
Abstract
The field of neuronal surface-directed antibody-mediated diseases of the central nervous system has dramatically expanded in the last few years and now forms an important cluster of treatable neurological conditions. In this review, we focus on three areas. First, we review the demographics, clinical features and treatment responses of these conditions. Second, we consider their pathophysiology and compare autoantibody mechanisms and their effects to genetic or pharmacological disruptions of the target antigens. Third, we discuss areas of controversy within the field, propose possible resolutions, and explore new directions for neuronal surface antibody-mediated diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Neuroimmunology is a rapidly developing field with increasing scope and relevance to multiple neurological presentations. Autoantibody-associated neurology has expanded since the discovery of pathogenic acetylcholine receptor autoantibodies in myasthenia gravis in the 1970s, and subsequently other neuromuscular and peripheral nerve targets.
The first antibodies associated with diseases of the central nervous system (CNS) were termed ‘onconeuronal’ antibodies due to their frequent cancer associations [1, 2]. These antibodies target intracellular proteins (such as Hu, Yo, Ma2, Ri, Tr and CV2/CRMP5), the antibody levels do not correlate with disease severity, and prognosis is poor despite tumour removal and immunotherapies. A cytotoxic T cell-mediated mechanism is thought to be central to their pathophysiology and the role of the antibodies is less clear.
These features contrast markedly with the neuronal surface-directed antibody (NSAb)-associated CNS disorders. The antibodies are much less frequently associated with tumours, and are directed against extracellular epitopes on surface antigens strongly expressed within the CNS, such as the N-methyl-d-aspartate receptor (NMDAR) [3] and leucine-rich glioma-inactivated 1 (LGI1) [4]. The discovery of these NSAbs has helped identify treatable neurological conditions, with retrospective evidence that earlier treatment improves patient outcomes [5]. Although research is rapidly evolving, the available data strongly support pathogenic roles for the NSAbs. The antibody targets can be divided empirically into three groups: excitatory neurotransmitter receptors, inhibitory neurotransmitter receptors, ion-channel subunits or cell adhesion molecules.
NSAbs and their clinical features
Antibodies directed against proteins involved in excitatory neurotransmission
NMDAR
Since their discovery in 2007 [3], NMDAR-antibodies now represent a more frequent cause of encephalitis than viruses in patients under the age of 30 [6]. This encephalitis shows a stereotyped evolution from a viral prodrome to a neuropsychiatric presentation, with psychosis, cognitive dysfunction and seizures, followed by a progression to a distinctive movement disorder, dysautonomia and coma [7].
Since its original description the spectrum has widened and this disease has been associated with fewer tumours (Fig. 1a), almost all ovarian teratomas [7, 8], increasing numbers of paediatric cases (with only a 6 % association with a tumour seen in those under 12 [5, 7]), and more male cases, particularly in younger and older age groups [5, 7, 9].

a Trends in NMDAR-antibody encephalitis. Demographics of published cases (series containing >3 patients) with NMDAR (N-methyl-d-aspartate receptor)-antibody encephalitis. Note the slightly decreasing median age (black line) and increasing male and falling female representation (green and red, respectively). Tumour frequencies (blue line) are falling, mainly due to the recent publications of many paediatric cases. Figure adapted from Irani et al. [31]. b The effect of immunotherapy on mortality, the percentage with a good recovery (modified rankin score 0–2) and relapse-free recovery at 24 months. Data derived from Titulaer et al. [5]. c Key features of a representative patient with faciobrachial dystonic seizures (FBDS). Note the increasing seizure frequency (red line), poor response to anti-epileptic drugs (AEDs), time of onset of cognitive impairment (quantified by fall in Addenbrooke’s cognitive examination-Revised score (ACE-R, green line)) and of hyponatraemia (orange line). IT results in dramatic improvement in all features. LGI1 leucine-rich glioma-inactivated, VGKC voltage-gated potassium channel—complex antibody titres are shown in purple and black, respectively
Mono- or oligo-symptomatic presentations in patients with NMDAR-antibodies have also been recognised with predominant seizures and psychosis [7, 8, 10, 11]. Other presentations seen in a small proportion of NMDAR-antibody-positive patients include longitudinally extensive transverse myelitis [12] and optic neuritis [13]. This overlap with demyelinating diseases may relate to the expression of NMDARs on oligodendrocytes. However, an overlap with neuropsychiatric lupus, where double-stranded DNA antibodies have been reported to cross-react with the NMDAR [14, 15], is yet to be confirmed using cell-based assay techniques (discussed below) [3, 7].
NMDAR-antibody encephalitis has an approximately 13 % untreated mortality, as compared to 9 % with immunotherapy (Fig. 1b) [5]. One large study showed that 50 % of patients responded to first-line therapy [with corticosteroids, plasma exchange (PLEX) and/or intravenous immunoglobulin (IVIG)]. Of the remaining 50 %, second-line therapies (with cyclophosphamide and rituximab) offered a good outcome in 37.5 % compared to the 12.5 % that did not receive such therapies. Immunotherapy administration was also associated with lower relapse rate, often seen in the natural history of this disease (Figs. 1b, 2) [5]. While it yet may transpire that immunotherapy has little effect on the long-term outcomes of the disease survivors, importantly it appears to hasten recovery at 2 years and reduce mortality.

The contrasting probable natural histories of three antibody-related encephalitidies. Key things to note are the relapsing course NMDAR (N-methyl-d-aspartate receptor)-antibody encephalitis, often with a good long-term outcome. The LGI1 (leucine-rich glioma-inactivated 1) or CASPR2 (contactin-associated protein 2)-associated encephalitis has a tendency to be more monophasic often with residual memory and functional deficits. GAD (glutamic acid decarboxylase)-antibody-associated LE has an insidious onset and tends to adopt a more chronic course with ongoing seizures and memory deficits
α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA-type (glutamate receptor)) receptor
Antibodies to the GluR1 and GluR2 AMPA-receptor subunits often associate with a rare limbic encephalitis (LE) in older females, typically with tumours of the thymus, breast and lung [16]. The syndrome of LE produces amnesia, disorientation and seizures and is also seen in patients with GAD, LGI1 and GABABR-antibodies, as discussed later. Some patients show a good response to immunotherapy [16]. All examined tumours expressed at least one of the antigens, and a predominance of one subunit in the tumour mirrored the antibody preference seen in the same patient [16].
AMPA receptors are usually tetramers of GluR subunits 1–4. GluR1/2 and GluR2/3 subunits are mostly post-synaptic and are expressed at especially high concentrations in limbic brain regions [17]. AMPAR-antibody-associated phenotypes have spread to include two patients with an acute psychosis-like illness [18] and antibodies to GluR2/3 receptors were found in two patients with Rasmussen’s encephalitis [Nibber et al. in preparation].
Antibodies to proteins involved in inhibitory neurotransmission
G AD
Glutamic acid decarboxylase (GAD) is a widely expressed intracellular enzyme which catalyses the synthesis of gamma aminobutyric acid (GABA), the major inhibitory CNS neurotransmitter. Antibodies to GAD are seen in type 1 diabetes mellitus, and usually at much higher titres in LE, cerebellar ataxia, epilepsy and the stiff person syndrome (SPS) spectrum [19].
