
Abstract
Acquired copper deficiency has been recognised as a rare cause of anaemia and neutropenia for over half a century. Copper deficiency myelopathy (CDM) was only described within the last decade, and represents a treatable cause of non-compressive myelopathy which closely mimics subacute combined degeneration due to vitamin B12 deficiency. Here, 55 case reports from the literature are reviewed regarding their demographics, aetiology, haematological and biochemical parameters, spinal imaging, treatment and outcome. The pathophysiology of disorders of copper metabolism is discussed. CDM most frequently presented in the fifth and sixth decades and was more common in women (F:M = 3.6:1). Risk factors included previous upper gastrointestinal surgery, zinc overload and malabsorption syndromes, all of which impair copper absorption in the upper gastrointestinal tract. No aetiology was established in 20% of cases. High zinc levels were detected in some cases not considered to have primary zinc overload, and in this situation the contribution of zinc to the copper deficiency state remained unclear. Cytopenias were found in 78%, particularly anaemia, and a myelodysplastic syndrome may have been falsely diagnosed in the past. Spinal MRI was abnormal in 47% and usually showed high T2 signal in the posterior cervical and thoracic cord. In a clinically compatible case, CDM may be suggested by the presence of one or more risk factors and/or cytopenias. Low serum copper and caeruloplasmin levels confirmed the diagnosis and, in contrast to Wilson’s disease, urinary copper levels were typically low. Treatment comprised copper supplementation and modification of any risk factors, and led to haematological normalisation and neurological improvement or stabilisation. Since any neurological recovery was partial and case numbers of CDM will continue to rise with the growing use of bariatric gastrointestinal surgery, clinical vigilance will remain the key to minimising neurological sequelae. Recommendations for treatment and prevention are made.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Copper is a trace metal which acts as a prosthetic group in several key enzymes and is thus essential for the structure and function of the bone marrow and nervous system. Acquired copper deficiency has long been recognised as a cause of anaemia and, less commonly, other cytopenias [11, 83]. It was only in 2001 that an association with myelopathy was reported [71]. The clinical and radiological picture is usually indistinguishable from subacute combined degeneration due to vitamin B12 (cobalamin) deficiency (SCD) [40, 44], and the syndrome is thought to represent the human counterpart of swayback or enzootic ataxia, a copper deficiency myelopathy (CDM) occurring in ruminants [3]. Other, less frequently reported and less clearly causally related neurological associations of acquired copper deficiency include isolated peripheral neuropathy [20, 86], motor neuron disease [84], myopathy [46], cerebral demyelination [65, 74], cognitive dysfunction [58, 77] and optic neuropathy [20, 74].
Patients typically present with gait difficulties, which arise through sensory ataxia due to dorsal column dysfunction and to a lesser extent through spasticity. Paraesthesia in upper and lower limbs is common, whilst urinary symptoms occur only in a minority. Examination usually shows a spastic paraparesis or tetraparesis with a truncal sensory level for dorsal column modalities. A sensory/motor neuropathy frequently coexists and manifests as depression of distal reflexes and superimposed sensory impairment in a glove and stocking distribution.
A previous review has addressed the clinical features, investigation findings and treatment outcomes in a series of 25 patients with CDM [38] (some of these cases were also reported elsewhere [21, 39, 41, 42, 44–47]). An additional 30 cases of myelopathy or myeloneuropathy have now been reported [1, 2, 4, 12, 13, 16, 18, 19, 21, 24, 28–30, 58, 63, 67, 71, 74, 77, 88, 89]. Here we review the published cases with regards to their demographics, aetiology, laboratory indices, spinal imaging, treatment and outcome. Clinical features and neurophysiological data were excluded as the mode and quality of description varied greatly between reports, and because they were already adequately addressed in the previous review.
Methods
The PubMed database was searched from its inception to August 2008 using the terms hypocupr* or copper in combination with myelopathy, myeloneuropathy, combined degeneration, subacute combined degeneration (MeSH) or spinal cord diseases (MeSH). References in relevant papers were reviewed for additional cases.
The following data were extracted for each case where available: age, sex, primary aetiology, haematological indices at presentation (haemoglobin, mean corpuscular volume (MCV), white cell count, platelet count), biochemical indices at presentation (serum copper, serum caeruloplasmin, serum zinc, 24 h urinary copper and zinc), spinal imaging findings, treatment and outcome.
The primary aetiology was classified as previous upper gastrointestinal surgery, malabsorption, zinc overload, iron supplements or unknown. Treatment was categorised by dose and route of administration of copper supplements, and any accompanying intervention such as cessation of exogenous zinc supply. Outcome was divided into neurologically improved, unchanged or unknown; there were no reports of deterioration with adequate copper replacement.
Units were standardised to μg/mL for serum copper and zinc and mg/dL for serum caeruloplasmin. Where figures were given in molar quantities, the standard atomic weights 63.546 (copper) and 65.049 (zinc) were used to convert into the corresponding mass unit. Urinary copper and zinc excretion were converted to mg/24 h, but this was not possible where figures were given as mass per unit volume.
Statistical analyses were performed using SPSS 14.0 (SPSS Inc., Chicago, Illinois).
Results
A total of 55 cases were found in the literature [1, 2, 4, 12, 13, 16, 18, 19, 21, 24, 28–30, 38, 39, 41, 42, 44–47, 58, 63, 67, 71, 74, 77, 88, 89]. Age at presentation ranged from 30 to 82 years with a peak in the fifth and sixth decades (mean 54 years, median 53 years; Fig. 1). There was a marked female preponderance (female:male ratio 3.6:1).

Age at presentation. Female and male cases are plotted in black and white, respectively
Previous upper gastrointestinal surgery was the commonest reported cause, being implicated in almost half the cases (47%, 26 cases; Table 1). Of these, 17 cases were attributable to non-bariatric surgery, typically partial gastrectomy for peptic ulcer disease, with the remaining 9 being due to bariatric interventions, mostly Roux-en-Y gastric bypass. The second commonest aetiology, zinc overload (16%, 9 cases), was thought to be caused by the use of denture cream (4 cases) or zinc supplements (2 cases), or by oversubstitution of zinc in the dialysate during chronic haemodialysis (1 case). In the remaining 2 cases of zinc overload no cause was found. Less common causes of copper deficiency were malabsorption (15%, 8 cases), with half of these being due to coeliac disease, and iron supplements (2%, 1 case). In the remaining 20% (11 cases), no cause of copper deficiency was identified.
