
Abstract
Antibodies to myelin oligodendrocyte glycoprotein (MOG-IgG) have been described in patients with neuromyelitis optica spectrum disorders (NMOSD) without aquaporin-4 antibodies (AQP4-IgG). We aimed to identify the proportion of AQP4-IgG-negative NMOSD patients who are seropositive for MOG-IgG. In a cross sectional study, we reviewed all patients seen in the National NMO clinic over the last 4 years (after the availability of MOG-IgG testing), including clinical information, MRI, and antibody tests. 261 unique patients were identified. 132 cases satisfied the 2015 NMOSD diagnostic criteria. Of these, 96 (73%) were AQP4-IgG positive and 36 (27%) were AQP4-IgG negative. These 36 patients were tested for MOG-IgG and 15/36 (42%) tested positive. 20% (25/125) of the patients who did not satisfy NMOSD criteria had MOG-IgG. Approximately half of seronegative NMOSD is MOG-Ig seropositive and one in five of non-NMOSD/non-MS demyelination is MOG-IgG positive. Since MOG-associated demyelinating disease is likely different from AQP4-IgG disease in terms of underlying disease mechanisms, relapse risk and possibly treatment, testing for MOG-IgG in patients with AQP4-IgG-negative NMOSD and other non-MS demyelination may have significant implications to management and clinical trials.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
73–90% of neuromyelitis optica spectrum disorder (NMOSD) patients diagnosed according to the 2015 International panel on NMO diagnosis have aquaporin-4 antibodies (AQP4-IgG) [1, 2]. It is presumed that at least a proportion of the remaining 10–27% of patients, classified as seronegative NMOSD have another disease specific antibody. Antibodies to myelin oligodendrocyte glycoprotein (MOG-IgG) have been increasingly reported in a variety of CNS neuroinflammatory conditions including patients with phenotypes typical for NMOSD [3]. We aimed to determine the prevalence of MOG-IgG in AQP4-IgG-negative NMOSD.
Methods
The Walton Centre Neurosciences NHS Trust in Liverpool, United Kingdom, is a tertiary neurology hospital that hosts one of the two national multidisciplinary specialist clinics for patients with NMOSD and non-MS demyelinating disorders as part of the UK NMOSD service. We systematically reviewed all patients seen in this clinic over the last 4 years (after the availability of MOG-IgG testing), including clinical information, MRI, and antibody tests. Both AQP4-IgG and MOG-IgG were tested using a validated live cell-based assay with high specificity (John Radcliffe Hospital, Oxford, UK) [4, 5]. This study was approved by Research Ethics Service, NRES Committee London—Hampstead, Ref. no. 15/LO/1433.
Results
261 unique patients with non-MS/atypical CNS inflammatory conditions attended the clinic and were assessed for NMOSD. All patients were tested for AQP4-IgG. 132 cases satisfied the 2015 NMOSD diagnostic criteria. Of these, 96 (73%) were AQP4-IgG positive and 36 (27%) AQP4-IgG negative. These 36 patients, were tested for MOG-IgG and 15/36 (42%) tested positive. This would account for 11% (15/132) of the total cohort of NMOSD patients (Fig. 1; Table 1). All MOG-IgG-negative patients were Caucasians with a median age of onset of 18 years (8–44 years) and median disease duration of 4.7 years (2–16 years). The predominant clinical phenotype of the demyelinating event was ON (60%), TM (21%), brain (12%), and brainstem (4%).

