
Abstract
It has been more than two decades since the original chromosome transmission fidelity (Ctf) screen of Saccharomyces cerevisiae was published. Since that time the spectrum of mutations known to cause Ctf and, more generally, chromosome instability (CIN) has expanded dramatically as a result of systematic screens across yeast mutant arrays. Here we describe a comprehensive summary of the original Ctf genetic screen and the cloning of the remaining complementation groups as efforts to expand our knowledge of the CIN gene repertoire and its mutability in a model eukaryote. At the time of the original screen, it was impossible to predict either the genes and processes that would be overrepresented in a pool of random mutants displaying a Ctf phenotype or what the entire set of genes potentially mutable to Ctf would be. We show that in a collection of 136 randomly selected Ctf mutants, >65% of mutants map to 13 genes, 12 of which are involved in sister chromatid cohesion and/or kinetochore function. Extensive screening of systematic mutant collections has shown that ~350 genes with functions as diverse as RNA processing and proteasomal activity mutate to cause a Ctf phenotype and at least 692 genes are required for faithful chromosome segregation. The enrichment of random Ctf alleles in only 13 of ~350 possible Ctf genes suggests that these genes are more easily mutable to cause genome instability than the others. These observations inform our understanding of recurring CIN mutations in human cancers where presumably random mutations are responsible for initiating the frequently observed CIN phenotype of tumors.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Genetic instability refers to circumstances that lead to alterations of the genetic content of a cell. This instability can be a regulated part of healthy cell growth, as in the case of somatic hypermutability and developmentally controlled rearrangement of immunoglobulin genes (Upton et al. 2011). Low level genetic instability in germ line cells is critical to the evolutionary process as it generates variability within species to support population adaptation to selective pressures. Nonetheless, many outcomes of genetic instability have a detrimental effect on the cell and/or the organism, and hence knowing the etiology of genetic instability is fundamental to understanding many human diseases. It has long been known that aberrant chromosome number and structure can cause developmental disorders such as Down syndrome (Lejeune et al. 1959). The role of microsatellite instability (MIN; increased mutation rate) and chromosomal instability (CIN; increased aneuploidy rate) in cancer development is well documented (Lengauer et al. 1997), and the identity of many cancer-causing somatic mutations is known (e.g., Cancer Gene Census; Futreal et al. 2004). Indeed, CIN is seen in the majority of human cancers and is thought to play a role early in oncogenesis by creating a mutational environment in which the necessary oncogenes or tumor-suppressor genes are more likely to be mutated and enable a malignant tumor phenotype (Stratton et al. 2009).
Analysis of genetic instability in model organisms has proven to be highly relevant to the mechanistic understanding of human genetic instability, as processes that control genetic instability are well conserved. Greater than 60% of baker’s yeast (Saccharomyces cerevisiae) genes are clearly conserved across diverse organisms including humans (Heinicke et al. 2007). Indeed, yeast has been instrumental in delineating many of the pathways that protect the genome from instability in large part due to its experimental accessibility using genetic, cytological, and biochemical tools. Genes identified several decades ago in screens for cell division cycle control (CDC), mini-chromosome maintenance (MCM), and mitotic spindle function were found to be critical for maintaining genome integrity in yeast and humans (Hartwell et al. 1970; Hoyt et al. 1990; Huffaker et al. 1988; Maine et al. 1984). Additionally, early work in yeast revealed much regarding the biology of important cis-acting chromosomal features required for genome integrity (e.g., centromeres, telomeres, autonomously replicating sequences) (Chan and Tye 1980; Clarke and Carbon 1980; Szostak and Blackburn 1982). Remarkably, recent work in yeast has also shown that the state of aneuploidy alone is sufficient to cause further genome instability and increased mutation rates independent of the specific chromosomal amplification (Sheltzer et al. 2011). Thus yeast genes, proteins and genome architecture itself have contributed to our understanding of eukaryotic genome integrity.
One of the first classical genetic screens whose sole criterion was chromosome transmission was the chromosome transmission fidelity (Ctf) screen conducted in the Hieter lab (Spencer et al. 1990). More recently, many groups have exploited the yeast deletion collection for systematic screens for defects in genome integrity by various criteria (Andersen et al. 2008; Smith et al. 2004; Kanellis et al. 2007; Yuen et al. 2007). Efforts to screen for genome instability phenotypes in genetically tractable model organisms are ultimately aimed at revealing the spectrum of mutability to CIN in a eukaryotic genome. Various factors confound such a complete description, for example redundancy of gene function may mask CIN phenotypes in strains where only a single gene is mutated. Moreover, while nonessential genes can be completely deleted for CIN studies, essential genes must be tested as hypomorphic or conditional alleles that may exhibit allele-specific effects on genome integrity. Furthermore, some genes tolerate a large spectrum of individual missense mutations throughout the protein sequence, while other genes do not (i.e., highly evolutionarily conserved genes presumably do not tolerate mutations well). In the face of these obstacles, it should be remembered that the relevant CIN targets for human disease are the pathways most likely to cause CIN while retaining cell viability, if mutated at random.
Here we review the efforts to elucidate the yeast CIN gene spectrum over the past two decades, particularly via Ctf screening, using random and systematic approaches, and describe successful examples of yeast CIN genes predicting candidate human CIN genes in cancer (Spencer et al. 1990; Yuen et al. 2007; Stirling et al. 2011; Barber et al. 2008). We also describe the use of tiling microarrays to identify the two remaining Ctf complementation groups CTF2 and CTF9 as TOF1 and SMC3, respectively. The complete analysis of the major targets of the Ctf screen provides a view of which genes and pathways are the most mutable to a Ctf phenotype by random mutational events in a model eukaryote. Comprehensive screening of the yeast gene deletion and essential mutant collections by loss of function or reduction in function (i.e., direct tests of mutants for each gene) provide a view of all genes whose mutation is sufficient to cause a Ctf phenotype. By defining the genes most mutable to Ctf at random and the identity of all possible Ctf genes, we arrive at an important result. Only 13 of several hundred possible Ctf genes account for 68% of the original Ctf alleles found in multimember complementation groups. Moreover, 12 of these 13 genes impact sister chromatid cohesion and/or kinetochore function. This indicates dramatic differences in mutability of target Ctf genes and has direct relevance to cancer where presumably random mutations can initiate the oncogenic process.
