
Abstract
Introduction
We performed 4-year follow-up neuropsychological assessment to investigate cognitive decline and the prognostic abilities from presymptomatic to symptomatic familial frontotemporal dementia (FTD).
Methods
Presymptomatic MAPT (n = 15) and GRN mutation carriers (n = 31), and healthy controls (n = 39) underwent neuropsychological assessment every 2 years. Eight mutation carriers (5 MAPT, 3 GRN) became symptomatic. We investigated cognitive decline with multilevel regression modeling; the prognostic performance was assessed with ROC analyses and stepwise logistic regression.
Results
MAPT converters declined on language, attention, executive function, social cognition, and memory, and GRN converters declined on attention and executive function (p < 0.05). Cognitive decline in ScreeLing phonology (p = 0.046) and letter fluency (p = 0.046) were predictive for conversion to non-fluent variant PPA, and decline on categorical fluency (p = 0.025) for an underlying MAPT mutation.
Discussion
Using longitudinal neuropsychological assessment, we detected a mutation-specific pattern of cognitive decline, potentially suggesting prognostic value of neuropsychological trajectories in conversion to symptomatic FTD.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Frontotemporal dementia (FTD) is a presenile neurodegenerative disorder, leading to a heterogeneous clinical presentation, involving behavioural (behavioural variant FTD; bvFTD) and/or language deterioration (primary progressive aphasia; PPA) [1]. FTD has an autosomal dominant pattern of inheritance in 30 percent of cases, with mutations in the progranulin (GRN) and microtubule-associated protein tau (MAPT) genes as its two main causes [2]. The cognitive profile of FTD varies depending on the clinical phenotype and the underlying genotype. Patients with bvFTD are characterized by deficits in executive function, social cognition and language, whereas memory and visuoconstruction are initially spared [3,4,5]. Non-fluent variant PPA (nfvPPA) patients show agrammatism and speech sound distortions, while semantic variant PPA (svPPA) patients experience deficits in confrontation naming and word comprehension [6]. GRN mutations often lead to a clinical diagnosis of bvFTD, nfvPPA or parkinsonism. In MAPT mutations, bvFTD is the main phenotype, and semantic and memory impairments can be prominent neuropsychological symptoms [7].
Research in familial FTD has demonstrated the presence of a presymptomatic stage in which subtle cognitive changes have been identified [8,9,10,11,12]. More specifically, cognitive decline can start as early as 8 years prior to estimated symptom onset and shows mutation-specific patterns, with GRN mutation carriers declining in memory, and MAPT mutation carriers declining in language, social cognition and memory [8, 10]. This suggests that cognitive measures could function as disease-tracking biomarkers in the presymptomatic stage. However, it is currently unknown what the long-term cognitive profiles of presymptomatic FTD mutations are, whether neuropsychological assessment can be used to track disease progression to the symptomatic stage, and what the prognostic value is of cognitive trajectories in the presymptomatic and early symptomatic stage of FTD.
In this study, we investigated longitudinal cognitive decline on neuropsychological assessment in presymptomatic mutation carriers (MAPT or GRN) and controls from the same families within our longitudinal presymptomatic Dutch familial FTD Risk Cohort (FTD-RisC). Second, we assessed the difference in cognitive course between converters’ genotypes (i.e. MAPT vs. GRN) and phenotypes (i.e. bvFTD vs. nfvPPA) versus non-converters. Lastly, we investigated the prognostic value of neuropsychological trajectories in predicting symptom onset within 2–4 years.
Methods
Participants
In FTD-RisC, we follow healthy 50% at-risk family members from genetic FTD families on a 2-year basis. In the current study, we included 87 participants from MAPT or GRN families with study entries between December 2009 and January 2013 [8, 9, 13]. The follow-up period was 4 years, in which we acquired neuropsychological assessments at study entry, follow-up after 2 years and follow-up after 4 years. DNA genotyping (see “Procedure”) assigned participants either to the presymptomatic mutation carrier (n = 46; 31 GRN, 15 MAPT), or control group (n = 39; 29 GRN, 10 MAPT family members). We excluded two controls as they had cognitive disorders (≥ 2 SD below mean) on multiple domains, ultimately including 85 participants (46 mutation carriers, 37 controls; Fig. 1).