The interval between upper gastrointestinal surgery and symptom onset ranged from 5 to 26 years in the bariatric group (mean 11.4 years, latencies reported in 8 of 9 cases), and from 10 to 46 years in the non-bariatric group (mean 22.2 years, latencies reported in 12 of 17 cases). The difference between the two groups was statistically significant (Mann–Whitney U test, p = 0.007). This analysis could not be extended to other aetiologies as onset latencies were less frequently reported and would have been impossible to determine where the predisposing factor was clinically silent.
Where haematological indices were reported, 78% (35 of 45 cases) showed at least one cytopenia. Anaemia was the commonest cytopenia, being seen in 68% (30 of 44 cases). Nine cases were macrocytic, three were normocytic and one was microcytic; no MCV was given in the remaining cases. Leukopenia was seen in 50% (22 of 44 cases). Platelet count was only infrequently reported, but was low in 3 of 15 cases.
Serum vitamin B12 levels at presentation were low in a single case [29], but no further details were provided. In the remaining cases, vitamin B12 levels at presentation were either normal or high (44 cases) or not specified (10 cases). These reports included 14 patients in whom B12 replacement had previously been instigated either for a documented deficiency state or empirically despite normal levels; in 10 of these, a lack of a haematological or neurological response was described.
By definition, serum copper was low in all patients and was below 0.1 μg/mL in 45% (25 cases). The distribution of serum copper showed a pronounced skew towards very low or undetectable serum copper (Fig. 2a). Caeruloplasmin was low in all 40 cases in which it was reported (mean reference range 21.5–48.1 mg/dL) and showed a similar distribution (Fig. 2b) supporting the copper deficiency state. A statistically significant correlation was seen between serum copper and serum caeruloplasmin (Pearson correlation coefficient 0.653, 2-tailed p < 0.001). There was a wide distribution of serum zinc values varying from 0.04 to 4.28 μg/mL (typical reference range 0.66–1.10 μg/mL). Serum zinc was low in 20% (10 cases), normal in 31% (15 cases) and high in 49% (24 cases), and tended to be higher in cases where zinc excess was reported as the primary aetiology (range 1.36–4.28 μg/mL, median 2.00 μg/mL, mean 1.12 μg/mL in 9 cases attributed to zinc excess; range 0.04–2.68 μg/mL, median 1.01 μg/mL, mean 1.12 μg/mL in 36 cases attributed to other aetiologies where a numerical figure was given; Student’s t test for different means, p < 0.001; a further 4 cases only had a qualitative description such as “normal” zinc levels).

Serum copper and caeruloplasmin. a In two cases, serum copper was reported to be low with no numerical value given (not shown). Typical reference ranges have a lower limit of normal of 0.75 μg/mL (range 0.65–0.85). b No readings were reported in 15 cases (not shown). The mean lower limit of normal was 21.5 mg/dL (range 14–26, excluding an outlier of 7.9)
There was a significant negative correlation between serum copper and zinc (Fig. 3; Pearson correlation coefficient −0.327, 45 cases, 2-tailed p = 0.028). When cases primarily attributed to zinc overload were excluded, this correlation remained highly significant (Pearson correlation coefficient −0.564, 36 cases, 2-tailed p < 0.001).

Serum zinc against serum copper in the 45 cases where both were reported. Different symbols represent different aetiologies
Urinary copper excretion was typically low (50%, 13 of 26 cases) or normal (42%, 11 cases), but was elevated in 2 cases (8%). Urinary zinc was elevated in 81% (13 of 16 cases), sometimes by more than an order of magnitude above the normal range. No correlation existed between serum copper and urinary copper excretion (Pearson’s correlation coefficient −0.083, 26 cases, 2-tailed p = 0.688).
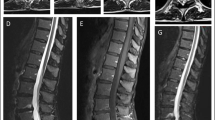
The results of spinal MRI scans were reported in 47 cases. These were abnormal in 47% (22 cases), typically showing increased T2 signal in the posterior cervical and thoracic cord (Fig. 4).

Sagittal and axial T2-weighted 3T MR images of the cervical cord. A typical high signal lesion is demonstrated in the dorsal columns (arrowheads)
Copper supplementation formed the mainstay of treatment and was typically given orally (71%, 32 of 45 cases) [1, 18, 19, 21, 24, 28, 30, 38, 39, 41, 42, 44–46, 63, 74, 77, 89] or intravenously with subsequent switching to the oral route (20%, 9 of 45 cases) [12, 38, 41, 44, 47, 58, 67]. The remaining patients received intravenous (4%, 2 cases) [71, 88], oral then intravenous (2%, 1 case) [38, 39, 44] or intramuscular supplements (2%, 1 case) [4]; in 10 cases (18%) the route of administration was not specified [13, 16, 29, 38, 39, 41, 44, 58]. Doses were usually equivalent to 2 mg/day of elemental copper, though administration of up to 9 mg/day was reported (ignoring extreme outliers which are likely to reflect reporting errors [89]). A variety of copper salts were used, including copper acetate [63], copper chloride [88], copper citrate [28], copper gluconate [2, 12, 38, 44, 45, 74, 89], copper histidine [4] and copper sulphate [1, 12, 38, 41, 44, 47, 67, 71, 77]. No adverse effects were reported. Where possible, the presumed underlying cause of copper deficiency was also treated. This included stopping any exogenous supply of zinc [30, 38, 44, 45, 58, 67, 89] or iron [38], gluten-free diet for coeliac disease [21, 38, 44, 46], gastric bypass revision [29], steroids for mesangioproliferative glomerulonephritis [2] and antibiotics for small bowel bacterial overgrowth [74].
Outcomes were stated in 47 cases. The neurological deficit improved in 49% (23 cases), though two cases continued to deteriorate neurologically until the dose of copper supplementation was escalated (see below). Descriptions of the improvements were usually scant and ranged from subjective reductions in sensory symptoms to objective improvements in spasticity, power and mobility; there were no reports of full neurological recovery. In three cases where spinal MRI had been abnormal at presentation, repeated imaging after treatment showed improved appearances [18, 40, 89]. In the other 51% (24 cases), the neurological deficit stabilised. Where a haematological outcome was reported, haematological recovery was rapid and complete.
Discussion
Copper metabolism
Dietary intake and total body content
Copper is an essential dietary micronutrient, with a Recommended Dietary Allowance (US) [14] and Reference Nutrient Intake (UK) [59] of 0.9 mg/day and 1.2 mg/day, respectively. The average total copper content of the human body is estimated at 75 mg (range 50–120 mg) [69]. Particularly high levels are found in the brain, and this probably applies to the spinal cord, too.