Classification of non-MS/atypical demyelination based on 2015 NMOSD criteria, AQP4-IgG, and MOG-IgG testing. NMOSD neuromyelitis optica spectrum disorder, AQP4 IgG Antibody to aquaporin 4, MOG-IgG antibody to myelin oligodendrocyte glycoprotein, OSD optico-spinal demyelination with normal brain MRI
While we tested all AQP4-IgG-negative patients for MOG-IgG (n = 36), only a proportion (33%) of AQP4-IgG-positive patients (n = 32) were tested (as double positives are exceptionally rare) (Fig. 1). None were definitely positive. However, one patient was ‘low positive/possibly negative. This patient with one episode of long myelitis also had antinuclear antibodies (1/80 titre with homogenous pattern (nuclear antigens all negative) and was ‘low positive’ for anti-glycine antibodies too. The significance of the MOG-IgG in the context of these additional antibodies is uncertain and may reflect a heightened humoral autoimmune response rather than truly pathogenic dual positivity. This patient has not been included in the MOG cohort in this paper.
We also tested the majority of patients with a demyelinating syndrome referred to the service who did not fulfill the NMOSD criteria (125/129, 97%). Twenty-five (20%) were positive for MOG-IgG. Details of these cases will be the subject of an upcoming separate research paper and are not discussed further here.
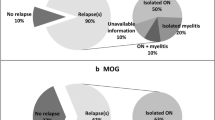
We also assessed how many of the MOG-IgG patients with NMOSD phenotype had a relapsing course. Thirteen patients (86%) had a relapsing course. However, a relapsing course was the reason for referral to the clinic in the first place (n = 13/13). The median duration of illness for the relapsing patients was 4.7 years (2–16 years). The median inter-attack interval was 1 year (0.16–17) and median EDSS in the relapsing MOG group at last follow-up was 3 (0–9, Table 1). All relapsing patient are on immunosuppressants (Table 1).
We also assessed the proportion of patients with optic neuritis and long myelitis who fulfill Wingerchuk 2006 criteria [6] that are MOG-IgG positive, as this is a clinical question often posed. Of the whole cohort of 261 patients, 75 patients had long myelitis and optic neuritis. Of these 49 were AQP4-IgG positive (66%) and 10 were MOG-IgG positive (13%, or 38% of AQP4-IgG-negative patients) and 16 remained seronegative (21%). Serial testing where done in 14/15 patients (13 relapsing); MOG-IgG was detected in all. Treatment with steroid or immunosuppression does not seem to have an effect on MOG-IgG serostatus in this cohort of predominantly relapsing patients (Table 2).
Discussion
In a cohort of well-characterised NMOSD patients (n = 132), 73% were AQP4-IgG and 11% were MOG-IgG seropositive and 16% remained seronegative. MOG-IgG disease accounts for 42% of the AQP4 IgG-negative seronegative cohort. MOG-IgG was present in 38% of patients with long myelitis and optic neuritis who do not have AQP4 IgG.
86% (13/15) of our patients who satisfy criteria for NMOSD who are MOG-IgG-positive patients have relapsing disease, similar to a recent study [7] who reported that 80% of their MOG-IgG-positive cohort (n = 50) followed a relapsing course. However, a relapsing course was the reason for referral to the clinic in the first place (n = 13/13) making this a biased sample. Long-term follow-ups of a cohort of MOG-IgG-positive patients after the very first event is required to obtain the true risk of relapse.
Importantly, 20% of patients with non-MS/atypical demyelination who do not satisfy criteria for NMOSD tested positive for MOG-IgG (Fig. 1). Double positive cases (both AQP4-IgG and MOG-IgG) are rare [8,9,10] with none of the tested patients were definite positives. Since we have tested only 52% (68/132) of the total NMOSD cohort for MOG-IgG, this requires further clarification in future studies.
In conclusion, our study provides the best possible answers at the current time on several questions on the frequency of MOG-IgG patients: NMOSD who are AQP4-IgG negative and MOG-IgG positive (42%), NMO (as per Wingerchuk 2006) with optic neuritis and long myelitis who are AQP4-IgG negative but MOG-IgG (13%). We also found that MOG-IgG is found in 20% of non-NMOSD/non-MS demyelination. It is also estimated that at least 11% of all NMOSD (as per 2015 criteria) is MOG-IgG positive.