Participant in- and exclusion and sample size per time point. Two controls were excluded as they had multiple cognitive disorders (≤ 2 SD below reference mean) on neuropsychological testing. Eight mutation carriers converted to clinical FTD within the study window. Their data were restructured, so that there were three time points: 4 years before symptom onset, 2 years before symptom onset and symptom onset. Four years before symptom onset, only data of six converters were available, as two mutation carriers converted between baseline and first follow-up. The data of converters were compared to, respectively, baseline, follow-up after 2 years and follow-up after 4 years in non-converters and healthy controls
Standard protocol approvals, registrations, and patient consents
Clinical investigators were blind for participants’ genetic status if they had not undergone predictive testing. In case of conversion to clinical FTD, we offered the patient and family members genetic counselling and unblinding of genetic status, to confirm the presence of the pathogenic mutation. At study entry, all participants gave written informed consent. The study was approved by the Medical and Ethical Review Committee of the Erasmus Medical Center.
Procedure
Every 2 years, participants underwent a standardized assessment consisting of a neuropsychological test battery, neurological examination, and MR imaging of the brain. DNA sequencing was performed at study entry. All participants were asymptomatic according to established diagnostic criteria for bvFTD [3] or PPA [6] at baseline. Knowledgeable informants were asked about cognitive and/or behavioural deterioration at each study visit by means of a structured interview and a well-validated questionnaire (Neuropsychiatric Inventory; NPI) [14].
Converters
Eight mutation carriers became symptomatic within the study time window (“converters”). Symptom onset was determined by means of the above mentioned assessment (anamnesis, MR imaging of the brain, neuropsychological assessment, heteroanamnestic information and unblinding of genetic status). Conversion was determined in a multidisciplinary consensus meeting of the Erasmus MC FTD Expertise Centre, involving neurologists (LDK, JCvsS), neuropsychologists (LCJ, JLP, SF, EvdB, JMP), medical doctors (LHM, ELvdE), as well as neuroradiologists, geriatricians, a clinical geneticist (RvM), and a care consultant. Six converters (5 MAPT, 1 GRN) presented with progressive behaviour deterioration, functional decline, and frontal and/or temporal lobe atrophy on MRI, fulfilling the international diagnostic consensus criteria of Rascovsky et al. [3] for bvFTD with definite FTLD pathology. Two converters (both GRN) presented with isolated language difficulties and no impairments in daily living activities, thereby fulfilling the diagnostic criteria for PPA of Gorno-Tempini et al. [6]. Both developed nfvPPA, as they showed a non-fluent, halting speech, with sound errors and agrammatism. See Supplementary Table 1 for demographic, clinical and neuropsychological data of the converters. We defined mutation carriers remaining without FTD symptoms as non-converters (n = 38; 28 GRN, 10 MAPT).
Neuropsychological assessment
We screened global cognitive functioning by means of the Mini-Mental State Examination [15] (MMSE) and frontal assessment battery [16] (FAB). Experienced neuropsychologists (LCJ, JLP, SF) administered neuropsychological tests within six cognitive domains: language, attention and mental processing speed, executive functioning, social cognition, memory, and visuoconstruction. We rated language with the 60-item Boston Naming Test (BNT) [17], verbal Semantic Association Test (SAT) [18], ScreeLing phonology [19], and categorical fluency [20]. We assessed attention and mental processing speed by means of Trail making Test (TMT)-A [21], Stroop Color-Word Test I and II [22], Wechsler Adult Intelligence Scale III (WAIS-III) Digit Span forwards [23], and Letter Digit Substitution Test (LDST) [24]. Executive functioning was evaluated using TMT-B [21], Stroop Color-Word Test III [22], WAIS-III Digit Span backwards [23], modified Wisconsin Card Sorting Test (WCST) concepts [25], letter fluency [20], and WAIS-III Similarities [23]. Happé cartoons [26] and Ekman Faces [27] measured social cognition. We assessed memory using the Dutch Rey Auditory Verbal Learning Test (RAVLT) [28] and Visual Association Test (VAT) [29]. We evaluated visuoconstruction by means of clock drawing [30] and WAIS-III Block Design [23]. Alternate forms were used at follow-up visits, when applicable (letter fluency, RAVLT, VAT). Depressive symptoms were rated with the Beck’s Depression Inventory (BDI) [31].
Study design
In converters, we restructured the three original time points within our study window (i.e. baseline, follow-up after 2 years, follow-up after 4 years) into the following three new time points (Fig. 1):
-
4 years before symptom onset: we used the data of the study visit 4 years before diagnosis. Analyses could were performed in six converters, as two (1 GRN, 1 MAPT—2 bvFTD) developed symptoms between baseline and first follow-up (i.e. at 2 years follow-up), and therefore no data 4 years prior to symptom onset were available.