Results and discussion
An early screen for highly mutable Ctf genes
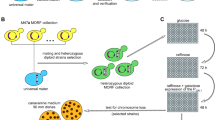
The haploid phase of the budding yeast life cycle allows the phenotypes of mutations to be revealed directly, without the need to backcross for homozygosity. This makes mutagenesis screens for nonlethal phenotypes highly efficient. As the loss of an endogenous chromosome from a haploid cell would be a lethal event, a screen for chromosome loss mutants required the design of a reporter strain containing a nonessential artificial chromosome fragment. Spencer et al. (1990) designed such a reporter to screen for mutants with defects in chromosome transmission fidelity by integrating a marker that would facilitate a visual colony color assay. The screen took advantage of plasmids with several important features to generate the reporter chromosome fragment (Fig. 1a). Each plasmid contained a telomeric Y′ element separated from a unique chromosomal sequence by a restriction site, a centromere, a selectable marker, and SUP11. When linearized, the plasmid recombines to create a partial disomy in which the telocentric chromosome fragment contains one short, plasmid-derived arm with a yeast telomere, and a long arm derived from yeast chromosomal DNA (Fig. 1a; (Spencer et al. 1990). The Ctf assay relies on the suppression of the ochre stop codon mutation in ade2-101 by the SUP11 gene, which encodes a suppressing tyrosyl-tRNA. While wild-type cells produce white colonies, ade2-101 cells give rise to red colonies due to the accumulation of an adenine precursor. The chromosome fragment carrying SUP11 allows reading through the ade2-101 mutation leading to an ADE2+ phenotype and suppression of the red color. Chromosome fragment-containing strains with an increased rate of chromosome loss grow as white colonies with red sectors derived from chromosome fragment loss events when grown on medium containing limiting adenine (for schematic see Fig. 1).

Features of the chromosome fragment and Ctf assay. a Schematic of chromosome fragment construction as described in the main text (Spencer et al. 1990). Briefly a plasmid with a unique restriction between Y′ and unique sequences (inverted triangle) is digested and used to transform a haploid strain. In this example, recombination (indicated by large X) of the Y′ element (i) fuses the chromosome fragment plasmid to the telomere on the right arm and coordinate recombination of a unique pericentromeric site (ii) fuses the left arm of the chromosome to the plasmid-telomere moiety to generate the independent chromosome fragment (iii). Diagonal lines indicate large chromosomal distances. b Schematic of the Ctf assay. ade2-101 cells form red colonies (left) which are suppressed to a white color by SUP11 (center). Mutagenesis of a Ctf gene leads to red sectors in a white colony. The colony on the far right is half-sectored. The frequency of half-sectored colonies, which are generated by loss of the CF in the first cell division on the plate, can be used to quantify chromosome loss rates
The visual Ctf assay system was the basis of the primary screen involving ethyl methanesulfonate (EMS) mutagenesis of yeast cells. This screen was somewhat unique in that the EMS concentration was reduced to permit ~100% cell viability and a modest tenfold increase in mutations as measured by de novo canavanine resistance (Spencer et al. 1990). Under these very light mutagenesis conditions, it could be predicted that the Ctf mutants identified would most likely be the result of mutations in single genes, thus simplifying follow-up experiments. The original Ctf screen yielded 136 Ctf mutants after screening ~600,000 independent single colonies (Spencer et al. 1990). Of the 136 Ctf mutants, 97 fell into 15 complementation groups ranging from 2 to 30 alleles (CTF1-12 and CTF17-19), and 39 were singletons. Subsequently, 26 Ctf genes, including representatives of all ≥2 member complementation groups and 11 singletons, were cloned either by complementation or, in two cases, the array-based methods described below (Fig. 2 and Table 1). In total, 108 of 136 Ctf alleles are mapped to mutations in these 26 cloned genes (Table 1).

Genomic tiling array analysis of CTF2 and CTF9 alleles. Prediction scores across the TOF1 (a) and SMC3 (b) open reading frames in the composite of four CTF2 and three CTF9 alleles respectively (“s” numbers indicate isolate number). A polymorphism in the strain background (asterisk) is also visible in all CTF2/TOF1 alleles. c, d Summary of sequencing data for TOF1 in 11 CTF2 alleles (c) and for SMC3 in three CTF9 alleles (d). Normal protein sequence is schematized as blue for Tof1p and orange for Smc3p.The amino acid number and identity of each mutation is noted. Three TOF1 frameshift mutations are indicated with additional amino acids schematized in red. SMC3 point mutations are indicated in green
Cloning of CTF2 and CTF9
The CTF2 and CTF9 allele groups remained uncloned following repeated attempts to “clone by complementation” using several yeast genomic libraries. A modern approach to identifying the genes responsible for a mutant phenotype is to use hybridization of mutant genomic DNA to microarrays consisting of overlapping, short oligonucleotides tiled over the entire yeast genome. Mismatches between the mutant genomic DNA and the reference sequence from which the array was designed lead to decreases in signal intensity at those probes. Subsequent Sanger sequencing is required to determine the nature of the base change. Of course, even more modern technologies—namely, next-generation sequencing—have also recently become economical ways to identify mutations. Both methods could prove useful for identifying the remaining singletons from the Ctf screen, and the availability of these techniques opens new possibilities for other genetic screens.
Tiling array analysis of genomic DNA from four CTF2 alleles and three CTF9 alleles identified single nucleotide polymorphisms (SNP) in the TOF1 and SMC3 genes, respectively (Fig. 2a, b). Each strain also contained additional high probability SNP predictions outside of TOF1 and SMC3. This was probably due to the use of EMS to generate these mutations coupled with spontaneous mutation accumulation during subsequent outgrowth and strain manipulation. By using several alleles from each complementation group, we were able to quickly identify the causative mutations. TOF1 encodes a 141-kDa protein that acts at stalled replication forks to promote chromatid cohesion after DNA damage (Katou et al. 2003; Mayer et al. 2004). Smc3p is a structural subunit of the cohesin complex and had a previously identified Ctf phenotype (Michaelis et al. 1997; Haering et al. 2002). Sequencing of each CTF2 and CTF9 allele revealed premature stop codons in the nonessential TOF1 gene and missense mutations in the essential SMC3 gene (Fig. 2c, d). These mutations presumably lead to complete or partial loss of function of TOF1, consistent with the results of a systematic Ctf screen that revealed a Ctf phenotype in tof1Δ cells (Yuen et al. 2007). The SMC3 mutations are likely hypomorphic as loss of function mutations in SMC3 would presumably be lethal. We identified SMC3 S75N mutations in two ctf9 alleles. Interestingly, SMC3 S75R mutations were found in a suppressor screen for a mutant allele of the cohesin acetyltransferase ECO1 (Rowland et al. 2009). These data suggest that SMC3 acetylation defects may play a role in the Ctf phenotype of these ctf9 alleles and highlights the importance of this region of Smc3 to genome stability (Rowland et al. 2009). Together CTF2/TOF1 and CTF9/SMC3 accounted for 14 of the original 136 Ctf alleles, completing analysis of all multimember complementation groups (≥3 independent alleles). The ability to clone most of the Ctf genes by complementation indicated that recessive alleles were identified in all of the classified Ctf alleles. After mapping CTF2/TOF1 and CTF9/SMC3 in this study (Fig. 2), only singletons from the original Ctf screen remain uncloned. Since most cloned Ctf alleles have been identified in other assays and subsequent direct tests, most of the remaining uncloned alleles have also likely been identified by subsequent studies of Ctf or other CIN phenotypes (Andersen et al. 2008; Smith et al. 2004; Kanellis et al. 2007; Yuen et al. 2007; Stirling et al. 2011).