Absorption and excretion
Copper absorption occurs predominantly in the duodenum [75, 76] and potentially also in the stomach [81]. Following uptake via the apical membrane transporter Ctr1, whose expression is regulated by copper status [48], copper is transferred across the basolateral membrane by a P-type ATPase (ATP7A), which is deficient in Menkes’ disease [53], an inherited disorder of copper absorption. Intestinal copper absorption can be inhibited by zinc and iron (see below). In the portal circulation, copper is transported bound to albumin and transcuprein. At the hepatocyte level, it is incorporated into the enzymes superoxide dismutase and cytochrome-c oxidase, or into the carrier protein caeruloplasmin for onward transport to systemic tissues, where it is integrated into the numerous other proteins described below [49]. Serum caeruloplasmin acts as a marker of copper status: although its rate of synthesis is unaffected by copper intake, caeruloplasmin lacking bound copper has a short half-life in the serum and is rapidly degraded [26].
When copper is found to be deficient, the serum levels are usually very low with around half of cases having serum copper levels below 0.1 μg/mL. This may reflect the increasing likelihood of clinical presentation with the degree of deficiency. The strong correlation between serum copper and caeruloplasmin levels was in keeping with caeruloplasmin being an effective marker of copper status. However, caeruloplasmin is not essential for copper metabolism, as a genetic deficiency (acaeruloplasminaemia) does not lead to impaired systemic delivery of copper.
Excess copper is bound by metallothioneins, which are upregulated to prevent toxicity. Copper is secreted into bile via a P-type ATPase (ATP7B), which is lacking in Wilson’s disease. Biliary excretion is the primary mode of homeostasis, and chronic copper toxicity is unlikely in the absence of a secretory deficit, either genetic (Wilson’s disease) or acquired (hepatic failure) [17]. Although much of the secreted copper is reabsorbed, 0.5–1.5 mg/day end up in the faeces [49] and only a minute amount of copper enters the urine (30–60 μg/day). In Wilson’s disease, genetic deficiency of ATP7B impairs copper export into bile and caeruloplasmin. Copper leaks into the serum and urinary copper excretion is increased, making 24 h urinary copper excretion a sensitive test for Wilson’s disease.
In 92% of cases, urinary copper was low or normal as might be expected in a copper deficiency state. However, two cases had an elevated urinary copper. In the first case [2], the patient was diagnosed with membranoproliferative glomerulonephritis in association with hepatitis C and had a proteinuria of 3.798 g/24 h. Both the cupriuria and a remote history of non-bariatric upper gastrointestinal surgery were felt to contribute to the copper deficiency state. The second case [12] had only a marginally elevated 24 h urinary copper. Renal function was not reported and there was no mention of renal impairment or conditions which could impair kidney function. Although the kidneys might be expected to conserve copper in a deficiency state, daily excretion of copper in the urine is an order of magnitude less than that in the faeces and thus normally has little bearing on serum copper. Hence, the lack of correlation between serum copper and urinary copper excretion is less surprising.
Systemic roles
Copper is a transition metal possessing two physiological oxidation states—reduced Cu1+ (cuprous) and oxidised Cu2+ (cupric)—and can thus act as an intermediary for electron transfer in redox reactions. It is involved in the catalytic function of several key enzymes [82], mostly oxidoreductases and monooxygenases, whose diverse metabolic roles are summarised in Table 2. Copper can regulate gene expression through copper-responsive transcription factors [79], and is known to upregulate the genes encoding superoxide dismutase and proteins involved in cellular copper storage and partitioning [82]. Moreover, metal-responsive elements have been found in all eukaryotic metallothionein promoters.
Mechanisms of acquired deficiency
Upper gastrointestinal surgery
Certain types of upper gastrointestinal surgery can reduce the effective absorption area for particular micronutrients. Deficiencies of iron and vitamin B12 are especially common [15, 54, 80], with a lack of intrinsic factor contributing to the latter. Whilst neurological complications of gastrectomy for peptic ulcer disease have long been recognised, the underlying aetiology often remained obscure [38, 39]. The association between a remote history of upper gastrointestinal surgery, copper deficiency and myelopathy was first described in 2001 [71], and has since been increasingly reported following both non-bariatric [2, 4, 12, 13, 28, 38, 39, 41, 44, 47, 63, 74, 77, 88] and bariatric intervention [16, 29, 38, 39, 44, 47].
Partial gastrectomy has rarely been used to treat peptic ulcer disease since the advent of effective pharmacological therapy. The number of cases of gastrectomy-related CDM is small relative to the number of past procedures. However, it is likely that many further cases will be diagnosed because undiagnosed hypocupraemia is common in patients with a history of gastrectomy [64], CDM may manifest over 40 years after the operation [28, 41] and awareness amongst physicians continues to increase.
The annual number of bariatric operations continues to grow [15, 55], with the commonest procedure being a Roux-en-Y gastric bypass. Post-operatively, food bypasses most of the stomach and the entire duodenum. CDM tends to develop sooner after bariatric than after non-bariatric operations, probably due to the greater reduction in the effective absorption area for copper. Case numbers of CDM will rise with the growing use of bariatric surgery and with the ageing of the present cohort.
Malabsorption
In half the cases of CDM attributed to malabsorption, coeliac disease was diagnosed; in the other half, no cause of malabsorption was established. In three cases of idiopathic copper malabsorption, no detailed investigation findings were reported [38, 44]. In the fourth case, malabsorption was not specific to copper: the patient was cachectic and exhibited concomitant deficiencies of iron, vitamin D and vitamin E. A d-xylose absorption test was compatible with proximal intestinal malabsorption, but upper gastrointestinal endoscopy, duodenal biopsies and faecal microbiology failed to identify an underlying cause [30].
Zinc excess
Zinc upregulates the expression of the chelator metallothionein in enterocytes. Copper has a higher affinity for metallothionein than zinc, and remains bound in the enterocytes which are sloughed into the lumen and eliminated. This mechanism is exploited therapeutically when using zinc to achieve a negative copper balance in Wilson’s disease [91].
It is estimated that 7–15% of denture wearers use adhesives [8], many of which contain zinc in significant concentrations, leading to potentially excessive daily zinc intakes [58]. The use of denture cream may not be covered in routine history taking and should be specifically excluded when a state of zinc excess has been established. In dialysis patients, oversubstitution of zinc through the dialysate should be considered, especially since this mechanism may be unknown to non-nephrologists. Where zinc overload was attributed to the use of supplements, the dose (200–400 mg/day) considerably exceeded the tolerable upper intake level of 40 mg/day (US) [14].