-
2 years before symptom onset: we used the data of the study visit 2 years before diagnosis. Analyses included all eight converters.
-
After symptom onset: we used the data of the diagnosis visit. Analyses included all eight converters.
In non-converters and controls, we used the original time points: baseline (data were compared to “4 years before symptom onset” data of converters), follow-up after 2 years (data were compared to “2 years before symptom onset data of converters) and follow-up after 4 years (data were compared to “after symptom onset data of converters).
Statistical analysis
Statistical analyses were performed using SPSS Statistics 21.0 (IBM Corp., Armonk, NY) and GraphPad Prism 7 (La Jolla, California, USA), with the significance level at p < 0.05 (two-tailed) across all comparisons. We compared demographic data between MAPT mutation carriers, GRN mutation carriers and controls, and between converters, non-converters and controls by means of one-way ANOVAs. We performed Pearson Χ2 tests to investigate differences in sex. Longitudinal comparisons of clinical data were performed with repeated measures ANOVAs. We standardized all raw neuropsychological test scores by converting them into z-scores (i.e. individual test score minus the baseline mean of the controls, divided by the baseline SD of the controls) per time point, after which we calculated composite z-scores for the respective six cognitive domains by averaging the z-scores of the individual tests per domain. For the longitudinal comparisons we used multilevel linear regression modeling. This analysis corrects for bias when data absence is dependent on characteristics present in the model, and can therefore efficiently handle missing and unbalanced time points. There were two levels in the models: the participants constituted the upper level; their repeated measures the lower level. We ran two analyses to assess cognitive decline per mutation (1) and clinical status (2):
-
1.
We entered mutation status (MAPT mutation carrier, GRN mutation carrier or control), time (4 years before symptom onset, 2 years before symptom onset, and after symptom onset), and first-order interactions, with age, gender and educational level as covariates. We reran the analyses excluding the converters to exclude converters driving the cognitive decline in the mutation carrier groups;
-
2.
We split the converter group according to genotype (MAPT or GRN) and phenotype (bvFTD or nfvPPA) to investigate specific profiles of cognitive decline over time. We then entered clinical status (converter, non-converter or control), time, and first-order interactions, with age, gender and educational level as covariates.
Third, to investigate the prognostic abilities of cognitive decline in discriminating between converters and non-converters, we determined the area under the curve (AUC) by receiver operating characteristic (ROC) analyses on the neuropsychological trajectories between visits. For this, we calculated deltas between test scores; one between 4 and 2 years before symptom onset and one between 2 years before symptom onset and symptom onset. Optimal cut-off levels were given by the highest Youden’s index [32]. Again, we split the converter group according to genotype (MAPT or GRN) and phenotype (bvFTD or nfvPPA). Next, we performed logistic regression analyses, taking group (converter vs. non-converter) as the dependent variable and the deltas (tests with significant diagnostic performance in abovementioned ROC analyses) as the independent variables. The models were selected with a forward stepwise method according to the likelihood ratio test and applying the standard p values for variable inclusion (0.05) and exclusion (0.10), with age, sex and education as covariates. Goodness of fit was evaluated with the HL Χ2 test. Nagelkerke R2 is reported as measure of effect size. We checked predictor variables for multicollinearity. All models were corrected for multiple comparisons (Bonferroni).
Results
Demographics
MAPT mutation carriers were significantly younger than GRN mutation carriers (p = 0.012; Table 1). The mean familial symptom onset age was lower in MAPT than in GRN mutation carriers and controls (both p < 0.001). There were no significant differences between groups regarding estimated years to symptom onset (p > 0.05). Longitudinal analyses demonstrated that MAPT mutation carriers declined significantly more than GRN mutation carriers and controls with regards to the MMSE (p = 0.014), and also developed more depressive symptoms (p = 0.028). FAB and NPI scores did not significantly change over time (p > 0.05). Converters, non-converters and controls did not differ regarding demographic variables, apart from a younger family onset in MAPT converters than GRN converters (p = 0.043) and non-converters (p = 0.001; Table 1). Both MAPT and GRN converters declined significantly with respect to MMSE score (p < 0.001) and they developed more neuropsychiatric symptoms in the form of higher BDI (p = 0.001) and NPI (p = 0.021) scores in comparison to non-converters and controls. FAB scores did not significantly change over time (p > 0.05).