SPS is characterised by rigidity, stimulus-induced spasms, anxiety, and more rarely, oculomotor and autonomic disturbances [19, 20]. By contrast, GAD-antibody-associated LE is predominantly a disease of young women and usually presents with AED-refractory epilepsy and amnesia, but without rigidity or spasms. The clinical features and GAD-antibody levels are often immunotherapy resistant, and the disease shows a chronic course (Fig. 2) [21]. However, serum and CSF IgGs from patients with GAD-antibodies do reproduce some of the clinical features of SPS in rodents [22]. The antibodies may access antigen upon its cell-surface exposure during exocytosis or programmed cell death [23, 24] or perhaps co-exist with pathogenic NSAbs. Indeed, antibodies to the AMPA, glycine, GABAB and GABAA receptors, in addition to novel/undefined NSAbs, have all been observed in patients with GAD-antibody-related neurology [25–31].
Glycine receptor
Progressive encephalomyelitis with rigidity and myoclonus (PERM) is at one end of the SPS spectrum with the poorest prognosis, and usually these patients have no GAD-antibodies [30]. In 2008, a patient with PERM without GAD-antibodies was found to have antibodies directed against the glycine-receptor (GlyR) alpha1 subunit [32]. Subsequently, GlyR-antibodies have been reported in patients with classical and variant SPS, brainstem encephalitis, a few with LE, many with PERM, and occasionally in patients with demyelinating disease. There is a good response to immunotherapy (median modified Rankin Scale scores fall from of 5 to 1) [27, 33, 34]. Tumour associations are infrequent but thymoma and lymphoma have been reported [27]. The GlyR is expressed in the upper and lower brainstem, diencephalon and the colliculi as well as the dorsal and ventral horns of the spinal cord: these localisations correlate well with the observed clinical features [27].
GABA B receptor
GABAB-antibodies, predominantly reacting with the GABAB1 subunit, have been associated with a form of LE, usually of later life, with prominent seizures [25, 26]. More recently the phenotype has expanded to include presentations with cerebellar ataxia, status epilepticus, and opsoclonus myoclonus, often in patients with cognitive impairment [25, 35]. There is a close association with small-cell lung cancers (SCLC) [25, 26, 36], which express the GABABR [25]. Mortality is high, especially in tumour-related cases, but 80 % of patients initially respond to immunotherapy, plus tumour removal where relevant [25].
GABAA receptor
Antibodies to the GABA α1/β3 subunits have recently been described in a small number of patients. When detected at high serum titres (>1:160) and in the CSF, these were associated with LE, status epilepticus or epilepsia partialis continua [28]. Patients have unusual cortical and subcortical imaging hyperintensities, a variable response to immunotherapy, and high mortality due to status epilepticus. Twelve patients with other neurological diseases had lower titre serum GABAA-antibodies, not detected in the CSF, with a broader spectrum of diseases including LE, SPS, and opsoclonus myoclonus [28]. Autoantibodies against the α1 and/or γ2 subunits were found in patients with seizures (47 %), memory impairment (47 %) and hallucinations (33 %); one had non-Hodgkin’s lymphoma (Pettingill et al. in press). In that study, however, many patients were not considered to have immune-mediated diseases, and immunotherapies were not used in most. Nevertheless, the antibodies internalised the GABAAR subunits in vitro, consistent with their pathogenic potential [37] (Pettingill et al. in press).
Antibodies directed against ion-channel-associated proteins and cell adhesion proteins
Voltage-gated potassium channel (VGKC) complex
Antibodies to the VGKC-complex were originally described in patients with peripheral nerve hyperexcitability (PNH) syndromes [38]. Since 2001, these antibodies have been recognised in patients with CNS features including Morvan’s syndrome (MoS) [39, 40], LE [4, 41, 42], faciobrachial dystonic seizures (FBDS) [43–45], a minority of patients with cryptogenic epilepsies [46], neuropathic pain syndromes [47] and some cerebellar ataxias [48]. VGKC-complex antibodies are detected by immunoprecipitation of iodinated alpha-dendrotoxin (α-DTX)-labelled VGKCs from digitonin-solubilised mammalian brain homogenates. α-DTX is known to bind with high affinity to the VGKC subunits Kv1.1, 1.2 and 1.6. Based on this, Kv1.1, 1.2 and 1.6 were considered the likely target epitopes [49]. However, only a minority of IgGs bind the Kv1 subunits themselves [4]. A much larger proportion bind to target cell-surface domains of proteins which are tightly associated with Kv1 subunits; most commonly LGI1 and contactin-associated protein-2 (CASPR2) [4, 39, 50] (Fig. 3). A smaller proportion were found to target contactin-2, which have been reported in association with LGI1- and CASPR2-antibodies [4].

Illustration of the VGKC-complexes: the association of Kv1s and CASPR2 (contactin-associated protein), LGI1 (leucine-rich glioma-inactivated and other components of the complexes. a Neuronal subcellular domains including the axon initial segment, presynaptic terminal, node of Ranvier (NoR), paranode (PN), juxtaparanode (JXTPN) and internode (IN). b Juxtaparanode: Kv1 channels (blue, alpha subunits = rectangle, beta subunit = circle), CASPR2 (pink oval), contactin-2 (black diamond), MAGUKs (membrane-associated guanylate-kinases) (semicircles), protein 4.1B/spectrins/ankyrins (green/blue triangles). c Synaptic Kv1 organisation. Kv1 s (blue, such as Kv1.1), LGI1 (red) and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPAR) and ADAM22/23 (a disintegrin and metalloproteinase 22/23) (brown) anchored at post-synaptic membranes
LGI1
LGI1 is a secreted protein that interacts in situ with Kv1.1, Kv1.2 and AMPARs. LGI1 forms a trans-synaptic protein complex with presynaptic ADAM23 (a disintegrin and metalloproteinase 23) and post-synaptic ADAM22 [51]. It is expressed throughout the brain, especially in the hippocampus and neocortex [4].
LGI1-antibodies are often found in LE [4, 52]. As the descriptions of LGI1-antibody-positive cohorts have grown, it has become increasingly clear that this is only rarely paraneoplastic, has an equal sex distribution, responds well to immunotherapy, and has a low overall mortality [4, 45, 50, 53] (see Table 1). Although there are descriptions of untreated partial recovery over around 2 years [54, 55], larger cohorts suggest that early immunotherapy offers the best short-term outcomes [42, 56], and recent data indicate that the addition of PLEX and/or IVIG to corticosteroids may not alter 4-year outcomes [31].
Several studies have described LGI1-antibodies in patients with isolated seizure syndromes of multiple semiologies, which are often immunotherapy-responsive [57–59]. A recent clinical observation has been the association of a highly distinctive seizure semiology—termed faciobrachial dystonic sseizures (FBDS)—in patients with LGI1-antibodies. These stereotyped events, characterised by their high frequency (median 50/day), short duration (usually <3 s) and their predilection for the hemiface and ipsilateral arm, are often refractory to anti-epileptic drugs but preferentially respond to the addition of immunotherapies (see Fig. 1c) [43–45, 60, 61]. Importantly, the onset of FBDS often precedes the onset of the cognitive impairment seen in patients with LE and one small prospective study has suggested that cognitive impairment may be avoided with early treatment of FBDS [43–45, 60]. In addition, ictal bradycardia and piloerection may be seizure semiologies enriched in patients with LGI1-antibodies [62, 63].