In two cases of CDM, zinc excess was evidenced by high zinc levels in serum samples and 24 h urine collections, but no cause could be identified [19, 24]. After instituting adequate copper supplementation, the patients stabilised neurologically and their serum copper levels improved or normalised, but zinc levels remained high in serum and urine. We are not aware that a similar neurological syndrome has been clearly attributed to hyperzincaemia without hypocupraemia. This suggests that the neurological deficit was caused by copper deficiency, which in turn was probably secondary to zinc excess.
The first case of CDM due to idiopathic zinc excess was found to have elevation of urinary zinc excretion (approx. 10 times the upper limit of normal, ULN) and serum zinc (approx. 1.5 times the ULN) although the zinc content of a liver biopsy specimen was normal [24]. Since no external source of zinc excess could be identified, the authors hypothesised that increased absorption or decreased excretion of zinc in the intestine might be responsible. To address a potential genetic cause, all living first degree relatives underwent neurological examination as well as serum copper and zinc assays, all of which were normal.
In the second case, the high ratio of urinary zinc excretion (up to 20 times the ULN) to serum zinc (approx. 1–2 times the ULN) was interpreted as indicating primary zinc toxicity [19]. Since 60–90% of zinc excretion is gastrointestinal [37], the continued urinary excretion of up to 20 mg/day implied either an ongoing intake of 50–200 mg/day, or previous massive body overload. As no external source of zinc was identified and zinc stool content could not be assayed, these possibilities could not be distinguished.
A negative correlation between serum zinc and serum copper would be expected in patients with zinc overload. However, high serum and/or urinary zinc levels were also reported in other aetiologies, including idiopathic cases. There was a negative correlation between serum zinc and serum copper across all patients, which persisted even when cases attributed to zinc overload were excluded. It remained unclear whether elevated zinc levels may have contributed to hypocupraemia in patients not considered to have primary zinc overload. High serum zinc concentrations can be seen as a heritable trait without any overt clinical manifestations [73], and might therefore represent an epiphenomenon.
Urinary zinc levels did not show a consistent pattern. Of those patients with high urinary zinc excretion, only seven had raised serum zinc levels with the other six having normal or even low serum zinc levels. It is possible that increased zinc excretion represents a homeostatic mechanism to maintain a normal serum zinc level in the face of external zinc intake.
Iron supplements
In one case [38, 41, 44], CDM was potentially related to the use of iron supplements. Neurological symptoms began 1 year after starting ferrous sulphate at 975 mg per day for anaemia of unclear cause. Vitamin B12 levels were borderline low, but methylmalonic acid levels were normal and neurological deterioration continued despite vitamin B12 replacement. When copper deficiency was detected, copper replacement was instituted and iron supplements were stopped. There was neurological improvement but the haematological outcome was not reported.
It was suggested that excess iron intake may have contributed to the hypocupraemia, and such an association has been described in animals [33, 72, 90] and in infants [23, 50, 57]. However, it is conceivable that her original anaemia could have been due to copper deficiency which was recognised only after neurological features developed.
Idiopathic
In two patients with idiopathic CDM, colonic biopsies were taken and copper content was found to be significantly increased [39]. This suggested that impaired mobilisation from the intestinal mucosa might contribute to the systemic deficiency state in some patients. Impaired copper transport across the basolateral membrane occurs in Menkes’ disease and leads to increased copper content in duodenal mucosa [10], but sequencing of the ATP7A gene in one case of idiopathic CDM did not show any abnormalities [43]. Although alterations in the promoter or other non-coding regions as well as large intragenic deletions could not be ruled out, this finding militates against ATP7A mutations contributing to hypocupraemia in idiopathic CDM. It remains possible that mutations in other copper transporters or chaperones play a role. Such mutations might also explain why CDM afflicts only a minority of patients with a comparatively prevalent risk factor such as previous upper gastrointestinal surgery.
Effects of copper deficiency
Copper deficiency may manifest in multiple organ systems.
Haematological effects
It was found in the 1930s that copper is vital for effective erythropoiesis [56]. The haematological manifestations of deficiency are typically anaemia or neutropenia, or more rarely pancytopenia [85]. The anaemia is usually normocytic or macrocytic, but may rarely be microcytic. A retrospective review of 40 patients with copper deficiency defined the typical bone marrow findings of hypocellularity, vacuolisation of granulocyte and erythroid precursors, iron-containing plasma cells and ring sideroblasts [22]. Owing to the similarity of the haematological findings, copper deficiency may be mistaken for a myelodysplastic syndrome. Some haematology textbooks do not list copper deficiency as a differential of myelodysplastic syndrome [20], and in cases with concomitant myelopathy the neurologist may be the first to consider copper deficiency as the unifying diagnosis.
The mechanisms underlying the haematological effects of copper deficiency have not been fully elucidated. The anaemia appears to be partly attributable to impaired iron metabolism. Copper is an essential component of both caeruloplasmin and hephaestin, which are necessary for normal iron homeostasis. Both have enzymatic activity as ferroxidases and are involved in iron export from non-intestinal [27] and intestinal cells [61], respectively. In copper deficiency, iron is seen to accumulate in the liver and can be released by intravenous caeruloplasmin. Although the literature suggests that copper deficiency typically causes a microcytic anaemia akin to iron deficiency, this pattern was reported in only one of the cases reviewed herein. The remaining cases where MCV was reported were either macrocytic (9 cases) or normocytic (3 cases). The difference between copper deficiency and iron deficiency anaemia may be due to iron utilisation being impaired only at certain steps or the contribution of other mechanisms. For example, copper deficiency seems to impair the production of haem from ferric iron and protoporphyrin, possibly because decreased energy production (cytochrome-c oxidase) in erythrocytes impinges on the synthetic pathway [49]. In addition, alterations in erythrocyte membrane fluidity and oxidative damage cause reduced red cell lifespan in copper-deficient rats [66].
Suggestions for the mechanism leading to neutropenia include destruction of myeloid precursors and impaired maturation within the bone marrow, impaired release of mature neutrophils from the bone marrow and increased serum clearance of neutrophils [60]. Indeed, the bone marrow of copper-deficient patients contains a decreased number of myeloid precursors which show vacuolisation [22].