Longitudinal cognitive decline in MAPT and GRN mutation carriers
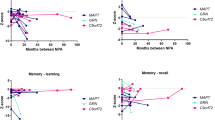
The whole group of MAPT mutation carriers declined significantly within the domains language, social cognition and memory compared with controls (Table 2; Fig. 1). This was reflected in lower scores on the BNT and categorical fluency, Happé cartoons, VAT and RAVLT delayed recall (Table 2). In the whole group of GRN mutation carriers, no longitudinal decline was found in comparison to controls. In comparison to GRN mutation carriers, MAPT mutation carriers declined significantly on the domains language (β = − 0.015, p < 0.001) and memory (β = − 0.016, p = 0.008), reflected in lower BNT (β = − 0.085, p = 0.01), SAT (β = − 0.027, p = 0.015), category fluency (β = − 0.107, p = 0.002), and RAVLT delayed recall (β = − 0.047, p = 0.001) scores. There were no cognitive domains or tests on which GRN mutation carriers declined more than MAPT mutation carriers (Table 2). By excluding the five MAPT converters from the analyses, none of the domain scores in MAPT mutation carriers continued to show significant decline over time in comparison to controls. Regarding individual tests, however, the decline on the RAVLT delayed recall remained significant (β = − 0.032, p = 0.023). The results did not change by excluding the three GRN converters from the analyses. In comparison to GRN, MAPT mutation carriers still declined more on language (β = − 0.010, p = 0.004), reflected in lower ScreeLing phonology (β = − 0.008, p = 0.024) and category fluency (β = − 0.007, p = 0.041). There was no cognitive decline in controls—but significant improvement was found on social cognition (Happé non-ToM and Ekman Faces) and memory (RAVLT immediate and delayed recall) (Table 2). The raw neuropsychological test scores per time point can be found in Supplementary Table 2.
Longitudinal cognitive decline in converters and non-converters
Converters with a MAPT mutation deteriorated significantly on all domains but visuoconstruction (Fig. 2a–d, f; Table 3). Within these domains, performances declined on BNT (p < 0.001), LDST (p = 0.035), Stroop I, II and III (I: p = 0.017; II: p < 0.001; III: p = 0.021), categorical fluency (p = 0.001), WAIS similarities (p < 0.001), Happé ToM (p = 0.011), and RAVLT immediate (p = 0.004) and delayed recall (p = 0.030). Converters with a GRN mutation deteriorated significantly on attention and mental processing speed, and executive function (Fig. 2b, c; Table 3). Within these domains, performances on TMT-B (p < 0.001), Stroop III (p < 0.001), WCST (p = 0.005), letter fluency (p = 0.012) and WAIS similarities (p < 0.001) deteriorated significantly over time. Converters with bvFTD had a similar pattern of cognitive decline as MAPT converters, with lower scores on social cognition, memory, language, attention and executive function (Table 3). Comparably, converters with nfvPPA had a similar pattern of cognitive decline as GRN converters, with lower scores on attention and executive function (Table 3). There were no differences in decline between converters with bvFTD and nfvPPA (Table 3). The raw neuropsychological test scores per time point can be found in Supplementary Table 3.

Multilevel linear regression model displaying longitudinal decline (4 years, 2 years and after symptom onset) in composite domain z-score in the total group of converters (light green), MAPT converters (light blue dotted line), GRN converters (dark blue dotted line), non-converters (dark green) and healthy controls (black). Models are displayed per cognitive domain: a social cognition, b attention and mental processing speed, c executive functioning, d memory, e visuoconstruction, and f language. NB: the healthy controls have a mean z-score of zero by default as the z-scores of mutation carriers were based on that (raw score minus mean score of healthy controls, divided by the standard deviation of healthy controls). MAPT microtubule-associated protein tau, GRN progranulin
Classification between converters and non-converters
Between 4 and 2 years before symptom onset, the delta domain and individual neuropsychological test scores failed to distinguish significantly between converters and non-converters. Between 2 years before symptom onset and symptom onset decline on categorical fluency was predictive of an underlying MAPT mutation (p = 0.025; Table 4). Decline on ScreeLing phonology (p = 0.046) and letter fluency (p = 0.046) was predictive of conversion to nfvPPA (Table 4).
