen receptor in SBMA patients. Brain 128:659–670
Huynh DP, Figueroa K, Hoang N, Pulst SM (2000) Nuclear localization or inclusion body formation of ataxin-2 are not necessary for SCA2 pathogenesis in mouse or human. Nat Genet 26:44–50
Koyano S, Yagishita S, Kuroiwa Y, Tanaka F, Uchihara T (2014) Neuropathological staging of spinocerebellar ataxia type 2 by semiquantitative 1C2-positive neuron typing. Nuclear translocation of cytoplasmic 1C2 underlies disease progression of spinocerebellar ataxia type 2. Brain Pathol 24:599–606
Corrado L, Mazzini L, Oggioni GD, Luciano B, Godi M, Brusco A, D’Alfonso S (2011) ATXN-2 CAG repeat expansions are interrupted in ALS patients. Hum Genet 130:575–580
Laffita-Mesa JM, Rodriguez Pupo JM, Moreno Sera R, Vazquez Mojena Y, Kouri V, Laguna-Salvia L, Martinez-Godales M, Valdevila Figueira JA, Bauer PO, Rodriguez-Labrada R, Gonzalez Zaldivar Y, Paucar M, Svenningsson P, Velazquez Perez L (2013) De novo mutations in ataxin-2 gene and ALS risk. PLoS ONE 8:e70560
Van Damme P, Veldink JH, van Blitterswijk M, Corveleyn A, van Vught PW, Thijs V, Dubois B, Matthijs G, van den Berg LH, Robberecht W (2011) Expanded ATXN2 CAG repeat size in ALS identifies genetic overlap between ALS and SCA2. Neurology 76:2066–2072
Isak B, Tankisi H, Johnsen B, Pugdahl K, Torvin MA, Finnerup NB, Christensen PB, Fuglsang-Frederiksen A (2016) Involvement of distal sensory nerves in amyotrophic lateral sclerosis. Muscle Nerve 54:1086–1092
Acknowledgments
We thank Dr. Pulst for providing wild-type and mutant GFP-ATXN2 plasmids, and Dr. Watabe of the Tokyo Metropolitan Institute of Medical Science for providing IMS32.
Funding
This study was partly supported by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan (MH19K07984 to MH).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Financial disclosures
None of the authors have any conflict of interest, including financial, personal, or other relationships with other people or organizations within 3 years since beginning the work submitted, that could have inappropriately influenced or biased the work presented here.
Statistical analysis
Not applicable.
Supplementary Information
Below is the link to the electronic supplementary material.
Appendix 1: Authors
Rights and permissions
About this article

Cite this article
Inada, R., Hirano, M., Oka, N. et al. Phenotypic and molecular diversities of spinocerebellar ataxia type 2 in Japan. J Neurol 268, 2933–2942 (2021). https://doi.org/10.1007/s00415-021-10467-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-021-10467-z