CASPR2
CASPR2 is a transmembrane protein localised to the juxtaparanode of myelinated axons. The extracellular domain of CASPR2 interacts with contactin-2 in both cis and trans (Fig. 3b), and in association with other proteins is responsible for concentrating Kv1.1 and Kv1.2 channels at the juxtaparanode [64]. Therefore, CASPR2 has cell adhesion and Kv1-partner functions. Patients with LE, PNH and subacute cerebellitis [48] have CASPR2-antibodies in around 10, 30 and 10 % of cases, respectively. However, CASPR2-antibodies are most consistently associated with MoS, in which about 50 % of patients also have LGI1-antibodies [39]. This combination may generate both the CNS and PNS features of MoS. MoS occurs almost exclusively in males, and interestingly, the prostate is one of the few non-neuronal sites of CASPR2 expression and CASPR2-antibody-associated MoS has been described post-scrotal hydrocele drainage [65]. Another potential immunisation mechanism is via the associated thymomas present in around 50 % of MoS and especially in patients with CASPR2-antibodies [39].
Other VGKC-complex proteins
Most VGKC-complex antibody-positive patients with higher VGKC-complex titres (>400 pM), have LGI1- or CASPR2-antibodies. Much more commonly at lower levels (100–400 pM), the targets of the antibodies are not yet known [4, 53]. Serum and CSFs from many of these patients do not show binding to the surface of live hippocampal neurons (Vincent, unpublished), suggesting that they may target intracellular VGKC-complex epitopes. While these may not be pathogenic, the antibodies may still be predictive of a neuroinflammatory syndrome and a response to immunotherapy, or an inflammatory component to a neurodegenerative disease [66, 67] (Hacohen et al. submitted).
Dipeptidyl-peptidase-like protein-6 (DPPX)
A subacute LE associated with tremor, myoclonus and diarrhoea was described in association with antibodies to DPPX, a cell-surface protein associated with the Kv4.2 potassium channel [68]. A more recent study has highlighted the brainstem focus of this condition and the multiorgan dysautonomia with bladder and cardiac involvement [69]. The condition is usually severe, with a gradual response to immunotherapy and relapses without immunotherapy [68].
IgLON5
Not all NSAbs are pathogenic. Antibodies to IgLON5, a neuronal cell adhesion protein involved in synapse formation, were described in patients with a progressive complex neurodegenerative sleep disorder with disordered breathing [70]. The most striking aspect of these patients was their lack of response to immunotherapy and atypical brainstem tau deposition [70]. The association of an NSAb with a neurodegenerative disease adds to evidence from CJD studies [71, 72] that NSAbs may not always play a primary role but can be secondary to neuronal damage with possible implications for disease progression or disease biomarkers.
Antibodies associated with glial damage, specifically to aquaporin-4 (AQP4) and myelin oligodendrocyte glycoprotein (MOG), are summarised in Table 1.
Pathogenic considerations
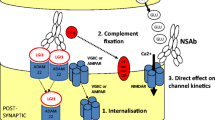
Exactly how these antibodies lead to the observed pathology is an area of active research (Fig. 4). For instance, many but not all NSAbs induce receptor internalisation in vitro resulting in receptor loss, as observed in myasthenia gravis. This applies to NMDAR [8], AMPAR [16], GABAAR [37] and GlyR [27] antibodies and can be demonstrated in cell cultures. Other NSAbs may mediate a direct effect on channel kinetics. LGI1- and DPPX-antibodies may indirectly induce channel modulation: for example, LGI1-antibodies appear to reduce Kv1 channel function [73] and to decrease AMPAR expression in vitro [74]. It is likely that the antibodies binding to LGI1 disrupt the trans-synaptic bridge between the pre- and post-synaptic membranes and this may affect the function of both VGKCs and AMPARs. In addition, however, antibodies of the IgG1 and IgG3 subclasses have the ability to fix complement. In biopsy studies, this has been shown to occur with AQP4- (and less so LGI1-) antibodies but not with NMDAR-antibodies [75, 76]. Mechanisms that appear to prevent complement fixation by the IgG1-subclass NMDAR-antibodies should be explored in future studies.

Potential pathogenic mechanisms of neuronal surface-directed antibodies (NSAbs). a Internalisation of receptors has been demonstrated in vitro using NMDAR (N-methyl-d-aspartate receptor), AMPAR (a-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor) and GABAAR (γ-aminobutyric acid A receptor)-antibodies. Here the LGI1–ADAM22 interaction is shown as a possible unit for co-internalisation. b Antibody-mediated complement fixation and complement-mediated membrane receptor disruption as seen with antibodies against AQP4 (aquaporin-4). c Direct alteration of ion-channel molecular function is an alternative mechanism
To further explore NSAb-pathogenicity, below we highlight features of genetic or pharmacological situations in which the target antigen is relatively specifically disrupted and compare this to the corresponding antibody-mediated process (Table 2).
Mutations, drugs and antibodies which target the NR1 subunit
NR1 homozygous null mice die 8 h after birth and hypoventilate, similar to patients with NMDAR-antibody encephalitis [77]. Mice with a 50 % NR1 genetic knockout exhibit both psychiatric and cognitive signs, similar to those seen in NMDAR-antibody encephalitis but without a movement disorder or seizures [78]. As described in Table 2, polymorphisms and de novo mutations in the human NR1 subunit gene (GRIN1), and NMDAR-antagonists such as ketamine and phencyclidine, recapitulate many aspects of NMDAR-antibody encephalitis. However, the ‘full’ syndrome appears to be unique to autoantibodies targeting the NR1 subunit.
GABAB receptor mutations and medications
Pathophysiologically, GABABR-antibody LE shows predominant seizures. This concurs with observations from murine genetic and pharmacological studies of GABABR downregulation [79, 80], but by contrast to most other antibodies GABABR-antibodies do not appear to internalise their target antigen. There are no documented GABAB1R human mutants, but GABAB1R polymorphisms have been associated with temporal lobe epilepsy [81], schizophrenia [82] and obsessive–compulsive disorder [83].
LGI1 mutations
Leucine-rich glioma-inactivated 1 homozygous null mice develop myoclonic seizures at days 12–18 of life, dying soon after [84]. Electrophysiological studies in both mutant LGI1 and LGI1 null mice demonstrate increased synaptic excitation [84, 85], thought to be mediated by increased glutamate efflux [84]. LGI1 mutations in humans cause autosomal dominant lateral temporal lobe epilepsy (ADLTE), with ictal auditory hallucinations [86]. Some patients have generalised tonic–clonic seizures, sensory aphasic seizures, and a few kindreds have ictal psychic phenomena [87].
Contrasts are stark between LGI1 human mutants and the corresponding antibody-mediated syndromes. In patients with LGI1 mutations, despite focal temporal lobe seizures, there are no cognitive or psychiatric manifestations, seizure semiology is predominantly auditory, and MRIs are normal. The opposites are true of patients with FBDS- and LGI1-antibodies. These differences may be accounted for by antibody access to only specific brain regions, the effect of complement-mediated neuronal damage, or genetic compensation in LGI1 mutants which may not occur in a rapid onset antibody-mediated disorder. Perhaps some effects of LGI1 mutations are via expression or function of VGKCs. Indeed, humans with Kv1.1 mutations have neuromyotonia and an increased rate of seizures [88, 89].