Discussion
This study examined a large cohort of at-risk participants from GRN and MAPT FTD families by means of neuropsychological assessment during a 4-year follow-up. Within the study time window, eight mutation carriers became symptomatic. Converters with a MAPT and GRN mutation had mutual as well as gene-specific profiles of cognitive decline. Cognitive decline on categorical fluency between 2 years before conversion and symptom onset was predictive for an underlying MAPT mutation, and decline on ScreeLing phonology and letter fluency was predictive for conversion to nfvPPA. These results suggest that neuropsychological assessment could provide sensitive clinical biomarkers to identify and track FTD mutation carriers at-risk of converting to the symptomatic stage. These findings hold potential for improving early clinical diagnosis by identifying the most sensitive neuropsychological tests for conversion, and use in upcoming disease-modifying clinical trials.
Following the MAPT mutation carriers over a 4-year period, we found significant decline in language, social cognition and memory. This is consistent with findings from previous presymptomatic familial FTD studies, in which both cross-sectional [9,10,11, 33] and longitudinal [8] decline was found. Specifically, in our first follow-up study [8], we demonstrated decline in the domains language, social cognition and memory 5–8 years before estimated symptom onset. It should be taken into account that this study made use of estimated onset as a proxy, instead of actual symptom onset as in the present study—but the similar profile of decline confirms the presence of early changes in these three domains. As in our previous study, the present results are largely driven by the converters. This could suggest that neuropsychological test scores remain static while mutation carriers are presymptomatic, and cognitive decline starts only near or at symptom onset [34,35,36], suggesting an explosive rather than gradual start of the symptomatic disease stage. Alternatively, we might be unable to pick up subtle cognitive changes in presymptomatic mutation carriers due to lack of power. Also, although well-validated, most of our neuropsychological tests were not developed for repeated administration in a preclinical population [37]. We therefore cannot rule out that familiarity and/or practice effects are obscuring subtle cognitive decline, a notion that seems to be underwritten by improvement in social cognition and memory in controls, but not mutation carriers.
In our exploratory analyses in converters, we discovered both common as well as mutation-specific profiles of cognitive decline in MAPT and GRN. In both mutations, decline in attention, mental processing speed and executive function was found—while only converters with a MAPT mutation demonstrated decline on language, memory and social cognition. Previous studies in familial FTD also point to distinct profiles for MAPT and GRN [8, 10,11,12], and are largely consistent with our present findings. Another important aspect is the longitudinal tracking of the different clinical phenotypes. The similar patterns of cognitive decline in bvFTD as MAPT, and nfvPPA as GRN are related to the dominant genotype in each group (e.g. all nfvPPA converters have a GRN mutation). These findings suggest that neuropsychological assessment can be used to track the different mutations and phenotypes from the presymptomatic to the symptomatic stage, which is advantageous considering the need for good clinical endpoints in future disease-modifying trials.
Extending the findings from our first follow-up study [8], we demonstrated significant decline on the RAVLT recall in presymptomatic MAPT mutation carriers. The additional finding that lower memory scores over time were also found in MAPT, and not GRN converters—suggesting a mutation-specific aetiology—corroborate this. Although memory loss has been described in GRN [38, 39], this is usually a later symptom, while episodic memory impairment has been found as the presenting and most prominent symptom in MAPT [7, 40, 41]. Interestingly, the Genetic Frontotemporal dementia Initiative (GENFI) consortium revealed hippocampal atrophy in presymptomatic MAPT from 15 years before estimated symptom onset [10], and as this medial temporal structure is critical for episodic memory processing [42] this offers a good explanation for our findings. In line with earlier studies [42, 43], we did find deficits in verbal recall but not visual associative memory. Semantically loaded tasks such as the RAVLT can be particularly more difficult than visual memory tasks like the VAT, as a result of the prominent semantic impairments seen early in MAPT-associated FTD [44]. Our results contribute to the present thinking that memory deficits can be an integral part of the clinical spectrum [42], and comprehensive memory tasks should therefore be incorporated in the standard diagnostic work-up.