Neurological effects
Whilst copper deficiency in utero leads to impaired neurodevelopment and neurodegeneration [31], acquired copper deficiency in adults has been reported to have a range of neurological manifestations. Myelopathy is by far the commonest association, and is clinically and radiologically indistinguishable from SCD [40, 44]. It has been suggested that CDM is caused by dysfunction of cytochrome-c oxidase [38], which is known to be copper-dependent. This is potentially supported by the high CSF lactate levels found in one case [30]. However, cytochrome-c oxidase is not vitamin B12-dependent, so this hypothesis fails to explain the phenotypic parallels between CDM and SCD. We have previously proposed that dysfunction of the methylation cycle is the first step in a final common pathway of CDM and SCD [87]. This cycle catalyses the net transfer of a methyl group from methyltetrahydrofolate to a variety of macromolecules, including myelin proteins. SCD is known to be caused by dysfunction of methionine synthase, which is a key part of the cycle. Methionine synthase and another enzyme involved in the cycle, S-adenosylhomocysteine hydrolase, may depend on copper. This theory could therefore explain the clinical and radiological congruence between CDM and SCD, but it remains to be proven with direct biochemical evidence.
It was recently noted that subacute myelo-optico-neuropathy (SMON) bears significant phenotypic similarities to CDM, and deranged copper homeostasis may have a dominant role in its pathogenesis [70]. Almost 10,000 cases of SMON in Japan are thought to have been caused by clioquinol until its use was banned in 1970. Clioquinol was originally developed as an antimicrobial and acts as a metal chelator and possibly an ionophore. Despite decades of intense research, the mechanism underlying SMON was never conclusively identified. There is renewed interest in clioquinol as a treatment for Alzheimer’s disease and cancer [5]. It would therefore be desirable to clarify the role of copper in SMON in an animal model [70], and a range of safety precautions has been proposed to minimise the risk of SMON in clinical studies [51].
Optic neuropathy in SMON was variable and often subclinical [70, 78]. Visual disturbance was not reported in cases of CDM, but optic neuropathy has been described in association with copper deficiency [20, 74]. It is therefore possible that subtle visual dysfunction might accompany some cases of CDM, and might only be detected on careful clinical examination, including assessment of colour vision.
A previous series of 25 cases of CDM found that spinal MRI was significantly abnormal in 44% [40], which is in agreement with the figure of 47% reported herein. Both studies also concur that the most consistent finding is a segment of high T2 signal in the dorsal midline cervical and thoracic cord.
Whilst neurophysiological data were not reviewed here, the largest series published to date reported an axonal peripheral neuropathy in 21 of 24 patients tested, with mixed sensory/motor involvement in 11 cases [38]. Somatosensory potentials were abnormal in all 20 patients who underwent testing, with impaired central conduction implicated in 19 of them.
Other effects
The first inherited defect of copper transport, Menkes’ disease, was described in the 1960s [53]. The low serum copper causes malfunction of essential copper-dependent enzymes manifesting as impaired growth, brittle or kinky hair (monoamine oxidase), skin and hair hypopigmentation (tyrosinase), hypothermia (cytochrome-c oxidase) and arterial fragility (lysyl oxidase) [52]. Affected infants develop severe neurodegeneration with deficient myelination and cerebellar and cerebral atrophy seen on MRI. The condition is fatal in infancy and does not respond to systemic copper treatment as ATP7A is necessary for copper uptake into the central nervous system [36].
Acquired copper deficiency in childhood may also cause skeletal abnormalities [25], hypopigmented hair and hypotonia [9], impaired growth [7] and immunity [6, 32]. In adults, copper deficiency can adversely affect cholesterol and glucose metabolism, thus acting as a vascular risk factor [34, 35].
Treatment and outcome
Copper supplementation
Nearly all aetiologies of CDM involve a reduction in gastrointestinal copper absorption. Nonetheless, oral copper supplementation is efficacious and affords an acceptable and practical mode of therapy. In some cases, oral maintenance therapy was preceded by parenteral supplementation, presumably in order to rapidly replenish copper stores and avert any further deterioration. Although some reports refer to parenteral treatment only [4, 71, 88], the efficacy of oral supplements should make prolonged parenteral therapy unnecessary.
Typical daily doses were equivalent to 2–4 mg of elemental copper. In one case attributed to non-bariatric gastrointestinal surgery, administration of 2 mg of oral copper per day normalised blood parameters and improved neurological symptoms. However, a subsequent symptomatic and biochemical relapse necessitated that the dose be incremented to 4 mg and then 6 mg, which achieved sustained remission [63]. Another patient with idiopathic zinc overload was poorly compliant with oral copper supplementation at 2 mg/day. His serum copper levels remained low and the neurological deficit progressed, though serum caeruloplasmin increased to a value at the low end of the normal range and the anaemia resolved. The copper dose was gradually escalated to 8 mg/day but compliance remained poor and he continued to deteriorate neurologically. Subsequently, the patient reported better compliance and his neurological status improved. However, his highest recorded serum copper level remained below the normal range, and it was unclear whether this reflected insufficient dosage, poor compliance or other factors. Copper doses of up to 9 mg per day have been reported [2].
Therapeutic efficacy does not appear to be influenced by the type of copper compound administered. Oral copper gluconate did not influence serum copper levels in normal volunteers [62]. Whilst this finding was previously interpreted as signifying poor bioavailability [38, 88], it appears to merely reflect intact copper homeostasis through biliary excretion in a copper-replete study population.
Since zinc can interfere with copper absorption, care must be taken to avoid copper preparations that also contain significant quantities of zinc, as may be the case with multivitamin tablets. We have encountered difficulties in sourcing pharmaceutical-grade copper-only supplements in the UK, and currently use an amino acid chelate of copper that can be obtained from health food retailers (Solgar copper amino acid chelate, available via http://www.solgar.co.uk) pending local availability of a dedicated formulation. We would suggest that the underlying cause should be treated if possible, and that oral supplementation equivalent to 8 mg of elemental copper per day should usually suffice. A brief course (e.g. for 5 days) of parenteral therapy may be considered to hasten normalisation of body copper stores before switching to the oral route. Lower doses are typically successful and may reduce the cost and number of tablets required, but carry a small risk of failing to prevent neurological progression. A dose of 8 mg/day, combined with an average diet, is unlikely to exceed the tolerable upper intake level of 10 mg/day (US) [14]) or the provisional maximum tolerable daily intake of 0.5 mg/kg/day (World Health Organisation [68]). Furthermore, bioavailability is lessened by the absorptive defects which underlie CDM, and the continued integrity of copper homeostasis through biliary excretion should prevent a state of chronic copper excess [17].