CASPR2 mutants
Contactin-associated protein-2 null mice were originally thought to have normal behaviour and neuronal growth despite loss of K+ channel juxtaparanode clustering [90]. More recent analyses have shown that they demonstrate hyperlocomotion, repetitive and inflexible behaviours, impaired socialising and seizures after 6 months of age [91].
The findings are reminiscent of those seen in autistic spectrum disorders, and mutations/polymorphisms in the CASPR2-encoding gene, CNTNAP2, have been linked to autism [92]. Interestingly, a recessive non-coding mutation for CNTNAP2 causes cortical dysplasia focal epilepsy syndrome (CDFE), characterised by seizures, intellectual disability, hyperactivity, and in two-thirds of cases, autism [93]. Mutations in CNTNAP2 have also been linked to schizophrenia, psychosis and other forms of epilepsy [94]. Therefore, there are clear similarities between patients with CASPR2-antibodies and mutations in CASPR2.
Controversies, possible resolutions and new directions
Antibody levels and assay methodologies
Early in these illnesses, the levels of serum autoantibodies are almost always higher than CSF autoantibodies. This seems intuitive in patients with a peripheral tumour, such as an ovarian teratoma, and also given patients positive clinical responses after plasma exchange. Therefore, it seems likely that reports of autoantibody detection in the CSF, but not serum, are due to differences in assay methodologies. One consideration is that the presence of intrathecal autoantibody synthesis, particularly seen with NMDAR-antibodies, and the constitutively low total IgG levels in CSF makes CSF easier to use than serum in diagnostic assays. As differences between antibody-detection methods have been discussed in detail elsewhere [31, 95, 96], here we summarise the main areas of controversy.
Autoantibodies with pathogenic potential recognise the extracellular domains of native membrane proteins. They are very rarely detected in denaturing western blots. However, assays utilised in the field do not always exclusively detect these autoantibodies. For example, the use of fixed tissue (where native epitopes may be destroyed) and the use of permeabilised antigen-transfected cells to detect antibodies may permit non-pathogenic autoantibody binding [8, 96]. Despite this possibility, the concurrent use of live hippocampal neurons [4, 8] and techniques of antibody absorption exclusively against the extracellular domain [4, 97] allay this concern [31]. In conclusion, differences between current assays suggest that both CSF and serum should be sent to laboratories whenever possible, and future cross-laboratory comparative assays should help understand the differences described above.
NSAbs in other neurological diseases and the healthy population
In a recent study by Dahm et al., sera from over 4,000 healthy and disease controls with varied neuropsychiatric presentations (including schizophrenia, ALS, Parkinson’s and stroke) were screened for a panel of NSAbs and intracellular-targeted antibodies. ~11 % of the combined cohort was found to be positive for IgM- (6 %), IgA- (5 %) and IgG (1 %)-NMDAR-antibodies at titres from 1:10 to 1:1,000 with equal proportions in disease and healthy controls. Other frequently detected antibodies were amphiphysin (2.0 %), CASPR2 (0.9 %), MOG (0.8 %), GAD65 (0.5 %), Ma2 (0.5 %), Yo (0.4 %) and Ma1 (0.4 %), also with similar frequencies in disease and healthy controls [98]. The use of a permeabilised cell-based assay without CSF testing may account for the lowered specificity. However, as these antibodies appear to be present in healthy controls, this study suggests that clinical syndrome classification remains key to defining disease-relevant autoantibodies with pathogenic potential. Indeed, the lack of gold standards for disease, independent of antibody positivity, for research purposes is a problem for future studies.
IgA and IgM autoantibodies
Antibodies of the IgG class associate with all of the NSAb-mediated diseases discussed thus far. There have, however, been reports of IgA- and IgM-NMDAR-antibodies associated with slow cognitive impairment [99], psychosis and bipolar disorder [100]. These IgM-NMDAR-antibodies caused a reduction in cell survival and NR1 expression in cultured rodent neurons [100], suggesting pathogenic potential. One study suggested that 31 % of patients with IgG-NMDAR-antibodies also had IgA-NMDAR-antibodies [99].
Autoantibody triggers: tumours, infections and neurodegeneration
As tumours often express the antigen of interest in paraneoplastic NSAb-associated conditions, they are the likely sites of antigen presentation. In addition, other paradigms for breaking immune tolerance have arisen.
After herpes simplex virus encephalitis (HSVE), children often suffer relapses which have recently been associated with NMDAR-antibodies [101, 102]. Relapses occurred a few weeks to months after the HSVE, phenotypically closely resemble classical NMDAR-antibody encephalitis, and appear to be immunotherapy-responsive. The NMDAR-antibodies were found alongside other novel NSAbs and may represent systemic immunisation after neuronal damage. Indeed in adults, generation of VGKC-complex, glycine receptor and NMDAR-antibodies has been observed in a small proportion (<5 %) of patients with rapid neurodegeneration as seen in CJD [72, 103]. A more elegant example of this is the observation that VGKC-complex antibodies (amongst others) are generated in abattoir workers after exposure to aerosolised porcine neural tissue [104]. Furthermore, experimental rodents exposed to inhaled brain tissue aerosol developed a similar clinical and serological profile to their human counterparts [105]. It is likely that these antibodies are secondary.
In summary, multiple triggers appear able to generate serum autoantibodies with pathogenic potential. Factors governing the antibody-pathogenicity may include their access to the brain/CSF compartments, concentrations, the duration of antibody production, and intrinsic individual patient thresholds.
Conclusions
Neuroimmunology has moved on from its position 15 years ago where it principally reflected research into multiple sclerosis and the pace of change and growth seems set to continue. There are increasing numbers of NSAbs associated with defined conditions. With the majority of discoveries being recent, much work will be needed to hone these phenotypes, their pathophysiological basis, optimal treatments, prognosis and longer term management. There are still methodological issues to settle, such as the best way to test for NSAbs, the independent gold standards for diagnosis of each condition and the relevance of low antibody levels in patients with non-classical syndromes. There are still antibodies to be found, for example in the VGKC-antibody-positive patients without LGI1- and CASPR2-antibodies. The underlying immunological mechanisms remain only partly characterised and much work will address this in coming years.
In the meantime, work must go on by the bedside, with clinicians recognising signs which suggest an underlying autoimmune condition and initiating the testing to detect an antibody, novel or otherwise. Only with such collaboration and high-quality clinical work can progress continue to be made in the laboratory to reduce the impact of these often devastating diseases.