Knowing the cognitive profile of decline indicative for conversion is important to get more insight into the timing of clinical changes in the earliest disease stage. We found that conversion can be predicted based on cognitive decline in the 2 years prior to symptom onset, but not earlier. As the cognitive decline was part of the diagnostic process of determining conversion, this is not a surprising finding. However, it does suggest a more explosive disease development with cognitive decline accelerating rapidly in proximity of symptom onset, which is in line with evidence from a large familial Alzheimer’s disease cohort [45]. By selectively choosing tests within the domains that have prognostic abilities, the neuropsychological battery can be shortened, which would benefit patient burden and helps cutting healthcare expenses. Especially fluency tasks seem to be promising candidates, as they were able to distinguish accurately between future phenotype and underlying genotype. The latter is essential for patient stratification in future clinical trials targeting specific pathologies, and ideally these interventions should be applied in the presymptomatic stage [46]. Reliable phenotypic prediction furthermore optimizes the diagnostic process by shortening the current diagnostic delay [47], and is helpful for the patient, caregiver and clinician in knowing what disease presentation and course to expect. Verbal fluency tests are widely used in dementia diagnosis setting [48], and are affected in both presymptomatic [8, 11] and symptomatic FTD [49, 50]. Future research could additionally investigate the use of qualitative assessment of verbal fluency (e.g. clustering, switching between clusters), as recent research [49] points to differences between FTD and PPA subtypes—making this a promising application of verbal fluency for a precise clinical differentiation in presymptomatic and early stage FTD.
Key strengths of our study constitute our longitudinal design, spanning a 4-year follow-up of at-risk participants from both MAPT and GRN families. Although our group of converters is currently small, this is the first study tracking FTD mutation carriers from the presymptomatic to symptomatic disease stage. Being aware of the caveats of small sample sizes and administering a large amount of neuropsychological tests with respect to statistical power, our results warrant replication in our cohort as well as larger international cohorts such as GENFI [10], in which with the passing of time more mutation carriers will approach symptom onset and/or convert to clinical FTD. The dropout rate is very low, creating balanced datasets across the three time points. Additionally, use of multilevel linear modeling further handles efficiently with missing data. Directions for future research entail the development of neuropsychological tasks more suited to administer in the presymptomatic phase (robust to ceiling effects) and repeated administration (robust to practice and able to measure small changes). More extensive quantification tools of behavioural functioning are also needed to capture the entire clinical spectrum of (presymptomatic) FTD, as well as assessment methods that rely less on the accuracy of informant report [37]. A disadvantage of the study is the fact that the neuropsychological assessment was part of the clinical assessment with which we determined conversion to the symptomatic stage. This has likely led to a circular reasoning, as we demonstrated that converters declined over time, while cognitive decline was considered a prerequisite for conversion. Ideally, the tests assessed in our study should not have been used in the diagnosis of conversion. However, in our multidisciplinary meeting, we followed the international consensus criteria for bvFTD [3] and PPA [6], using all available clinical information—e.g. MR imaging of the brain, anamnestic and heteroanamnestic information, behavioural and neuropsychiatric questionnaires, unblinding of genetic status—so that symptom onset did not solely depend on the neuropsychological assessment. Furthermore, as the multilevel model assumes a linear relationship between genetic status and cognitive decline over time, we could have missed non-linear effects over time. Lastly, the analyses on the non-converters and controls were performed using the original baseline and follow-up visits, regardless of, e.g. age and time to estimated symptom onset. It is possible that these analyses therefore lost some sensitivity to detect cognitive decline over time. However, as between-group analyses on age and estimated years to symptom onset in converters, non-converters, and controls did not show significant differences (respectively, p = 0.99 and p = 0.19), we believe this effect is minimal.
Our study investigates longitudinal neuropsychological performance in a large cohort of at-risk individuals from genetic FTD families. We provide evidence of mutation-specific cognitive decline when moving from the presymptomatic into symptomatic stage, and of neuropsychological trajectories predicting symptom onset. These results suggest the potential biomarker value of neuropsychological assessment in both disease-monitoring and predicting conversion to clinical FTD.