The original Ctf genes should represent those genes most easily mutable to a Ctf phenotype because they were identified through a random screen. Table 1 lists the original Ctf genes, the molecular function of the gene product, and where known, the mutational status of the human ortholog in tumors. The original Ctf screen identified some different biological pathways but revealed a remarkable enrichment for genes involved in sister chromatid cohesion and separation (e.g., with one exception, CDC6, all genes in multimember complementation groups are annotated as cohesion or kinetochore related; Table 1). These include cohesin subunits (SMC1, SMC3, and SCC3), a cohesin loader (SCC2), kinetochore components (CHL3, MCM21, CTF13, NDC10, CHL4, CTF19, and MCM16), and regulatory factors for these processes (i.e., cohesion: ECO1, PDS5, RAD61, and CHL1, and kinetochore mitotic checkpoint: MAD1). A presumably analogous situation exists in precancerous cells where random mutations could lead to genome instability. By identifying the Ctf genes that are the most sensitive to random mutagenesis, we predicted that human orthologs of the yeast Ctf genes may represent CIN genes mutated in tumors. Subsequent studies of human tumors, including candidate gene sequencing studies derived from the yeast Ctf gene list, have identified human orthologs with mutations in 9 of the 18 original Ctf genes (see Table 1 and below).
Secondary screening: Ctf as an entry point to chromosome biology
Subsequent to the original screen, other groups have used the Ctf assay as a starting point to characterize specific biological pathways. An elegant example of this approach identified the cohesin complex (Michaelis et al. 1997). The authors engineered a Ctf assay strain in which the essential anaphase-promoting complex subunit CDC16 was mutated to allow conditional cell cycle arrest at anaphase. They mutagenized their reporter strain with EMS to 40–60% survival, a considerably heavier mutagenesis than the original Ctf screen (Spencer et al. 1990) and identified 377 Ctf alleles. The CDC16 ts allele enabled them to arrest each Ctf mutant at anaphase via temperature shift and to visually screen for the ability of the mutants to separate their nuclei and elongate their spindles (Michaelis et al. 1997). This directed secondary screen identified four complementation groups corresponding to the cohesin genes SMC1, SMC3, and SCC1 and the cohesin loader SCC2. By engineering a secondary screening tool into their Ctf screen, the authors were able to illuminate an important biological process.
Secondary screens have been used repeatedly to assign functions to Ctf mutants in the past two decades. For example, monitoring the integrity of kinetochores in the Ctf mutant collection identified two of the first kinetochore proteins, Ctf13 and Ctf14/Ndc10 (Doheny et al. 1993). This development was followed by the discovery of roles for Spt4 and nucleoporins in kinetochore function using the same secondary assay (Basrai et al. 1996; Iouk et al. 2002; Kerscher et al. 2001). Subsequent development of the synthetic dosage lethality concept (Kroll et al. 1996) was used to identify other kinetochore proteins including Ctf19p and Ctf3p (Hyland et al. 1999; Measday et al. 2002). More recently, a small-scale survey of telomere length in uncharacterized essential CIN and Ctf mutants led to the functional characterization of the yeast TTT (Tel2-Tti1-Tti2) complex, which has a conserved role in the biogenesis of phosphatidyl inositol 3-kinase-related kinases (Stirling et al. 2011; Hurov et al. 2010; Takai et al. 2010). In each case the choice of secondary assay determined the functions discovered for the Ctf genes. As the number of Ctf and CIN genes expands, it is increasingly clear that there are many mechanisms that stabilize the genome. In this light, choosing an appropriate secondary assay is critical to delineating the pathways that lead to genome instability in a particular mutant background.
Systematic screening for Ctf mutants in ordered mutant arrays
The development of the systematic yeast deletion collection and the synthetic genetic array technology made possible the systematic screening of single yeast gene deletions for Ctf phenotypes (Tong et al. 2001; Winzeler et al. 1999). These genome scale “reverse” genetic approaches provide the opportunity to answer the question “what are all genes mutable to Ctf in yeast?” This approach has several advantages over random mutagenesis, primarily the high coverage rate that comes with the ability to directly test an individual null mutant and knowledge of the gene deletion being tested, which removes the need for subsequent cloning steps (although it is still advisable to confirm the deletion is causing your phenotype of interest). In addition, direct comparison of Ctf chromosome loss rates can be informative when complete null alleles are tested as opposed to comparisons of different hypomorphic alleles. However, screening the systematic gene deletion array does remove the potential of finding dominant or neomorphic alleles. Such a screen also erases any information on which genes are the most easily mutable to the Ctf phenotype and does not interrogate essential genes.
A systematic Ctf screen of nonessential gene deletions was published in 2007 and identified 89 deletion mutants with detectable Ctf phenotypes (Yuen et al. 2007). More recently, it has become possible to test hypomorphic or temperature-sensitive alleles of essential genes systematically (Ben-Aroya et al. 2008; Breslow et al. 2008; Li et al. 2011). The spectrum of essential Ctf genes has also been expanded by testing ~2,000 alleles of more than 1,000 essential genes (Stirling et al. 2011). When combined with previous work, this study identified a list of 354 mutants with Ctf phenotypes, comprising 237 essential and 117 nonessential genes (Yuen et al. 2007; Stirling et al. 2011; Measday et al. 2005). The large proportional excess of essential Ctf genes may reflect the critical nature of faithful chromosome segregation. The essential Ctf genes are involved in a small number of cellular processes and often encode proteins that function in complexes with many subunits; for example, the kinetochore, the replication apparatus, the proteasome, and the general transcription machinery together account for a large percentage of the Ctf genes and each is composed of many essential subunits. Together these systematic Ctf screens should have identified the large majority of single genes mutable to a Ctf phenotype. One implication of this hypothesis is the ability to confidently ascribe Ctf mutability to entire cellular pathways. This approach gains even more power when combined with the various other CIN phenotypic screens that have been conducted on a genome-wide scale (Fig. 3; Stirling et al. 2011).