References
Leypoldt F, Wandinger KP (2014) Paraneoplastic neurological syndromes. Clin Exp Immunol 175(3):336–348
McKeon A, Pittock SJ (2011) Paraneoplastic encephalomyelopathies: pathology and mechanisms. Acta Neuropathol 122(4):381–400
Dalmau J, Tuzun E, Wu HY, Masjuan J, Rossi JE et al (2007) Paraneoplastic anti-N-methyl-d-aspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol 61(1):25–36
Irani SR, Alexander S, Waters P, Kleopa KA, Pettingill P et al (2010) Antibodies to Kv1 potassium channel-complex proteins leucine-rich, glioma inactivated 1 protein and contactin-associated protein-2 in limbic encephalitis, Morvan’s syndrome and acquired neuromyotonia. Brain 133(9):2734–2748
Titulaer MJ, McCracken L, Gabilondo I, Armangue T, Glaser C et al (2013) Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet Neurol 12(2):157–165
Gable MS, Sheriff H, Dalmau J, Tilley DH, Glaser CA (2012) The frequency of autoimmune N-methyl-d-aspartate receptor encephalitis surpasses that of individual viral etiologies in young individuals enrolled in the California Encephalitis Project. Clin Infect Dis 54(7):899–904
Irani SR, Bera K, Waters P, Zuliani L, Maxwell S et al (2010) N-methyl-d-aspartate antibody encephalitis: temporal progression of clinical and paraclinical observations in a predominantly non-paraneoplastic disorder of both sexes. Brain 133(Pt 6):1655–1667
Dalmau J, Gleichman AJ, Hughes EG, Rossi JE, Peng X et al (2008) Anti-NMDA-receptor encephalitis: case series and analysis of the effects of antibodies. Lancet Neurol 7(12):1091–1098
Florance NR, Davis RL, Lam C, Szperka C, Zhou L et al (2009) Anti-N-methyl-d-aspartate receptor (NMDAR) encephalitis in children and adolescents. Ann Neurol 66(1):11–18
Zandi MS, Irani SR, Lang B, Waters P, Jones PB et al (2011) Disease-relevant autoantibodies in first episode schizophrenia. J Neurol 258(4):686–688
Niehusmann P, Dalmau J, Rudlowski C, Vincent A, Elger CE et al (2009) Diagnostic value of N-methyl-d-aspartate receptor antibodies in women with new-onset epilepsy. Arch Neurol 66(4):458–464
Outteryck O, Baille G, Hodel J, Giroux M, Lacour A et al (2013) Extensive myelitis associated with anti-NMDA receptor antibodies. BMC Neurol 13:211
Cobo-Calvo A, Izquierdo Gracia C, Quinones SM, Torro CM, Saiz A et al (2014) Optic neuritis in the setting of NMDA receptor encephalitis. J Neuro-ophthalmol 34(3):316–319
ter Borg EJ, Horst G, Hummel EJ, Limburg PC, Kallenberg CG (1990) Measurement of increases in anti-double-stranded DNA antibody levels as a predictor of disease exacerbation in systemic lupus erythematosus. A long-term, prospective study. Arthritis Rheum 33(5):634–643
Hanly JG (2014) Diagnosis and management of neuropsychiatric SLE. Nat Rev Rheumatol 10(6):338–347
Lai M, Hughes EG, Peng X, Zhou L, Gleichman AJ et al (2009) AMPA receptor antibodies in limbic encephalitis alter synaptic receptor location. Ann Neurol 65(4):424–434
Sprengel R (2006) Role of AMPA receptors in synaptic plasticity. Cell Tissue Res 326(2):447–455
Graus F, Boronat A, Xifro X, Boix M, Svigelj V et al (2010) The expanding clinical profile of anti-AMPA receptor encephalitis. Neurology 74(10):857–859
Saiz A, Blanco Y, Sabater L, Gonzalez F, Bataller L et al (2008) Spectrum of neurological syndromes associated with glutamic acid decarboxylase antibodies: diagnostic clues for this association. Brain 131(Pt 10):2553–2563
Mata S, Muscas GC, Naldi I, Rosati E, Paladini S et al (2008) Non-paraneoplastic limbic encephalitis associated with anti-glutamic acid decarboxylase antibodies. J Neuroimmunol 199(1–2):155–159
Malter MP, Helmstaedter C, Urbach H, Vincent A, Bien CG (2010) Antibodies to glutamic acid decarboxylase define a form of limbic encephalitis. Ann Neurol 67(4):470–478
Manto MU, Laute MA, Aguera M, Rogemond V, Pandolfo M et al (2007) Effects of anti-glutamic acid decarboxylase antibodies associated with neurological diseases. Ann Neurol 61(6):544–551
Christgau S, Aanstoot HJ, Schierbeck H, Begley K, Tullin S et al (1992) Membrane anchoring of the autoantigen GAD65 to microvesicles in pancreatic beta-cells by palmitoylation in the NH2-terminal domain. J Cell Biol 118(2):309–320
Chavez-Valdez R, Martin LJ, Flock DL, Northington FJ (2012) Necrostatin-1 attenuates mitochondrial dysfunction in neurons and astrocytes following neonatal hypoxia-ischemia. Neuroscience 219:192–203
Lancaster E, Lai M, Peng X, Hughes E, Constantinescu R et al (2010) Antibodies to the GABA(B) receptor in limbic encephalitis with seizures: case series and characterisation of the antigen. Lancet Neurol 9(1):67–76
Boronat A, Sabater L, Saiz A, Dalmau J, Graus F (2011) GABA(B) receptor antibodies in limbic encephalitis and anti-GAD-associated neurologic disorders. Neurology 76(9):795–800
Carvajal-Gonzalez A, Leite MI, Waters P, Woodhall M, Coutinho E et al (2014) Glycine receptor antibodies in PERM and related syndromes: characteristics, clinical features and outcomes. Brain 137(Pt 8):2178–2192
Petit-Pedrol M, Armangue T, Peng X, Bataller L, Cellucci T et al (2014) Encephalitis with refractory seizures, status epilepticus, and antibodies to the GABAA receptor: a case series, characterisation of the antigen, and analysis of the effects of antibodies. Lancet Neurol 13(3):276–286
Chang T, Alexopoulos H, McMenamin M, Carvajal-Gonzalez A, Alexander SK et al (2013) Neuronal surface and glutamic acid decarboxylase autoantibodies in nonparaneoplastic stiff person syndrome. JAMA Neurol 70(9):1140–1149
Meinck HM, Thompson PD (2002) Stiff man syndrome and related conditions. Mov Disord 17(5):853–866
Irani SR, Gelfand JM, Al-Diwani A, Vincent A (2014) Cell-surface central nervous system autoantibodies: clinical relevance and emerging paradigms. Ann Neurol 76(2):168–184
Hutchinson M, Waters P, McHugh J, Gorman G, O’Riordan S et al (2008) Progressive encephalomyelitis, rigidity, and myoclonus: a novel glycine receptor antibody. Neurology 71(16):1291–1292
Damasio J, Leite MI, Coutinho E, Waters P, Woodhall M et al (2013) Progressive encephalomyelitis with rigidity and myoclonus: the first pediatric case with glycine receptor antibodies. JAMA Neurol 70(4):498–501