References
Snowden JS, Pickering-Brown SM, Mackenzie IR et al (2006) Progranulin gene mutations associated with frontotemporal dementia and progressive non-fluent aphasia. Brain 129:3091–3102
Warren JD, Rohrer JD, Rossor MN (2013) Clinical review. Frontotemporal dementia. BMJ 347:f4827
Rascovsky K, Hodges JR, Knopman D et al (2011) Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 134:2456–2477
Seelaar H, Rohrer JD, Pijnenburg Y et al (2011) Clinical, genetic and pathological heterogeneity of frontotemporal dementia: a review. J Neurol Neurosurg Psychiatry 82:476–486
Adenzato M, Cavallo M, Enrici I (2010) Theory of mind ability in the behavioural variant of frontotemporal dementia: an analysis of the neural, cognitive, and social levels. Neuropsychologia 48:2–12
Gorno-Tempini ML, Hillis AE, Weintraub S et al (2011) Classification of primary progressive aphasia and its variants. Neurology 76(11):1006–1014
Rohrer JD, Warren JD (2011) Phenotypic signatures of genetic frontotemporal dementia. Curr Opin Neurol 24(6):542–549
Jiskoot LC, Dopper EGP, den Heijer T et al (2016) Presymptomatic cognitive decline in familial frontotemporal dementia: a longitudinal study. Neurology 87:384–391
Dopper EG, Rombouts SA, Jiskoot LC et al (2014) Structural and functional brain connectivity in presymptomatic familial frontotemporal dementia. Neurology 83:e19–e26
Rohrer JD, Nicholas JM, Cash DM et al (2015) Presymptomatic cognitive and neuroanatomical changes in genetic frontotemporal dementia in the genetic frontotemporal dementia initiative (GENFI) study: a cross-sectional analysis. Lancet Neurol 14:253–262
Geschwind DH, Robidoux J, Alarcón M et al (2001) Dementia and neurodevelopmental predisposition: cognitive dysfunction in presymptomatic subjects precedes dementia by decades in frontotemporal dementia. Ann Neurol 50:741–746
Hallam BJ, Jacova C, Hsiung GYR et al (2014) Early neuropsychological characteristics of progranulin mutation carriers. J Int Neuropsychol Soc 20:694–703
Dopper EG, Chalos V, Ghariq E et al (2016) Cerebral blood flow in presymptomatic MAPT and GRN mutation carriers: a longitudinal arterial spin labeling study. Neuroimage Clin 12:460–465
Kaufer DI, Cummings JL, Ketchel P et al (2000) Validation of the NPI-Q, a brief clinical form of the neuropsychatric inventory. J Neuropsychiatry Clin Neurosci 12(2):233–239
Folstein MF, Folstein SE, McHugh PR (1975) “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 12(3):189–198
Dubois B, Slachevsky A, Litvan I, Pillon B (2000) The FAB: a frontal assessment battery at bedside. Neurology 55(11):1621–1626
Kaplan E, Goodglass H, Weintraub S (1978) The Boston Naming Test. Lea & Febiger, Philadelphia
Visch-Brink E, Stronks D, Denes G (2005) Semantische Associatie test. Swets & Zeitlinger, Lisse
Doesborgh SJ, van de Sandt-Koenderman WM, Dippel DW et al (2003) Linguistic deficits in the acute phase of stroke. J Neurol 250(8):977–982
Thurstone LLT, Thurstone TG (1962) Primary mental abilities. Science Research Associates, Chicago
Battery Army Individual Test (1994) Manual of directions and scoring. War Department, Adjutant General’s office, Washington, DC
Stroop JR (1935) Studies of interference in serial verbal reactions. J Exp Psychol 18:643–662
Wechsler D (2005) WAIS-III nederlandse bewerking, technische handleiding. Harcourt Test Publishers, Lisse
Jolles J, Houx PJ, van Boxtel MPJ, Ponds RWHM (1995) Maastricht aging study: determinants of cognitive aging. Neuropsych Publishers, Maastricht
Nelson HE (1976) A modified card sorting test sensitive to frontal lobe defects. Cortex 12:313–324
Happe F, Brownell H, Winner E (1999) Acquired ‘theory of mind’ impairments following stroke. Cognition 70(3):211–240
Ekman P, Friesen WV (1976) Pictures of facial affect. Consulting Psychologists Press, Palo Alto
Rey A (1958) L’examen clinique en psychologie. Presses Universitaires de France, Paris
Lindeboom J, Schmand B, Tulner L et al (2002) Visual association test to detect early dementia of the Alzheimer type. J Neurol Neurosurg Psychiatry 73(2):126–133
Royall DR, Cordes JA, Polk M (1998) CLOX: an executive clock drawing task. J Neurol Neurosurg Psychiatry 64:588–594
Beck AT, Ward CH, Mendelson M, Mock J, Erbaugh J (1961) An inventory for measuring depression. Arch Gen Psychiatry 4:561–571
Youden WJ (1950) Index for rating diagnostic tests. Cancer 3:32–35
Barandiaran M, Estanga A, Moreno F et al (2012) Neuropsychological features of asymptomatic c.709-1G > A progranulin mutation carriers. JINS 18:1086–1090