A network of CIN-associated gene ontology terms. GO terms associated with ≥20 CIN genes were included and then filtered for redundant terms to retain the most genes while simplifying the network. Node size is set to number of genes annotated to a term and edge weight is set to the number of genes overlapping between terms. The figure encompasses >80% of the 692 CIN genes reported (Stirling et al. 2011). Blue node color indicates ≥2 genes from the 13 multimember Ctf complementation groups from Spencer et al. (1990) (CTF1–CTF12, CTF18) are annotated to that GO term. Darker blue indicates more genes annotated to that term (i.e., establishment of/mitotic sister chromatid cohesion—12 genes; DNA replication—6 genes; DNA repair, meiosis—4 genes; mitosis—3 genes; chromosome, replication fork protection complex, kinetochore—2 genes)
Evaluation of diverse CIN phenotypes reveals common themes
Systematic screening for CIN phenotypes in yeast has been conducted using at least four other assays in addition to Ctf: namely, haploid a-like faker (ALF), diploid bi-mater (BiM), diploid loss of heterozygosity (LOH), and haploid gross chromosomal rearrangements (GCR) (Andersen et al. 2008; Smith et al. 2004; Kanellis et al. 2007; Yuen et al. 2007). The ALF and BiM assays measure loss of an endogenous locus (MAT) in haploid MATα or diploid MATa/MATα cells, respectively (Haber 1974; Yuen et al. 2007). LOH measures nonconservative rearrangements within or between homologous chromosomes that lead to hemi- or homozygosity of the met15Δ locus starting from a met15Δ/MET15 heterozygous diploid (Andersen et al. 2008). The GCR assay measures loss of a distal portion of a chromosome arm, typically by terminal deletion and de novo telomere addition (Smith et al. 2004; Kanellis et al. 2007). Each assay reports on loss of a genetic marker that can originate from several possible mechanisms. Thus, the CIN gene lists from each phenotypic screen are only partially overlapping (Yuen et al. 2007; Stirling et al. 2011). The aggregate output of the various CIN screens creates an organism-wide view of cellular structures and pathways comprising 692 genes that ensure the faithful transmission of the genome to subsequent generations.
Perhaps unsurprisingly, analysis of CIN genes en masse indicates that typical CIN genes encode nuclear proteins with functions closely linked to DNA or mitosis (Stirling et al. 2011). However, the compilation of CIN genes does identify >100 genes with peripheral or poorly understood functions in genome integrity, and for many of these pathways the mechanism of CIN is not clear (Fig. 3). A mechanistic understanding of CIN in each mutant is not currently available; however, in many cases a simple hypothesis can be generated to explain the role of a particular cellular pathway implicated in genome integrity. This is because each CIN mutation must ultimately cause or fail to correct one of only a few cellular events (i.e., DNA damage/DNA repair or chromosome non-disjunction). For example, >100 CIN gene products function directly in either DNA replication or DNA repair and it is rational to hypothesize that CIN arises due to faulty replication or repair of endogenous DNA damage (Stirling et al. 2011; Lange et al. 2011). Similarly, the ~70 CIN genes involved in kinetochore and spindle function could lead to aneuploidy as a result of mitotic defects and chromosome non-disjunction (Stirling et al. 2011; Thompson et al. 2010). In addition, many seemingly peripheral CIN pathways have been mechanistically linked to canonical genome stability pathways (i.e., the nuclear pore and proteasome function directly in DNA repair (Kerscher et al. 2001; Nagai et al. 2008; Ben-Aroya et al. 2010)). Another large class of CIN genes pertains to the production of RNA, either at the level of transcription or RNA processing including mRNA, tRNA, and rRNA (Stirling et al. 2011). Multiple hypotheses on the role of RNA production or processing and its interplay with genome stability have been put forward and differ depending on the mutant in question. Transcription can lead to genome instability through collision with DNA replication forks (Sikdar et al. 2008; Prado and Aguilera 2005; Takeuchi et al. 2003) but the transcriptional apparatus also plays a direct role in DNA repair (i.e., transcription-coupled repair; reviewed by Hanawalt and Spivak 2008). Splicing of a single transcript, alpha tubulin (TUB1), has been shown to affect genome stability due to dysregulated α/β-tubulin levels and subsequent mitotic spindle defects (Biggins et al. 2001; Burns et al. 2002). Finally, mRNA processing defects can also cause genome instability through the production of R-loops, which are RNA:DNA hybrids normally removed co-transcriptionally by processing and export factors (PCS, PH, and Yujia A. Chan, unpublished observation; D. Koshland personal communication; Gomez-Gonzalez et al. 2009; Mischo et al. 2011). Thus, even seemingly related CIN genes (e.g., spliceosome and mRNA processing) could have different mechanisms leading to CIN that require direct experimental validation.
Mutability and the value of classical screens
Systematic analysis has revealed the spectrum of genes that are potentially mutable to a Ctf phenotype but gives no information on the relative mutability of individual genes or pathways. In the case of deletions, direct tests give a “yes” or “no” answer, whereas hypomorphic alleles of essential genes provide a “yes” or “probably not” answer because allele specificity and incomplete loss of function cannot eliminate the possibility that other alleles may show Ctf. While the strength of phenotypes may provide some information regarding the importance of individual genes or pathways to chromosome segregation, systematic screens cannot answer which genes are the most highly mutable to Ctf. Naively, one might think that nonessential genes would be more mutable since they are dispensable for viability. However, the frequency of Ctf alleles in nonessential genes is similar to the frequency of nonessential genes in the yeast genome (~80% in each case; Table 1), suggesting that no such bias exists. The cloning of CTF2 and CTF9 demonstrates how different mutations can be selected in essential versus nonessential genes; all of the CTF2/TOF1 mutants are truncations, whereas only point mutations are found in the essential CTF9/SMC3 gene (Fig. 2). Prior studies have demonstrated the wide variation in mutability of specific genes in random mutation collections. For example, extensive CDC screening did not identify mutations in β-tubulin (TUB2), whereas subsequent direct mutagenesis of the cloned TUB2 gene, and introduction into the yeast genome, led to the identification of tub2-ts alleles that cause a CDC phenotype (Hartwell et al. 1970; Huffaker et al. 1988). TUB2 is clearly mutable to a CDC phenotype when directly tested but evidently dozens of other genes are more mutable (Hartwell et al. 1973). The most important factors controlling the relative mutability of genes to a phenotype are poorly understood.