Turner MR, Irani SR, Leite MI, Nithi K, Vincent A et al (2011) Progressive encephalomyelitis with rigidity and myoclonus: glycine and NMDA receptor antibodies. Neurology 77(5):439–443
Mundiyanapurath S, Jarius S, Probst C, Stocker W, Wildemann B et al (2013) GABA-B-receptor antibodies in paraneoplastic brainstem encephalitis. J Neuroimmunol 259(1–2):88–91
Kim TJ, Lee ST, Shin JW, Moon J, Lim JA et al (2014) Clinical manifestations and outcomes of the treatment of patients with GABAB encephalitis. J Neuroimmunol 270(1–2):45–50
Ohkawa T, Satake S, Yokoi N, Miyazaki Y, Ohshita T et al (2014) Identification and characterization of GABA(A) receptor autoantibodies in autoimmune encephalitis. J Neurosci 34(24):8151–8163
Hart IK, Waters C, Vincent A, Newland C, Beeson D et al (1997) Autoantibodies detected to expressed K+ channels are implicated in neuromyotonia. Ann Neurol 41(2):238–246
Irani SR, Pettingill P, Kleopa KA, Schiza N, Waters P et al (2012) Morvan syndrome: clinical and serological observations in 29 cases. Ann Neurol 72(2):241–255
Liguori R, Vincent A, Clover L, Avoni P, Plazzi G et al (2001) Morvan’s syndrome: peripheral and central nervous system and cardiac involvement with antibodies to voltage-gated potassium channels. Brain 124(Pt 12):2417–2426
Thieben MJ, Lennon VA, Boeve BF, Aksamit AJ, Keegan M et al (2004) Potentially reversible autoimmune limbic encephalitis with neuronal potassium channel antibody. Neurology 62(7):1177–1182
Vincent A, Buckley C, Schott JM, Baker I, Dewar BK et al (2004) Potassium channel antibody-associated encephalopathy: a potentially immunotherapy-responsive form of limbic encephalitis. Brain 127(Pt 3):701–712
Barajas RF, Collins DE, Cha S, Geschwind MD (2010) Adult-onset drug-refractory seizure disorder associated with anti-voltage-gated potassium-channel antibody. Epilepsia 51(3):473–477
Irani SR, Michell AW, Lang B, Pettingill P, Waters P et al (2011) Faciobrachial dystonic seizures precede Lgi1 antibody limbic encephalitis. Ann Neurol 69(5):892–900
Irani SR, Stagg CJ, Schott JM, Rosenthal CR, Schneider SA et al (2013) Faciobrachial dystonic seizures: the influence of immunotherapy on seizure control and prevention of cognitive impairment in a broadening phenotype. Brain 136(Pt 10):3151–3162
McKnight K, Jiang Y, Hart Y, Cavey A, Wroe S et al (2005) Serum antibodies in epilepsy and seizure-associated disorders. Neurology 65(11):1730–1736
Klein CJ, Lennon VA, Aston PA, McKeon A, Pittock SJ (2012) Chronic pain as a manifestation of potassium channel-complex autoimmunity. Neurology 79(11):1136–1144
Becker EB, Zuliani L, Pettingill R, Lang B, Waters P et al (2012) Contactin-associated protein-2 antibodies in non-paraneoplastic cerebellar ataxia. J Neurol Neurosurg Psychiatry 83(4):437–440
Kleopa KA, Elman LB, Lang B, Vincent A, Scherer SS (2006) Neuromyotonia and limbic encephalitis sera target mature Shaker-type K+ channels: subunit specificity correlates with clinical manifestations. Brain 129(Pt 6):1570–1584
Lai M, Huijbers MG, Lancaster E, Graus F, Bataller L et al (2010) Investigation of LGI1 as the antigen in limbic encephalitis previously attributed to potassium channels: a case series. Lancet Neurol 9(8):776–785
Fukata Y, Lovero KL, Iwanaga T, Watanabe A, Yokoi N et al (2010) Disruption of LGI1-linked synaptic complex causes abnormal synaptic transmission and epilepsy. Proc Nat Acad Sci USA 107(8):3799–3804
Malter MP, Frisch C, Schoene-Bake JC, Helmstaedter C, Wandinger KP et al (2014) Outcome of limbic encephalitis with VGKC-complex antibodies: relation to antigenic specificity. J Neurol 261(9):1695–1705
Klein CJ, Lennon VA, Aston PA, McKeon A, O’Toole O et al (2013) Insights from LGI1 and CASPR2 potassium channel complex autoantibody subtyping. JAMA Neurol 70(2):229–234
Buckley C, Oger J, Clover L, Tuzun E, Carpenter K et al (2001) Potassium channel antibodies in two patients with reversible limbic encephalitis. Ann Neurol 50(1):73–78
Szots M, Marton A, Kover F, Kiss T, Berki T et al (2014) Natural course of LGI1 encephalitis: 3–5 years of follow-up without immunotherapy. J Neurol Sciences 343(1–2):198–202
Shin YW, Lee ST, Shin JW, Moon J, Lim JA et al (2013) VGKC-complex/LGI1-antibody encephalitis: clinical manifestations and response to immunotherapy. J Neuroimmunol 265(1–2):75–81
Lilleker JB, Jones MS, Mohanraj R (2013) VGKC complex antibodies in epilepsy: diagnostic yield and therapeutic implications. Seizure 22(9):776–779
Plantone D, Renna R, Grossi D, Plantone F, Iorio R (2013) Teaching NeuroImages: basal ganglia involvement in facio-brachial dystonic seizures associated with LGI1 antibodies. Neurology 80(17):e183–e184
Quek AM, Britton JW, McKeon A, So E, Lennon VA et al (2012) Autoimmune epilepsy: clinical characteristics and response to immunotherapy. Arch Neurol 69(5):582–593
Irani SR, Buckley C, Vincent A, Cockerell OC, Rudge P et al (2008) Immunotherapy-responsive seizure-like episodes with potassium channel antibodies. Neurology 71(20):1647–1648
Yoo JY, Hirsch LJ (2014) Limbic encephalitis associated with anti-voltage-gated potassium channel complex antibodies mimicking Creutzfeldt-Jakob disease. JAMA Neurol 71(1):79–82
Rocamora R, Becerra JL, Fossas P, Gomez M, Vivanco-Hidalgo RM et al (2014) Pilomotor seizures: an autonomic semiology of limbic encephalitis? Seizure 23(8):670–673
Naasan G, Irani SR, Bettcher BM, Geschwind MD, Gelfand JM (2014) Episodic bradycardia as neurocardiac prodrome to voltage-gated potassium channel complex/leucine-rich, glioma inactivated 1 antibody encephalitis. JAMA Neurol 71(10):1300–1304
Gu C, Gu Y (2011) Clustering and activity tuning of Kv1 channels in myelinated hippocampal axons. J Biol Chem 286(29):25835–25847
Sharma S, Sharma P (2013) Morvan syndrome: after scrotal sac drainage and chemical instillation in hydrocele. Neurol India 61(3):300–302
Olberg H, Haugen M, Storstein A, Vedeler CA (2013) Neurological manifestations related to level of voltage-gated potassium channel antibodies. J Neurol Neurosurg Psychiatry 84(8):941–943
Paterson RW, Zandi MS, Armstrong R, Vincent A, Schott JM (2014) Clinical relevance of positive voltage-gated potassium channel (VGKC)-complex antibodies: experience from a tertiary referral centre. J Neurol Neurosurg Psychiatry 85(6):625–630