Janssen JC, Schott JM, Cipolotti L et al (2005) Mapping the onset and progression of atrophy in familial frontotemporal lobar degeneration. JNNP 76(2):162–168
Rohrer JD, Warren JD, Barnes J et al (2008) Mapping the progression of progranulin-associated frontotemporal lobar degeneration. Nat Clin Pract Neurol 4(8):455–460
Ferman TJ, McRae CA, Arvanitakis Z et al (2003) Early and pre-symptomatic neuropsychological dysfunction in the PPND family with the N279K tau mutation. Parkinsonism Relat Disord 9(5):265–270
Miller JB, Banks SJ, Léger GC, Cummings JL (2014) Randomized controlled trials in frontotemporal dementia: cognitive and behavioral outcomes. Transl Neurodegener 3:12
Whitwell JL, Jack CR, Boeve BF et al (2009) Voxel-based morphometry patterns of atrophy in FTLD with mutations in MAPT or PGRN. Neurology 72:813–820
Rohrer JD, Ridgway GR, Modat M et al (2010) Distinct profiles of brain atrophy in frontotemporal lobar degeneration caused by progranulin and tau mutations. Neuroimage 53:1070–1076
Smith R, Puschmann A, Schöll M et al (2017) 18F-AV-1451 tau PET imaging correlates strongly with tau neuropathology in MAPT mutation carriers. Brain 139(9):2372–2379
Tolboom N, Koedam ELGE, Schott JM et al (2010) Dementia mimicking Alzheimer’s disease owing to a tau mutation: CSF and PET findings. Alzheimers Dis Assoc Disord 24:303–307
Hornberger M, Piguet O (2012) Episodic memory in frontotemporal dementia: a critical review. Brain 135:678–692
Spina S, Schonhaut D, Boeve BF et al (2017) Frontotemporal dementia with the V337M MAPT mutation. Neurology 88(8):758–766
Snowden JS, Adams J, Harris J et al (2015) Distinct clinical and pathological phenotypes in frontotemporal dementia associated with MAPT, PGRN and C9orf72 mutations. Amyotroph Lateral Scler Frontotemp Degener 16:497–505
Hassenstab J, Aschenbrenner AJ, Balota DA et al (2016) Cognitive trajectories in DIAN: relationships with symptom onset, mutation types and clinical status. Alzheimers Dement 12(7):368
Meeter LH, Donker Kaat L, Rohrer JD, van Swieten JC (2017) Imaging and fluid biomarkers in frontotemporal dementia. Nat Rev Neurol 13(7):406–419
Rosness TA, Engedal K, Chemali Z (2016) Frontotemporal dementia: an updated clinician’s guide. J Geriatr Psychiatry 29(5):271–280
Pakhomov SVS, Hemmy LS (2014) A computational linguistic measure of clustering behavior on semantic verbal fluency task predicts risk of future dementia in the Nun study. Cortex 55:97–106
van den Berg E, Jiskoot LC, Grosveld MJH et al (2017) Qualitative assessment of verbal fluency performance in frontotemporal dementia. Dement Geriatr Cogn Disord 44:35–44
Laisney M, Matuszewski V, Mézenge F et al (2009) The underlying mechanisms of verbal fluency deficit in frontotemporal dementia and semantic dementia. J Neurol 256:1083
Acknowledgements
We would like to thank all the participants and their families for taking part in our study. This work was supported by Dioraphte Foundation Grant 09-02-03-00, the Association for Frontotemporal Dementias Research Grant 2009, Alzheimer Nederland and Memorabel ZonMw Grant 733050102 (Deltaplan Dementie).
Author information
Authors and Affiliations
Contributions
LCJ contributed to the conception and design of the study, acquired and analysed data, and drafted the manuscript, figures and tables. JLP acquired data. LvA acquired and analysed data. SF acquired data. LHM acquired data and contributed to the design of the figures. LDK acquired data. ELvdE acquired data. EGPD contributed to the conception of the study and acquired data. RT contributed to the design of the study and data analysis. RvM is the genetic guardian of the study. JvS contributed to the conception and design of the study and is PI of the project. EvdB contributed to the design and data interpretation of the study. JMP contributed to the design of the study, and drafting the manuscript, figures and tables. All authors were involved in copyediting and approval of the final draft of the manuscript.
Corresponding author
Ethics declarations
Conflicts of interest
LCJ, JLP, LvA, LHM, LDK, ELvdE, EGPD, RT, RvM, JvS, EvdB, JMP report no conflicts of interest.
Ethical standard
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and national research committee, and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Jiskoot, L.C., Panman, J.L., van Asseldonk, L. et al. Longitudinal cognitive biomarkers predicting symptom onset in presymptomatic frontotemporal dementia. J Neurol 265, 1381–1392 (2018). https://doi.org/10.1007/s00415-018-8850-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-018-8850-7