Our data suggest striking differences in mutability within the Ctf genes—if the 354 Ctf genes were equally mutable the probability of finding three alleles of the same gene from the 136 original Ctf alleles would be P = 0.0069, yet more than two thirds of the original Ctf alleles are accounted for by 13 genes in complementation groups with greater than or equal to three members (Table 1). Clearly, the structural elements of a gene (e.g., size, genomic position, number of codons mutable to stop by single base substitutions, etc. discussed in Lang and Murray 2008) influence its mutability, in addition to the effect of the gene’s impact on cellular fitness and the strength of the relevant phenotype. The question of mutability to a phenotype in a pool of variants is particularly relevant to Ctf/CIN phenotypes because of their functional relationship to oncogenesis, where a presumably random mutation initiates a CIN phenotype. The original screen for Ctf clearly pointed to cohesion as the most mutable process including mutations in the cohesin subunits themselves (Table 1; Spencer et al. 1990). Subsequent work has suggested that chromatid cohesion is critical to preventing CIN in cancer and that cohesin subunits are mutated in colorectal tumors (Barber et al. 2008; Schvartzman et al. 2010; Xu et al. 2011). A recent study has demonstrated that a mutation in a cohesin subunit in human cancer causes aneuploidy (Solomon et al. 2011). Because of obvious advantages (i.e., coverage, mutant identification), most genetic screens are now done on a systematic basis using arrayed or pooled collections of known mutants. However, with the proliferation of next-generation sequencing, the ability of classical screens to detect the most labile genes in a phenotypic assay could be revisited as an alternative to systematic screens in certain circumstances. Perhaps most importantly, the issue of mutability impacts human cancers, where recurring mutations of particular CIN genes are probably due both to selection and preferential mutability of certain targets.
Implications for CIN in human health and disease
Driver mutations in cancer can be subdivided into two broad classes: oncogenes (i.e., gain of function mutations that induce proliferative and/or survival phenotypes) and tumor suppressors (i.e., loss of function mutations in genes whose normal function counteracts proliferative and survival signals). CIN mutations could be considered a third class or perhaps as a subcategory of tumor suppressors. CIN typically arises from the loss of function of a gene that protects genome integrity, similar to the protective effects of tumor suppressors. However, genetic instability itself is insufficient to drive tumor formation; rather it creates an environment in which the mutations, translocations, or copy number alterations needed for a proliferative cancer phenotype are more likely to occur (Stratton et al. 2009; Schvartzman et al. 2010; Cahill et al. 1999). A possible role for CIN or mutator mutations as early events in tumorigenesis is illustrated in Fig. 4 (similar concepts are portrayed in Stratton et al. 2009; Loeb 2011). Somatic cells are exposed both to the perils of normal mitosis and to potentially mutagenic environmental factors. At some frequency, this mutational pressure will randomly mutate a CIN gene, which in turn dramatically increases the rate of mutations/chromosomal abnormalities in subsequent mitoses (Fig. 4). Ultimately, oncogenic mutations are selected from a mutated population. Recent research has confirmed, at least in a childhood acute lymphoblastic leukemia, that tumor cell populations have variegated genetic content and follow complex and nonlinear evolutionary paths (Anderson et al. 2011). The genetic variation created by CIN or MIN mutations within a tumor population could also facilitate adaptation to the stresses intrinsic to tumor growth (e.g., apoptotic signals, hypoxia, and nutrient restriction) and extrinsic pressures such as chemotherapy or radiation (Anderson et al. 2011; Pavelka et al. 2010).

Possible role of CIN in human tumors. CIN mutations may facilitate the evolutionary process underlying tumorigenesis. This concept is also illustrated for mutator phenotypes in the literature (Stratton et al. 2009; Loeb 2011). From left to right, a cellular mutational pathway is fed by intrinsic and extrinsic factors and can lead to a CIN mutation (blue diamond). CIN creates a mutant population in which oncogenic mutations driving proliferation are more likely to occur (red star). The proliferative phenotype and increased CIN facilitate variation (green star) that can help tumors respond to the environmental challenges (e.g., hypoxia, chemotherapy)
One approach to utilize yeast Ctf genes to identify cancer genes has been candidate gene sequencing. One such study sequenced the homologs of 96 yeast CIN genes in a colorectal tumor sample and found 11 genes with somatic mutations, 4 in cohesin subunits (Barber et al. 2008). Resequencing a panel of colorectal tumors determined that several cohesin subunits are mutated in ~3% of tumors and thus together may underlie a significant proportion of colorectal tumors (Barber et al. 2008). An earlier study used genome instability phenotypes in both yeast and Drosophila to identify human candidate CIN genes for sequencing (Wang et al. 2004). With the emergence of next-generation sequencing, the days of candidate gene sequencing may be limited. Instead, researchers will opt to sequence the genome of a tumor and matched normal tissue to identify all possible somatic mutations. This has already led to an abundance of data regarding somatic variants with limited phenotypic information for most of them. For example, at the time of writing, the COSMIC database contained records of >550 whole cancer genomes and >200,000 mutations (http://www.sanger.ac.uk/genetics/CGP/cosmic/) (Forbes et al. 2010). Presumably, most of the mutations in a tumor are “passenger” mutations that do not contribute significantly to the disease phenotype. It is our opinion that model organisms can contribute to the dissection of the functions of these somatic variants. This viewpoint spurred our recent effort to catalog CIN phenotypes in essential yeast genes and compile a master list of CIN pathways (Stirling et al. 2011). The phenotypic blueprints provided by systematic screens in model organisms can help guide the search for functional variants among tumor somatic mutations. In other words, we predict that a candidate gene for CIN in a tumor will typically belong to a cellular pathway known to cause CIN in model organisms. Thus, having high-quality surveys of tumor-relevant phenotypes in model organisms should create a functional filter for somatic variants in tumors (i.e., Table 1; Stirling et al. 2011).