Boronat A, Gelfand JM, Gresa-Arribas N, Jeong HY, Walsh M et al (2013) Encephalitis and antibodies to dipeptidyl-peptidase-like protein-6, a subunit of Kv4.2 potassium channels. Ann Neurol 73(1):120–128
Tobin WO, Lennon VA, Komorowski L, Probst C, Clardy SL et al (2014) DPPX potassium channel antibody: Frequency, clinical accompaniments, and outcomes in 20 patients. Neurology 83(20):1797–1803
Sabater L, Gaig C, Gelpi E, Bataller L, Lewerenz J et al (2014) A novel non-rapid-eye movement and rapid-eye-movement parasomnia with sleep breathing disorder associated with antibodies to IgLON5: a case series, characterisation of the antigen, and post-mortem study. Lancet Neurol 13(6):575–586
Angus-Leppan H, Rudge P, Mead S, Collinge J, Vincent A (2013) Autoantibodies in sporadic Creutzfeldt-Jakob disease. JAMA Neurol 70(7):919–922
Rossi M, Mead S, Collinge J, Rudge P, Vincent A (2014) Neuronal antibodies in patients with suspected or confirmed sporadic Creutzfeldt-Jakob disease. J Neurol Neurosurg Psychiatry (in press)
Lalic T, Pettingill P, Vincent A, Capogna M (2011) Human limbic encephalitis serum enhances hippocampal mossy fiber-CA3 pyramidal cell synaptic transmission. Epilepsia 52(1):121–131
Ohkawa T, Fukata Y, Yamasaki M, Miyazaki T, Yokoi N et al (2013) Autoantibodies to epilepsy-related LGI1 in limbic encephalitis neutralize LGI1-ADAM22 interaction and reduce synaptic AMPA receptors. J Neurosci 33(46):18161–18174
Bien CG, Vincent A, Barnett MH, Becker AJ, Blumcke I et al (2012) Immunopathology of autoantibody-associated encephalitides: clues for pathogenesis. Brain 135(Pt 5):1622–1638
Misu T, Fujihara K, Kakita A, Konno H, Nakamura M et al (2007) Loss of aquaporin 4 in lesions of neuromyelitis optica: distinction from multiple sclerosis. Brain 130(Pt 5):1224–1234
Forrest D, Yuzaki M, Soares HD, Ng L, Luk DC et al (1994) Targeted disruption of NMDA receptor 1 gene abolishes NMDA response and results in neonatal death. Neuron 13(2):325–338
Belforte JE, Zsiros V, Sklar ER, Jiang Z, Yu G et al (2010) Postnatal NMDA receptor ablation in corticolimbic interneurons confers schizophrenia-like phenotypes. Nat Neurosci 13(1):76–83
McNamara RK, Skelton RW (1996) Baclofen, a selective GABAB receptor agonist, dose-dependently impairs spatial learning in rats. Pharmacol Biochem Behav 53(2):303–308
Schuler V, Luscher C, Blanchet C, Klix N, Sansig G et al (2001) Epilepsy, hyperalgesia, impaired memory, and loss of pre- and postsynaptic GABA(B) responses in mice lacking GABA(B(1)). Neuron 31(1):47–58
Xi B, Chen J, Yang L, Wang W, Fu M et al (2011) GABBR1 gene polymorphism(G1465A)isassociated with temporal lobe epilepsy. Epilepsy Res 96(1–2):58–63
Zai G, King N, Wong GW, Barr CL, Kennedy JL (2005) Possible association between the gamma-aminobutyric acid type B receptor 1 (GABBR1) gene and schizophrenia. Eur Neuropsychopharmacol 15(3):347–352
Zai G, Arnold P, Burroughs E, Barr CL, Richter MA et al (2005) Evidence for the gamma-amino-butyric acid type B receptor 1 (GABBR1) gene as a susceptibility factor in obsessive-compulsive disorder. Am J Med Genet Part B Neuropsychiatric Genet 134B(1):25–29
Yu YE, Wen L, Silva J, Li Z, Head K et al (2010) Lgi1 null mutant mice exhibit myoclonic seizures and CA1 neuronal hyperexcitability. Hum Mol Genet 19(9):1702–1711
Zhou YD, Lee S, Jin Z, Wright M, Smith SE et al (2009) Arrested maturation of excitatory synapses in autosomal dominant lateral temporal lobe epilepsy. Nat Med 15(10):1208–1214
Nobile C, Michelucci R, Andreazza S, Pasini E, Tosatto SC et al (2009) LGI1 mutations in autosomal dominant and sporadic lateral temporal epilepsy. Hum Mutat 30(4):530–536
Gu W, Brodtkorb E, Steinlein OK (2002) LGI1 is mutated in familial temporal lobe epilepsy characterized by aphasic seizures. Ann Neurol 52(3):364–367
Kinali M, Jungbluth H, Eunson LH, Sewry CA, Manzur AY et al (2004) Expanding the phenotype of potassium channelopathy: severe neuromyotonia and skeletal deformities without prominent Episodic Ataxia. Neuromuscul Disord 14(10):689–693
Zuberi SM, Eunson LH, Spauschus A, De Silva R, Tolmie J et al (1999) A novel mutation in the human voltage-gated potassium channel gene (Kv1.1) associates with episodic ataxia type 1 and sometimes with partial epilepsy. Brain 122(Pt 5):817–825
Poliak S, Salomon D, Elhanany H, Sabanay H, Kiernan B et al (2003) Juxtaparanodal clustering of Shaker-like K+ channels in myelinated axons depends on Caspr2 and TAG-1. J Cell Biol 162(6):1149–1160
Penagarikano O, Abrahams BS, Herman EI, Winden KD, Gdalyahu A et al (2011) Absence of CNTNAP2 leads to epilepsy, neuronal migration abnormalities, and core autism-related deficits. Cell 147(1):235–246
Alarcon M, Abrahams BS, Stone JL, Duvall JA, Perederiy JV et al (2008) Linkage, association, and gene-expression analyses identify CNTNAP2 as an autism-susceptibility gene. Am J Hum Genet 82(1):150–159
Strauss KA, Puffenberger EG, Huentelman MJ, Gottlieb S, Dobrin SE et al (2006) Recessive symptomatic focal epilepsy and mutant contactin-associated protein-like 2. N Engl J Med 354(13):1370–1377
Friedman JI, Vrijenhoek T, Markx S, Janssen IM, van der Vliet WA et al (2008) CNTNAP2 gene dosage variation is associated with schizophrenia and epilepsy. Mol Psychiatry 13(3):261–266
Leypoldt F, Armangue T, Dalmau J (2014) Autoimmune encephalopathies. Ann N Y Acad Sci (in press)
Zandi MS, Paterson RW, Ellul MA, Jacobson L, Al-Diwani A et al. (2014) Clinical relevance of serum antibodies to extracellular N-methyl-d-aspartate receptor epitopes. J Neurol Neurosurg Psychiatry (in press)
Hughes EG, Peng X, Gleichman AJ, Lai M, Zhou L et al (2010) Cellular and synaptic mechanisms of anti-NMDA receptor encephalitis. J Neurosci 30(17):5866–5875
Dahm L, Ott C, Steiner J, Stepniak B, Teegen B et al (2014) Seroprevalence of autoantibodies against brain antigens in health and disease. Ann Neurol 76(1):82–94
Pruss H, Holtje M, Maier N, Gomez A, Buchert R et al (2012) IgA NMDA receptor antibodies are markers of synaptic immunity in slow cognitive impairment. Neurology 78(22):1743–1753
Choe CU, Karamatskos E, Schattling B, Leypoldt F, Liuzzi G et al (2013) A clinical and neurobiological case of IgM NMDA receptor antibody associated encephalitis mimicking bipolar disorder. Psychiatry Res 208(2):194–196