Our study has important practical implications. First, the definite diagnosis of MOG-IgG-associated disease offers patients and physicians a better diagnostic label than seronegative NMOSD. Second, as nearly one in every two of seronegative NMOSD, and 1/5 of atypical non-MS demyelination is MOG-Ig positive, testing for these cohorts will be of high yield and worthwhile, compared to testing every demyelination (which in most Caucasian predominant populations is likely to be MS) with attendant costs and risk of false-positive results. Third, it is likely that the long-term disease course and therefore treatment strategies of AQP4-IgG and MOG-IgG is different. If this is the case, MOG-IgG status, should be part of inclusion/exclusion criteria or a variable for stratification in clinical trials. The latter issue may have importance for currently recruiting trials that include seronegative NMOSD.
References
Hamid SH, Elsone L, Mutch K, Solomon T, Jacob A (2017) The impact of 2015 neuromyelitis optica spectrum disorders criteria on diagnostic rates. Mult Scler 23(2):228–233
Hyun JW, Jeong IH, Joung A, Kim SH, Kim HJ (2016) Evaluation of the 2015 diagnostic criteria for neuromyelitis optica spectrum disorder. Neurology 86(19):1772–1779
Ramanathan S, Dale RC, Brilot F (2016) Anti-MOG antibody: the history, clinical phenotype, and pathogenicity of a serum biomarker for demyelination. Autoimmun Rev 15(4):307–324
Waters PJ, McKeon A, Leite MI, Rajasekharan S, Lennon VA, Villalobos A et al (2012) Serologic diagnosis of NMO: a multicenter comparison of aquaporin-4-IgG assays. Neurology 78(9):665–671 (discussion 9)
Waters P, Woodhall M, O’Connor KC, Reindl M, Lang B, Sato DK et al (2015) MOG cell-based assay detects non-MS patients with inflammatory neurologic disease. Neurol Neuroimmunol Neuroinflamm 2(3):e89
Wingerchuk DM, Lennon VA, Pittock SJ, Lucchinetti CF, Weinshenker BG (2006) Revised diagnostic criteria for neuromyelitis optica. Neurology 66(10):1485–1489
Jarius S, Ruprecht K, Kleiter I, Borisow N, Asgari N, Pitarokoili K et al (2016) MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 1: frequency, syndrome specificity, influence of disease activity, long-term course, association with AQP4-IgG, and origin. J Neuroinflamm 13(1):279
Jarius S, Ruprecht K, Kleiter I, Borisow N, Asgari N, Pitarokoili K et al (2016) MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 2: epidemiology, clinical presentation, radiological and laboratory features, treatment responses, and long-term outcome. J Neuroinflamm 13(1):280
Hoftberger R, Sepulveda M, Armangue T, Blanco Y, Rostasy K, Cobo Calvo A et al (2015) Antibodies to MOG and AQP4 in adults with neuromyelitis optica and suspected limited forms of the disease. Mult Scler 21(7):866–874
Sato DK, Callegaro D, Lana-Peixoto MA, Waters PJ, de Haidar Jorge FM, Takahashi T et al (2014) Distinction between MOG antibody-positive and AQP4 antibody-positive NMO spectrum disorders. Neurology 82(6):474–481
Author information
Authors and Affiliations
Contributions
Dr. SHMH collected and compiled the data and wrote the manuscript. KM, MB, KD, SL collected data. Prof. TS critically reviewed the manuscript. DW collected data and reviewed the manuscript. Dr. AJ collected the data and critically reviewed the manuscript. Simple statistical analysis done by authors.
Corresponding author
Ethics declarations
Funding
The study is not industry sponsored.
Conflicts of interest
All authors declare no conflict of interest.
Ethical standards
This study meets UK ethical standards.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Hamid, S.H.M., Whittam, D., Mutch, K. et al. What proportion of AQP4-IgG-negative NMO spectrum disorder patients are MOG-IgG positive? A cross sectional study of 132 patients. J Neurol 264, 2088–2094 (2017). https://doi.org/10.1007/s00415-017-8596-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-017-8596-7