Understanding the spectrum of CIN genes will allow us to understand the etiology of human diseases associated with genetic instability and ultimately help design strategies to combat these diseases. One powerful approach to designer therapeutics that has gained significant attention in recent years is to exploit the concept of synthetic lethality. (For a recent review see Brough et al. 2011). Synthetic lethality describes a situation where two viable single mutations lead to cell death when they occur in the same cell/organism. The application of this concept to cancer was first articulated nearly 15 years ago by Hartwell et al. (1997). Since cancers are genetically distinct from surrounding tissues, there exists an opportunity to leverage synthetic genetic interactions to selectively kill tumors based on the very genetic differences that permit oncogenesis. In principle, synthetic lethality can target oncogenes or tumor suppressors, and there are now numerous large-scale efforts to define genetic interactions both in human cancer cells and in model organisms (e.g., Costanzo et al. 2010, Luo et al. 2009). Since CIN phenotypes are common to diverse tumors, we suggest that synthetic lethal targeting of CIN mutations is a viable therapeutic approach. In fact, we have already exploited conserved genetic interactions from yeast to identify conditions that selectively kill CIN gene-deficient tumor cells (McManus et al. 2009). Accordingly, the synthetic lethal partners of the most highly mutable human cancer CIN genes would be predicted to be the best and broadest therapeutic targets. The combination of tumor genome sequence data and mutant identification with genetic interaction networks and insights into relative mutability should continue to present novel therapeutic opportunities in the future.
Ctf in the next two decades
With the advent of next-generation sequencing, human tumor genome sequencing data are currently leading to the identification of a huge number of variants with potential relevance to cancer. Determining which of these variants is responsible for the CIN phenotype seen in various tumors is a challenge and a daunting task for biologists. One future direction that yeast Ctf and, more broadly, CIN phenotypes will play a role is in predicting functionality for human CIN candidate genes mutated in tumors. Given the considerable evolutionary distance between yeast and humans, it is likely that other model systems will contribute to this effort as technologies improve. We predict that model organisms will continue to directly contribute to efficient functional analysis of tumor-associated somatic variants. For example, yeast has proven to be a useful system to test the function of a complementing human cDNA or assess the function of conserved residues in human disease proteins (e.g., Gammie et al. 2007, Jeong et al. 2007, Marini et al. 2008). In principle these complementation or homology-based approaches could be applied to assess the role of human tumor variants in genome integrity.
One approach that will be used more widely in the future is screening cultured mammalian cells using genome-wide siRNA or shRNA libraries. While a screen for increased aneuploidy in siRNA/shRNA-treated cells has yet to be published, there have been several relevant screens conducted. For example, mammalian genome-wide screens for increased sensitivity to ionizing radiation or spontaneous γ-H2Ax phosphorylation have been conducted (Hurov et al. 2010; Paulsen et al. 2009). The expense and lack of facile genetics make an RNAi screening approach in tissue culture impractical for many labs and highlight the importance of model organisms for biological validation. However, screening of CIN and other phenotypes in high throughput directly in human cells has obvious advantages and immediate relevance to human disease. Indeed, efforts by the MitoCheck consortium have generated impressive datasets of physical interactions within the mammalian chromosome segregation pathway and siRNAs that induce various mitotic defects in human cells (Hutchins et al. 2010; Neumann et al. 2010). These datasets have already implicated many new proteins in chromosome segregation and will certainly have an impact on our understanding of CIN in cancer.
The rate of genome instability should itself be a trait subject to variation among individuals. Certainly, the study of genome instability in model organisms demonstrates that a highly significant variation in this trait is compatible with viability. Interestingly, early documentation of variation in mutation rate among humans has been recently revealed in whole genome sequence analysis of family trios (the 1000 Genomes Project et al. 2011). How variable is the CIN rate among individuals in the current human population? Which pathways contribute most strongly to natural variation in CIN, for humans or for organisms occupying other niches? The past two decades have elucidated many, if not most, of the players. Perhaps the next two decades will translate the Ctf data to therapeutic use in humans and reveal how cells and organisms both preserve and enrich their genomic repertoires in the context of ongoing evolution.
Materials and methods
Comparative genome hybridization
Four CTF2 (s1, s29, s38, and s59) and three CTF9 (s16, s24, and s104) alleles were chosen from the original Ctf mutant collection (Spencer et al. 1990). DNA preparation, microarray hybridization, and data analysis were performed as described (Gresham et al. 2006).
Sequencing of TOF1 and SMC3
TOF1 from 11 CTF2 alleles and SMC3 from 3 CTF9 alleles were amplified using Platinum PCR SuperMix High Fidelity (Invitrogen #12532-016) according to the manufacturer’s instructions with primers flanking the gene of interest. Sequencing was performed by Retrogen using 5-ng/uL template DNA and 100-pmol primer. Primers for sequencing included regions 100–150 bp upstream and downstream of the ORF. Where tiling microarray hybridization was employed, the base pair changes matched closely with the chromosomal coordinates of the predicted SNP (Fig. 2).