Treatment may need to be continued indefinitely where the underlying risk factor cannot be eliminated. Regular follow-up is necessary to ensure normalisation of biochemical and haematological parameters and stabilisation or improvement of neurological features.
Prevention
Following bariatric surgery, patients remain under follow-up for years, and if copper were included amongst the micronutrients that are already assayed at regular intervals, any deficiency could potentially be corrected at a pre-symptomatic stage. Patients with most other risk factors, including past non-bariatric upper gastrointestinal surgery, are unlikely to have ongoing follow-up arrangements. Physician vigilance therefore remains paramount for making an early diagnosis. There is probably no role for prophylactic copper supplementation, since most risk factors for copper deficiency are relatively prevalent yet only carry a low risk of CDM. Except for the avoidance of excessive zinc supplementation, primary risk factor modification does not appear realistic.
Outcome
Whilst treatment causes prompt and full resolution of haematological abnormalities, the neurological deficit merely stabilises or partially improves. Hence, it is crucial to avoid delays in diagnosis and treatment to avert potentially irreversible neurological deterioration.
We would suggest that serum copper and caeruloplasmin levels should be first line tests in patients presenting with a phenotype suggestive of SCD, especially if risk factors for copper deficiency or cytopenias are present. In other non-compressive myelopathies, these tests should be considered early as they may identify copper deficiency as a treatable aetiology. Due to the lack of a gold standard test, the sensitivity and specificity of these tests is unknown.
Future avenues
Advances in the understanding of the pathophysiology of CDM may yield further therapeutic approaches in the future. For example, if dysfunction of the methylation cycle were confirmed as the pathological mechanism underlying CDM, methionine and betaine supplementation might help to restore normal function of this cycle [87].
Conclusion
CDM should be considered as a differential in the work-up of non-compressive myelopathies, and a compatible clinical picture combined with low serum copper and caeruloplasmin establishes the diagnosis. Diagnostic pointers include cytopenias and predisposing factors. Anaemia and leukopenia are seen in 68 and 50% of cases, respectively, and a diagnosis of myelodysplastic syndrome may have been made previously. Risk factors for CDM reduce the effective absorption area for copper and include previous upper gastrointestinal surgery (47% of cases); zinc overload (16%) through denture cream, supplements or haemodialysis; and malabsorption (15%). The role of iron supplements in causing CDM (2%) is unclear, as is the role of high serum or urinary zinc in patients not deemed to have zinc overload.
The clinical and radiological phenotype closely mimics SCD. Spinal MRI is abnormal in 47% of cases, typically showing a longitudinal high T2 signal lesion in the dorsal cervical and thoracic cord. Dysfunction of the methylation cycle is a potential pathophysiological substrate explaining the phenotypic similarity between SCD and CDM. Reported associations of copper deficiency with other neurological manifestations require further study.
Treatment involves copper supplementation and management of any underlying risk factors. With replenishment of copper, haematological abnormalities resolve rapidly and completely. By contrast, neurological deficits merely stabilise (51% of cases) or partially improve (49%). Hence, early recognition of CDM is vital to avert irreversible deterioration. After bariatric surgery, which is likely to become the commonest risk factor, surveillance of copper levels or prophylactic supplementation could also help to avoid copper deficiency and thus CDM.
References
Allred JW, Aulino JM (2007) Hypocupremia-associated myelopathy. J Comput Assist Tomogr 31:157–159
Bartner R, Will M, Conrad J, Engelhardt A, Schwarz-Eywill M (2005) Pancytopenia, arthralgia and myeloneuropathy due to copper deficiency. Med Klin (Munich) 100:497–501
Bennetts HW, Chapman FE (1937) Copper deficiency in sheep in Western Australia: a preliminary account of the aetiology of enzootic ataxia of lambs and anaemia of ewes. Aust Vet J 13:138–149
Bertfield DL, Jumma O, Pitceathly RD, Sussman JD (2008) Copper deficiency: an unusual case of myelopathy with neuropathy. Ann Clin Biochem 45:434–435
Cahoon L (2009) The curious case of clioquinol. Nat Med 15:356–359
Castillo-Duran C, Fisberg M, Valenzuela A, Egana JI, Uauy R (1983) Controlled trial of copper supplementation during the recovery from marasmus. Am J Clin Nutr 37:898–903
Castillo-Duran C, Uauy R (1988) Copper deficiency impairs growth of infants recovering from malnutrition. Am J Clin Nutr 47:710–714
Coates AJ (2000) Usage of denture adhesives. J Dent 28:137–140
Danks DM (1988) Copper deficiency in humans. Annu Rev Nutr 8:235–257
Danks DM, Cartwright E, Stevens BJ, Townley RR (1973) Menkes’ kinky hair disease: further definition of the defect in copper transport. Science 179:1140–1142
Dunlap WM, James GW 3rd, Hume DM (1974) Anemia and neutropenia caused by copper deficiency. Ann Intern Med 80:470–476
Everett CM, Matharu M, Gawler J (2006) Neuropathy progressing to myeloneuropathy 20 years after partial gastrectomy. Neurology 66:1451
Ferrara JM, Skeen MB, Edwards NJ, Gray L, Massey EW (2007) Subacute combined degeneration due to copper deficiency. J Neuroimaging 17:375–377
Food and Nutrition Board, Institute of Medicine, National Academy of Sciences (2000) Dietary reference intakes for vitamin A, vitamin K, arsenic, boron, chromium, copper, iodine, iron, manganese, molybdenum, nickel, silicon, vanadium, and zinc. National Academy Press, Washington, DC