Armangue T, Titulaer MJ, Malaga I, Bataller L, Gabilondo I et al (2013) Pediatric anti-N-methyl-d-aspartate receptor encephalitis-clinical analysis and novel findings in a series of 20 patients. J Pediatr 162(4):850–856
Hacohen Y, Deiva K, Pettingill P, Waters P, Siddiqui A et al (2014) N-methyl-d-aspartate receptor antibodies in post-herpes simplex virus encephalitis neurological relapse. Mov Disord 29(1):90–96
Mackay G, Ahmad K, Stone J, Sudlow C, Summers D et al (2012) NMDA receptor autoantibodies in sporadic Creutzfeldt-Jakob disease. J Neurol 259(9):1979–1981
Lachance DH, Lennon VA, Pittock SJ, Tracy JA, Krecke KN et al (2010) An outbreak of neurological autoimmunity with polyradiculoneuropathy in workers exposed to aerosolised porcine neural tissue: a descriptive study. Lancet Neurol 9(1):55–66
Meeusen JW, Klein CJ, Pirko I, Haselkorn KE, Kryzer TJ et al (2012) Potassium channel complex autoimmunity induced by inhaled brain tissue aerosol. Ann Neurol 71(3):417–426
Sato DK, Callegaro D, Lana-Peixoto MA, Waters PJ, de Haidar Jorge FM et al (2014) Distinction between MOG antibody-positive and AQP4 antibody-positive NMO spectrum disorders. Neurology 82(6):474–481
Lancaster E, Huijbers MG, Bar V, Boronat A, Wong A et al (2011) Investigations of caspr2, an autoantigen of encephalitis and neuromyotonia. Ann Neurol 69(2):303–311
Hoftberger R, Titulaer MJ, Sabater L, Dome B, Rozsas A et al (2013) Encephalitis and GABAB receptor antibodies: novel findings in a new case series of 20 patients. Neurology 81(17):1500–1506
Cree BA, Lamb S, Morgan K, Chen A, Waubant E et al (2005) An open label study of the effects of rituximab in neuromyelitis optica. Neurology 64(7):1270–1272
Watanabe S, Nakashima I, Misu T, Miyazawa I, Shiga Y et al (2007) Therapeutic efficacy of plasma exchange in NMO-IgG-positive patients with neuromyelitis optica. Multiple Scler 13(1):128–132
Jacob A, Weinshenker BG, Violich I, McLinskey N, Krupp L et al (2008) Treatment of neuromyelitis optica with rituximab: retrospective analysis of 25 patients. Arch Neurol 65(11):1443–1448
Wingerchuk DM, Hogancamp WF, O’Brien PC, Weinshenker BG (1999) The clinical course of neuromyelitis optica (Devic’s syndrome). Neurology 53(5):1107–1114
Brodtkorb E, Gu W, Nakken KO, Fischer C, Steinlein OK (2002) Familial temporal lobe epilepsy with aphasic seizures and linkage to chromosome 10q22-q24. Epilepsia 43(3):228–235
Hamdan FF, Gauthier J, Araki Y, Lin DT, Yoshizawa Y et al (2011) Excess of de novo deleterious mutations in genes associated with glutamatergic systems in nonsyndromic intellectual disability. Am J Hum Genet 88(3):306–316
Manahan-Vaughan D, von Haebler D, Winter C, Juckel G, Heinemann U (2008) A single application of MK801 causes symptoms of acute psychosis, deficits in spatial memory, and impairment of synaptic plasticity in rats. Hippocampus 18(2):125–134
Curran HV, Morgan C (2000) Cognitive, dissociative and psychotogenic effects of ketamine in recreational users on the night of drug use and 3 days later. Addiction 95(4):575–590
Prosser HM, Gill CH, Hirst WD, Grau E, Robbins M et al (2001) Epileptogenesis and enhanced prepulse inhibition in GABA(B1)-deficient mice. Mol Cell Neurosci 17(6):1059–1070
Wernicke C, Samochowiec J, Schmidt LG, Winterer G, Smolka M et al (2003) Polymorphisms in the N-methyl-d-aspartate receptor 1 and 2B subunits are associated with alcoholism-related traits. Biol Psychiatry 54(9):922–928
Zhao X, Li H, Shi Y, Tang R, Chen W et al (2006) Significant association between the genetic variations in the 5′ end of the N-methyl-d-aspartate receptor subunit gene GRIN1 and schizophrenia. Biol Psychiatry 59(8):747–753
Chanasong R, Thanoi S, Watiktinkorn P, Reynolds GP, Nudmamud-Thanoi S (2013) Genetic variation of GRIN1 confers vulnerability to methamphetamine-dependent psychosis in a Thai population. Neurosci Lett 551:58–61
Epi KC, Phenome Epilepsy, Genome P, Allen AS, Berkovic SF, Cossette P et al (2013) De novo mutations in epileptic encephalopathies. Nature 501(7466):217–221
Bennett DR, Madsen JA, Jordan WS, Wiser WC (1973) Ketamine anesthesia in brain-damaged epileptics. Electroencephalographic and clinical observations. Neurology 23(5):449–460
Badran S, Schmutz M, Olpe HR (1997) Comparative in vivo and in vitro studies with the potent GABAB receptor antagonist, CGP 56999A. Eur J Pharmacol 333(2–3):135–142
Taittonen MT, Kirvela OA, Aantaa R, Kanto JH (1998) The effect of clonidine or midazolam premedication on perioperative responses during ketamine anesthesia. Anesth Analg 87(1):161–167
McCarron MM, Schulze BW, Thompson GA, Conder MC, Goetz WA (1981) Acute phencyclidine intoxication: incidence of clinical findings in 1,000 cases. Ann Emerg Med 10(5):237–242
Marco LA, Joshi RS (1992) A ketamine-induced rat model of tardive dyskinesia. Prog Neurobiol 38(6):571–600
Tricklebank MD, Singh L, Oles RJ, Preston C, Iversen SD (1989) The behavioural effects of MK-801: a comparison with antagonists acting non-competitively and competitively at the NMDA receptor. Eur J Pharmacol 167(1):127–135
Acknowledgments
SRI is supported by the Wellcome Trust, the BMA Grant—Vera Down, Epilepsy Research UK and is a member of the Oxford Epilepsy Research Group. Work in the laboratory is funded by the NIHR Oxford BRC. We thank Dr Al-Diwani for his assistance with Fig. 1a.
Conflicts of interest
AV and the Nuffield Department of Clinical Neurosciences in Oxford receive royalties and payments for antibody assays and AV is the named inventor on patent application WO/2010/046716 entitled ‘Neurological Autoimmune Disorders’. The patent has been licensed to Euroimmun AG for the development of assays for LGI1 and other VGKC-complex antibodies. SI is a co-inventor and has received royalties.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Varley, J., Vincent, A. & Irani, S.R. Clinical and experimental studies of potentially pathogenic brain-directed autoantibodies: current knowledge and future directions. J Neurol 262, 1081–1095 (2015). https://doi.org/10.1007/s00415-014-7600-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-014-7600-8