References
Andersen MP, Nelson ZW, Hetrick ED et al (2008) A genetic screen for increased loss of heterozygosity in Saccharomyces cerevisiae. Genetics 179:1179–1195
Anderson K, Lutz C, van Delft FW et al (2011) Genetic variegation of clonal architecture and propagating cells in leukaemia. Nature 469:356–361
Barber TD, McManus K, Yuen KW et al (2008) Chromatid cohesion defects may underlie chromosome instability in human colorectal cancers. Proc Natl Acad Sci USA 105:3443–3448
Basrai MA, Kingsbury J, Koshland D et al (1996) Faithful chromosome transmission requires Spt4p, a putative regulator of chromatin structure in Saccharomyces cerevisiae. Mol Cell Biol 16:2838–2847
Ben-Aroya S, Coombes C, Kwok T et al (2008) Toward a comprehensive temperature-sensitive mutant repository of the essential genes of Saccharomyces cerevisiae. Mol Cell 30:248–258
Ben-Aroya S, Agmon N, Yuen K et al (2010) Proteasome nuclear activity affects chromosome stability by controlling the turnover of Mms22, a protein important for DNA repair. PLoS Genet 6:e1000852
Biggins S, Bhalla N, Chang A et al (2001) Genes involved in sister chromatid separation and segregation in the budding yeast Saccharomyces cerevisiae. Genetics 159:453–470
Breslow DK, Cameron DM, Collins SR et al (2008) A comprehensive strategy enabling high-resolution functional analysis of the yeast genome. Nat Methods 5:711–718
Brough R, Frankum JR, Costa-Cabral S et al (2011) Searching for synthetic lethality in cancer. Curr Opin Genet Dev 21:34–41
Burns CG, Ohi R, Mehta S et al (2002) Removal of a single alpha-tubulin gene intron suppresses cell cycle arrest phenotypes of splicing factor mutations in Saccharomyces cerevisiae. Mol Cell Biol 22:801–815
Cahill DP, Kinzler KW, Vogelstein B et al (1999) Genetic instability and Darwinian selection in tumours. Trends Cell Biol 9:M57–60
Chan CS, Tye BK (1980) Autonomously replicating sequences in Saccharomyces cerevisiae. Proc Natl Acad Sci USA 77:6329–6333
Clarke L, Carbon J (1980) Isolation of a yeast centromere and construction of functional small circular chromosomes. Nature 287:504–509
Costanzo M, Baryshnikova A, Bellay J et al (2010) The genetic landscape of a cell. Science 327:425–431
Doheny KF, Sorger PK, Hyman AA et al (1993) Identification of essential components of the S. cerevisiae kinetochore. Cell 73:761–774
Forbes SA, Tang G, Bindal N et al (2010) COSMIC (the Catalogue of Somatic Mutations in Cancer): a resource to investigate acquired mutations in human cancer. Nucleic Acids Res 38:D652–7
Futreal PA, Coin L, Marshall M et al (2004) A census of human cancer genes. Nat Rev Cancer 4:177–183
Gammie AE, Erdeniz N, Beaver J et al (2007) Functional characterization of pathogenic human MSH2 missense mutations in Saccharomyces cerevisiae. Genetics 177:707–721
Gomez-Gonzalez B, Felipe-Abrio I, Aguilera A (2009) The S-phase checkpoint is required to respond to R-loops accumulated in THO mutants. Mol Cell Biol 29:5203–5213
Gresham D, Ruderfer DM, Pratt SC et al (2006) Genome-wide detection of polymorphisms at nucleotide resolution with a single DNA microarray. Science 311:1932–1936
Haber JE (1974) Bisexual mating behavior in a diploid of Saccharomyces cerevisiae: evidence for genetically controlled non-random chromosome loss during vegetative growth. Genetics 78:843–858
Haering CH, Lowe J, Hochwagen A et al (2002) Molecular architecture of SMC proteins and the yeast cohesin complex. Mol Cell 9:773–788
Hanawalt PC, Spivak G (2008) Transcription-coupled DNA repair: two decades of progress and surprises. Nat Rev Mol Cell Biol 9:958–970
Hartwell LH, Culotti J, Reid B (1970) Genetic control of the cell-division cycle in yeast. I. Detection of mutants. Proc Natl Acad Sci USA 66:352–359
Hartwell LH, Mortimer RK, Culotti J et al (1973) Genetic control of the cell division cycle in yeast: V. Genetic analysis of cdc mutants. Genetics 74:267–286
Hartwell LH, Szankasi P, Roberts CJ et al (1997) Integrating genetic approaches into the discovery of anticancer drugs. Science 278:1064–1068
Heinicke S, Livstone MS, Lu C et al (2007) The Princeton Protein Orthology Database (P-POD): a comparative genomics analysis tool for biologists. PLoS One 2:e766
Hoyt MA, Stearns T, Botstein D (1990) Chromosome instability mutants of Saccharomyces cerevisiae that are defective in microtubule-mediated processes. Mol Cell Biol 10:223–234
Huffaker TC, Thomas JH, Botstein D (1988) Diverse effects of beta-tubulin mutations on microtubule formation and function. J Cell Biol 106:1997–2010
Hurov KE, Cotta-Ramusino C, Elledge SJ (2010) A genetic screen identifies the Triple T complex required for DNA damage signaling and ATM and ATR stability. Genes Dev 24:1939–1950
Hutchins JR, Toyoda Y, Hegemann B et al (2010) Systematic analysis of human protein complexes identifies chromosome segregation proteins. Science 328:593–599
Hyland KM, Kingsbury J, Koshland D et al (1999) Ctf19p: a novel kinetochore protein in Saccharomyces cerevisiae and a potential link between the kinetochore and mitotic spindle. J Cell Biol 145:15–28
Iouk T, Kerscher O, Scott RJ et al (2002) The yeast nuclear pore complex functionally interacts with components of the spindle assembly checkpoint. J Cell Biol 159:807–819
Jeong H, Herskowitz I, Kroetz DL et al (2007) Function-altering SNPs in the human multidrug transporter gene ABCB1 identified using a Saccharomyces-based assay. PLoS Genet 3:e39
Kanellis P, Gagliardi M, Banath JP et al (2007) A screen for suppressors of gross chromosomal rearrangements identifies a conserved role for PLP in preventing DNA lesions. PLoS Genet 3:e134
Katou Y, Kanoh Y, Bando M et al (2003) S-phase checkpoint proteins Tof1 and Mrc1 form a stable replication-pausing complex. Nature 424:1078–1083
Kerscher O, Hieter P, Winey M et al (2001) Novel role for a Saccharomyces cerevisiae nucleoporin, Nup170p, in chromosome segregation. Genetics 157:1543–1553
Kroll ES, Hyland KM, Hieter P et al (1996) Establishing genetic interactions by a synthetic dosage lethality phenotype. Genetics 143:95–102
Lang GI, Murray AW (2008) Estimating the per-base-pair mutation rate in the yeast Saccharomyces cerevisiae. Genetics 178:67–82
Lange SS, Takata K, Wood RD (2011) DNA polymerases and cancer. Nat Rev Cancer 11:96–110
Lejeune J, Gautier M, Turpin R (1959) Study of somatic chromosomes from 9 Mongoloid children. C R Hebd Seances Acad Sci 248:1721–1722
Lengauer C, Kinzler KW, Vogelstein B (1997) Genetic instability in colorectal cancers. Nature 386:623–627