Fujioka K (2005) Follow-up of nutritional and metabolic problems after bariatric surgery. Diabetes Care 28:481–484
Goldberg ME, Laczek J, Napierkowski JJ (2008) Copper deficiency: a rare cause of ataxia following gastric bypass surgery. Am J Gastroenterol 103:1318–1319
Gollan JL, Deller DJ (1973) Studies on the nature and excretion of biliary copper in man. Clin Sci 44:9–15
Goodman BP, Chong BW, Patel AC, Fletcher GP, Smith BE (2006) Copper deficiency myeloneuropathy resembling B12 deficiency: partial resolution of MR imaging findings with copper supplementation. Am J Neuroradiol 27:2112–2114
Greenberg SA, Briemberg HR (2004) A neurological and hematological syndrome associated with zinc excess and copper deficiency. J Neurol 251:111–114
Gregg XT, Reddy V, Prchal JT (2002) Copper deficiency masquerading as myelodysplastic syndrome. Blood 100:1493–1495
Halfdanarson TR, Kumar N, Hogan WJ, Murray JA (2009) Copper deficiency in celiac disease. J Clin Gastroenterol 43:162–164
Halfdanarson TR, Kumar N, Li CY, Phyliky RL, Hogan WJ (2008) Hematological manifestations of copper deficiency: a retrospective review. Eur J Haematol 80:523–531
Haschke F, Ziegler EE, Edwards BB, Fomon SJ (1986) Effect of iron fortification of infant formula on trace mineral absorption. J Pediatr Gastroenterol Nutr 5:768–773
Hedera P, Fink JK, Bockenstedt PL, Brewer GJ (2003) Myelopolyneuropathy and pancytopenia due to copper deficiency and high zinc levels of unknown origin: further support for existence of a new zinc overload syndrome. Arch Neurol 60:1303–1306
Heller RM, Kirchner SG, O’Neill JA Jr, Hough AJ Jr, Howard L, Kramer SS, Green HL (1978) Skeletal changes of copper deficiency in infants receiving prolonged total parenteral nutrition. J Pediatr 92:947–949
Hellman NE, Gitlin JD (2002) Ceruloplasmin metabolism and function. Annu Rev Nutr 22:439–458
Hentze MW, Muckenthaler MU, Andrews NC (2004) Balancing acts: molecular control of mammalian iron metabolism. Cell 117:285–297
Jaiser SR, Duddy M (2007) Copper deficiency masquerading as subacute combined degeneration of the cord and myelodysplastic syndrome. Adv Clin Neurosci Rehabil 7:20–21
Juhasz-Pocsine K, Rudnicki SA, Archer RL, Harik SI (2007) Neurologic complications of gastric bypass surgery for morbid obesity. Neurology 68:1843–1850
Jung A, Marziniak M (2008) Copper deficiency as a treatable cause of myelopathy. Nervenarzt 79:421–425
Keen CL, Uriu-Hare JY, Hawk SN, Jankowski MA, Daston GP, Kwik-Uribe CL, Rucker RB (1998) Effect of copper deficiency on prenatal development and pregnancy outcome. Am J Clin Nutr 67:1003S–1011S
Kelley DS, Daudu PA, Taylor PC, Mackey BE, Turnlund JR (1995) Effects of low-copper diets on human immune response. Am J Clin Nutr 62:412–416
Klevay LM (2001) Iron overload can induce mild copper deficiency. J Trace Elem Med Biol 14:237–240
Klevay LM, Canfield WK, Gallagher SK, Henriksen LK, Lukaski HC, Bolonchuk W, Johnson LK, Milne DB (1986) Decreased glucose tolerance in two men during experimental copper depletion. Nutr Rep Int 33:371–382
Klevay LM, Inman L, Johnson LK, Lawler M, Mahalko JR, Milne DB, Lukaski HC, Bolonchuk W, Sandstead HH (1984) Increased cholesterol in plasma in a young man during experimental copper depletion. Metabolism 33:1112–1118
Kodama H, Murata Y, Kobayashi M (1999) Clinical manifestations and treatment of Menkes disease and its variants. Pediatr Int 41:423–429
Krebs NF (2000) Overview of zinc absorption and excretion in the human gastrointestinal tract. J Nutr 130:1374S–1377S
Kumar N (2006) Copper deficiency myelopathy (human swayback). Mayo Clin Proc 81:1371–1384
Kumar N, Ahlskog JE, Gross JB Jr (2004) Acquired hypocupremia after gastric surgery. Clin Gastroenterol Hepatol 2:1074–1079
Kumar N, Ahlskog JE, Klein CJ, Port JD (2006) Imaging features of copper deficiency myelopathy: a study of 25 cases. Neuroradiology 48:78–83
Kumar N, Crum B, Petersen RC, Vernino SA, Ahlskog JE (2004) Copper deficiency myelopathy. Arch Neurol 61:762–766
Kumar N, Elliott MA, Hoyer JD, Harper CM Jr, Ahlskog JE, Phyliky RL (2005) “Myelodysplasia”, myeloneuropathy, and copper deficiency. Mayo Clin Proc 80:943–946
Kumar N, Gross JB Jr (2006) Mutation in the ATP7A gene may not be responsible for hypocupraemia in copper deficiency myelopathy. Postgrad Med J 82:416
Kumar N, Gross JB Jr, Ahlskog JE (2004) Copper deficiency myelopathy produces a clinical picture like subacute combined degeneration. Neurology 63:33–39
Kumar N, Gross JB Jr, Ahlskog JE (2003) Myelopathy due to copper deficiency. Neurology 61:273–274
Kumar N, Low PA (2004) Myeloneuropathy and anemia due to copper malabsorption. J Neurol 251:747–749
Kumar N, McEvoy KM, Ahlskog JE (2003) Myelopathy due to copper deficiency following gastrointestinal surgery. Arch Neurol 60:1782–1785
Kuo YM, Gybina AA, Pyatskowit JW, Gitschier J, Prohaska JR (2006) Copper transport protein (Ctr1) levels in mice are tissue specific and dependent on copper status. J Nutr 136:21–26
Linder MC, Hazegh-Azam M (1996) Copper biochemistry and molecular biology. Am J Clin Nutr 63:797S–811S
Lonnerdal B, Hernell O (1994) Iron, zinc, copper and selenium status of breast-fed infants and infants fed trace element fortified milk-based infant formula. Acta Paediatr 83:367–373
Mao X, Schimmer AD (2008) The toxicology of clioquinol. Toxicol Lett 182:1–6
Menkes JH (1988) Kinky hair disease: twenty-five years later. Brain Dev 10:77–79
Menkes JH, Alter M, Steigleder GK, Weakley DR, Sung JH (1962) A sex-linked recessive disorder with retardation of growth, peculiar hair, and focal cerebral and cerebellar degeneration. Pediatrics 29:764–779
Meyer JH (1994) Nutritional outcomes of gastric operations. Gastroenterol Clin North Am 23:227–260
Miller K (2004) Obesity: surgical options. Best Pract Res Clin Gastroenterol 18:1147–1165
Mills ES (1930) The treatment of idiopathic (hypochromic) anemia with iron and copper. Can Med Assoc J 22:175–178
Morais MB, Fisberg M, Suzuki HU, Amancio OM, Machado NL (1994) Effects of oral iron therapy on serum copper and serum ceruloplasmin in children. J Trop Pediatr 40:51–52
Nations SP, Boyer PJ, Love LA, Burritt MF, Butz JA, Wolfe GI, Hynan LS, Reisch J, Trivedi JR (2008) Denture cream: an unusual source of excess zinc, leading to hypocupremia and neurologic disease. Neurology 71:639–643
Panel on dietary reference values, committee on medical aspects of food policy (1991) Dietary reference values for food energy and nutrients for the United Kingdom. Department of Health, London
Percival SS (1995) Neutropenia caused by copper deficiency: possible mechanisms of action. Nutr Rev 53:59–66
Petrak J, Vyoral D (2005) Hephaestin—a ferroxidase of cellular iron export. Int J Biochem Cell Biol 37:1173–1178
Pratt WB, Omdahl JL, Sorenson JR (1985) Lack of effects of copper gluconate supplementation. Am J Clin Nutr 42:681–682
Prodan CI, Bottomley SS, Holland NR, Lind SE (2006) Relapsing hypocupraemic myelopathy requiring high-dose oral copper replacement. J Neurol Neurosurg Psychiatry 77:1092–1093
Prodan CI, Bottomley SS, Vincent AS, Cowan LD, Greenwood-Van Meerveld B, Holland NR, Lind SE (2009) Copper deficiency after gastric surgery: a reason for caution. Am J Med Sci 337:256–258
Prodan CI, Holland NR, Wisdom PJ, Burstein SA, Bottomley SS (2002) CNS demyelination associated with copper deficiency and hyperzincemia. Neurology 59:1453–1456
Rock E, Gueux E, Mazur A, Motta C, Rayssiguier Y (1995) Anemia in copper-deficient rats: role of alterations in erythrocyte membrane fluidity and oxidative damage. Am J Physiol 269:C1245–C1249
Rowin J, Lewis SL (2005) Copper deficiency myeloneuropathy and pancytopenia secondary to overuse of zinc supplementation. J Neurol Neurosurg Psychiatry 76:750–751
International programme on chemical safety (1982) The 26th report of the joint FAO/WHO expert committee on food additives, Toxicological evaluation of certain food additives and contaminants. WHO food additive series: 17
Sass-Kortsak A (1965) Copper metabolism. In: Sobotka H, Stewart CP (eds) Advances in clinical chemistry. Academic Press, New York and London, pp 1–68
Schaumburg H, Herskovitz S (2008) Copper deficiency myeloneuropathy: a clue to clioquinol-induced subacute myelo-optic neuropathy? Neurology 71:622–623
Schleper B, Stuerenburg HJ (2001) Copper deficiency-associated myelopathy in a 46-year-old woman. J Neurol 248:705–706
Smith CH, Bidlack WR (1980) Interrelationship of dietary ascorbic acid and iron on the tissue distribution of ascorbic acid, iron and copper in female guinea pigs. J Nutr 110:1398–1408
Smith JC, Zeller JA, Brown ED, Ong SC (1976) Elevated plasma zinc: a heritable anomaly. Science 193:496–498
Spinazzi M, De Lazzari F, Tavolato B, Angelini C, Manara R, Armani M (2007) Myelo-optico-neuropathy in copper deficiency occurring after partial gastrectomy. Do small bowel bacterial overgrowth syndrome and occult zinc ingestion tip the balance? J Neurol 254:1012–1017
Sternlieb I, Janowitz HD (1964) Absorption of copper in malabsorption syndromes. J Clin Invest 43:1049–1055
Sternlier I, Morell AG, Bauer CD, Combes B, De Bobes-Sternberg S, Schein-Berg IH (1961) Detection of the heterozygous carrier of the Wilson’s disease gene. J Clin Invest 40:707–715
Tan JC, Burns DL, Jones HR (2006) Severe ataxia, myelopathy, and peripheral neuropathy due to acquired copper deficiency in a patient with history of gastrectomy. JPEN J Parenter Enteral Nutr 30:446–450
Tateishi J (2000) Subacute myelo-optico-neuropathy: clioquinol intoxication in humans and animals. Neuropathology 20(Suppl):S20–S24
Thiele DJ (1992) Metal-regulated transcription in eukaryotes. Nucleic Acids Res 20:1183–1191
Tovey FI, Hobsley M (2000) Post-gastrectomy patients need to be followed up for 20–30 years. World J Gastroenterol 6:45–48
Turnlund JR (1999) Copper. In: Shils ME, Olson JA, Shike M, Ross AC (eds) Modern nutrition in health and disease. Williams & Wilkins, Baltimore, pp 241–252
Uauy R, Olivares M, Gonzalez M (1998) Essentiality of copper in humans. Am J Clin Nutr 67:952S–959S
Wasa M, Satani M, Tanano H, Nezu R, Takagi Y, Okada A (1994) Copper deficiency with pancytopenia during total parenteral nutrition. JPEN J Parenter Enteral Nutr 18:190–192
Weihl CC, Lopate G (2006) Motor neuron disease associated with copper deficiency. Muscle Nerve 34:789–793
Williams DM (1983) Copper deficiency in humans. Semin Hematol 20:118–128
Willis MS, Monaghan SA, Miller ML, McKenna RW, Perkins WD, Levinson BS, Bhushan V, Kroft SH (2005) Zinc-induced copper deficiency: a report of three cases initially recognized on bone marrow examination. Am J Clin Pathol 123:125–131
Winston GP, Jaiser SR (2008) Copper deficiency myelopathy and subacute combined degeneration of the cord—why is the phenotype so similar? Med Hypotheses 71:229–236
Wu J, Ricker M, Muench J (2006) Copper deficiency as cause of unexplained hematologic and neurologic deficits in patient with prior gastrointestinal surgery. J Am Board Fam Med 19:191–194
Yaldizli O, Johansson U, Gizewski ER, Maschke M (2006) Copper deficiency myelopathy induced by repetitive parenteral zinc supplementation during chronic hemodialysis. J Neurol 253:1507–1509
Yu S, West CE, Beynen AC (1994) Increasing intakes of iron reduce status, absorption and biliary excretion of copper in rats. Br J Nutr 71:887–895
Yuzbasiyan-Gurkan V, Grider A, Nostrant T, Cousins RJ, Brewer GJ (1992) Treatment of Wilson’s disease with zinc: X. Intestinal metallothionein induction. J Lab Clin Med 120:380–386
Acknowledgments
No funding was received for this study.
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Jaiser, S.R., Winston, G.P. Copper deficiency myelopathy. J Neurol 257, 869–881 (2010). https://doi.org/10.1007/s00415-010-5511-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-010-5511-x