Li Z, Vizeacoumar FJ, Bahr S et al (2011) Systematic exploration of essential yeast gene function with temperature-sensitive mutants. Nat Biotechnol 29:361–367
Loeb LA (2011) Human cancers express mutator phenotypes: origin, consequences and targeting. Nat Rev Cancer 11:450–457
Luo J, Emanuele MJ, Li D et al (2009) A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell 137:835–848
Maine GT, Sinha P, Tye BK (1984) Mutants of S. cerevisiae defective in the maintenance of minichromosomes. Genetics 106:365–385
Marini NJ, Gin J, Ziegle J et al (2008) The prevalence of folate-remedial MTHFR enzyme variants in humans. Proc Natl Acad Sci USA 105:8055–8060
Mayer ML, Pot I, Chang M et al (2004) Identification of protein complexes required for efficient sister chromatid cohesion. Mol Biol Cell 15:1736–1745
McManus KJ, Barrett IJ, Nouhi Y et al (2009) Specific synthetic lethal killing of RAD54B-deficient human colorectal cancer cells by FEN1 silencing. Proc Natl Acad Sci USA 106:3276–3281
Measday V, Hailey DW, Pot I et al (2002) Ctf3p, the Mis6 budding yeast homolog, interacts with Mcm22p and Mcm16p at the yeast outer kinetochore. Genes Dev 16:101–113
Measday V, Baetz K, Guzzo J et al (2005) Systematic yeast synthetic lethal and synthetic dosage lethal screens identify genes required for chromosome segregation. Proc Natl Acad Sci USA 102:13956–13961
Michaelis C, Ciosk R, Nasmyth K (1997) Cohesins: chromosomal proteins that prevent premature separation of sister chromatids. Cell 91:35–45
Mischo HE, Gomez-Gonzalez B, Grzechnik P et al (2011) Yeast Sen1 helicase protects the genome from transcription-associated instability. Mol Cell 41:21–32
Nagai S, Dubrana K, Tsai-Pflugfelder M et al (2008) Functional targeting of DNA damage to a nuclear pore-associated SUMO-dependent ubiquitin ligase. Science 322:597–602
Neumann B, Walter T, Heriche JK et al (2010) Phenotypic profiling of the human genome by time-lapse microscopy reveals cell division genes. Nature 464:721–727
Oikawa K, Ohbayashi T, Kiyono T et al (2004) Expression of a novel human gene, human wings apart-like (hWAPL), is associated with cervical carcinogenesis and tumor progression. Cancer Res 64:3545–3549
Paulsen RD, Soni DV, Wollman R et al (2009) A genome-wide siRNA screen reveals diverse cellular processes and pathways that mediate genome stability. Mol Cell 35:228–239
Pavelka N, Rancati G, Li R (2010) Dr Jekyll and Mr Hyde: role of aneuploidy in cellular adaptation and cancer. Curr Opin Cell Biol 22:809–815
Prado F, Aguilera A (2005) Impairment of replication fork progression mediates RNA polII transcription-associated recombination. EMBO J 24:1267–1276
Rowland BD, Roig MB, Nishino T et al (2009) Building sister chromatid cohesion: smc3 acetylation counteracts an antiestablishment activity. Mol Cell 33:763–774
Schvartzman JM, Sotillo R, Benezra R (2010) Mitotic chromosomal instability and cancer: mouse modelling of the human disease. Nat Rev Cancer 10:102–115
Sheltzer JM, Blank HM, Pfau SJ et al (2011) Aneuploidy drives genomic instability in yeast. Science 333:1026–1030
Sikdar N, Banerjee S, Zhang H et al (2008) Spt2p defines a new transcription-dependent gross chromosomal rearrangement pathway. PLoS Genet 4:e1000290
Smith S, Hwang JY, Banerjee S et al (2004) Mutator genes for suppression of gross chromosomal rearrangements identified by a genome-wide screening in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A 101:9039–9044
Solomon DA, Kim T, Diaz-Martinez LA et al (2011) Mutational inactivation of STAG2 causes aneuploidy in human cancer. Science 333:1039–1043
Spencer F, Gerring SL, Connelly C et al (1990) Mitotic chromosome transmission fidelity mutants in Saccharomyces cerevisiae. Genetics 124:237–249
Stirling PC, Bloom MS, Solanki-Patil T et al (2011) The complete spectrum of yeast chromosome instability genes identifies candidate CIN cancer genes and functional roles for ASTRA complex components. PLoS Genet 7:e1002057
Stratton MR, Campbell PJ, Futreal PA (2009) The cancer genome. Nature 458:719–724
Szostak JW, Blackburn EH (1982) Cloning yeast telomeres on linear plasmid vectors. Cell 29:245–255
Takai H, Xie Y, de Lange T et al (2010) Tel2 structure and function in the Hsp90-dependent maturation of mTOR and ATR complexes. Genes Dev 24:2019–2030
Takeuchi Y, Horiuchi T, Kobayashi T (2003) Transcription-dependent recombination and the role of fork collision in yeast rDNA. Genes Dev 17:1497–1506
The 1000 Genomes Project, Conrad DF, Keebler JE et al (2011) Variation in genome-wide mutation rates within and between human families. Nat Genet 43:712–714
Thompson SL, Bakhoum SF, Compton DA (2010) Mechanisms of chromosomal instability. Curr Biol 20:R285–95
Tong AH, Evangelista M, Parsons AB et al (2001) Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science 294:2364–2368
Upton DC, Gregory BL, Arya R et al (2011) AID: a riddle wrapped in a mystery inside an enigma. Immunol Res 49:14–24
Wang Z, Cummins JM, Shen D et al (2004) Three classes of genes mutated in colorectal cancers with chromosomal instability. Cancer Res 64:2998–3001
Winzeler EA, Shoemaker DD, Astromoff A et al (1999) Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science 285:901–906
Xu H, Tomaszewski JM, McKay MJ (2011) Can corruption of chromosome cohesion create a conduit to cancer? Nat Rev Cancer 11:199–210
Yuen KW, Warren CD, Chen O et al (2007) Systematic genome instability screens in yeast and their potential relevance to cancer. Proc Natl Acad Sci U S A 104:3925–3930
Acknowledgments
PCS is a fellow of the Terry Fox Foundation (#700044) and acknowledges the additional support of the Michael Smith Foundation for Health Research. MJD is a Rita Allen Scholar and was supported in part by National Institutes of Health (NIH) grant P50 GM071508 to the Lewis Sigler Institute at Princeton University. MB is supported by the Intramural Research Program of the National Cancer Institute, NIH. FS is supported by NIH GM077456. PH is supported by grants from the NIH CA016519 and Canadian Institutes of Health Research MOP-38096.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Erich Nigg
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Stirling, P.C., Crisp, M.J., Basrai, M.A. et al. Mutability and mutational spectrum of chromosome transmission fidelity genes. Chromosoma 121, 263–275 (2012). https://doi.org/10.1007/s00412-011-0356-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00412-011-0356-3