
Abstract
Homogeneous single crystals of synthetic monticellite with the composition \({\text{Ca}}_{0.88}{\text{Mg}}_{1.12}{\text{SiO}}_4\) (Mtc I) were annealed in a piston-cylinder apparatus at temperatures between 1000 and \(1200\,^{\circ }\hbox {C}\), pressures of 1.0–1.4 GPa, for run durations from 10 min to 24 h and applying bulk water contents ranging from 0.0 to 0.5 wt% of the total charge. At these conditions, Mtc I breaks down to a fine-grained, symplectic intergrowth. Thereby, two types of symplectites are produced: a first symplectite type (Sy I) is represented by an aggregate of rod-shaped forsterite immersed in a matrix of monticellite with end-member composition (Mtc II), and a second symplectite type (Sy II) takes the form of a lamellar merwinite–forsterite intergrowth. Both symplectites may form simultaneously, where the formation of Sy I is favoured by the presence of water. Sy I is metastable with respect to Sy II and is successively replaced by the latter. For both symplectite types, the characteristic spacing of the symplectite phases is independent of run duration and is only weeakly influenced by the water content, but it is strongly temperature dependent. It varies from about 400 nm at \(1000\,^{\circ }\hbox {C}\) to 1200 nm at \(1100\,^{\circ }\hbox {C}\) in Sy I, and from 300 nm at \(1000\,^{\circ }\hbox {C}\) to 700 nm at \(1200\,^{\circ }\hbox {C}\) in Sy II. A thermodynamic analysis reveals that the temperature dependence of the characteristic spacing of the symplectite phases is due to a relatively high activation energy for chemical segregation by diffusion within the reaction front as compared to the activation energy for interface reactions at the reaction front. The temperature dependence of the characteristic lamellar spacing and the temperature-time dependence of overall reaction progress have potential for applications in geo-thermometry and geo-speedometry.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Symplectites are relatively fine-grained, two- or polyphase mineral aggregates with characteristic vermicular or lamellar intergrowth replacing a homogeneous precursor phase (Spry 1969; Vernon 2004). Typically, replacement of a primary mineral by a symplectite occurs at a sharp reaction front (Fig. 1). The vermiculae or lamellae of the symplectite are usually oriented approximately perpendicular to the replacement front (Obata 2011), and the different symplectite phases alternate at close to regular intervals.

Schematic drawing of a symplectite replacement microstructure. A lamellar intergrowth of phases \(\beta\) and \(\gamma\) replaces the homogeneous precursor phase \(\alpha\); u is the velocity of the reaction front relative to a material point in phase \(\alpha\), \(\delta\) is the width of the reaction front, and \(\lambda\) is the characteristic lamellar spacing. Arrows indicate redistribution of chemical components by diffusion within the reaction front
Symplectites are frequently observed in magmatic and metamorphic rocks and are known from silicate, oxide, carbonate and sulfide systems (Vernon 2004). Replacement of the precursor phase may occur isochemically or with a change in bulk composition on the scale of the replacement structure. For example, partial replacement of garnet by various symplectic intergrowths has been interpreted to result from decompression-induced breakdown of garnet, where plagioclase and scapolite may constitute the felsic, and pyroxene, amphibole, cordierite, spinel or olivine the mafic phases in the symplectite (Carlswell et al. 1989; Messiga and Bettini 1990; Mukhopadhyay 1991; Harley and Buick 1992; Altherr and Kalt 1996; Godard and Martin 2000; Keankeo et al. 2000; Foley et al. 2006; Degi et al. 2009; Obata 2011; Obata et al. 2013; Spacek et al. 2013). Familiar examples of symplectite formation involving composition change are myrmekites, where a vermicular intergrowth of sodic plagioclase and quartz replaces K-feldspar (Phillips 1974; Abart et al. 2014) pseudoleucites, where vermicular nepheline–K-feldspar intergrowths pseudomorphically replace leucite (Gittins et al. 1980); scapolite–quartz symplectites replacing K-feldspar (Harley and Santosh 1995) or scapolite–clinopyroxene symplectites replacing garnet (Abart et al. 2001).
A striking feature of symplectites is that the characteristic spacing of phase alternation may vary by three orders of magnitude. Intergrowths with characteristic spacing from 400 nm for orthopyroxene–anorthite–spinel symplectites (Degi et al. 2009) up to 300 μm for scapolite–clinopyroxene intergrowths replacing garnet (Abart et al. 2001) have been reported. It is generally noted that for a given type of symplectite the characteristic spacing of the symplectite phases increases with increasing temperature (Abart et al. 2014), but the factors controlling the characteristic spacing have remained elusive.
Replacement of a homogeneous precursor by a vermicular or lamellar intergrowth of product phases has long been recognised in materials—(Vaks 2004) and polymer science (Oswald et al. 1987). The underlying process is referred to as cellular segregation reaction (Cahn 1959; Christian 2002). Two types of cellular segregation reactions are discerned (Cahn 1959; Gaidies et al. 2017): (1) in cellular precipitation a solid solution, which is supersaturated with respect to some component, segregates out this component through precipitation of a second phase, which forms rods or lamellae in the matrix of the formerly supersaturated solid solution. (2) In eutectoidal decomposition, a homogeneous precursor phase decomposes into two new phases, which both differ in structure and composition from the reactant and form a vermicular or lamellar intergrowth. Irrespective of the reaction type, symplectite formation requires two processes to occur simultaneously: On the one hand, bonds must be broken at the reacting surface of the reactant phase, material must be transferred across the reaction front, and new bonds must be built at the growth front of the symplectite. The bulk of these processes is referred to as the interface reaction. On the other hand, the homogeneous reactant phase separates into two or more phases with different compositions, and chemical components must be segregated on the scale of the characteristic spacing of the symplectite phases. This chemical segregation occurs by diffusion within the reaction front. The coupling between the interface reaction and the diffusion within the reaction front determines the overall kinetics and microstructure evolution during symplectite formation (Cahn 1959).
Theoretical models for describing cellular segregation reactions have been developed in materials- and polymer science. More specifically, relations among the thermodynamic driving force, the mobility of the reaction front, the propagation velocity of the reaction front, the degree of chemical separation by diffusion, and the characteristic spacing of the separated phases have been derived (Zener 1958; Cahn 1959). More recently, phase field models have been developed that address cellular segregation reactions (Steinbach and Apel 2007). Cahn’s (1959) model accounts for the contributions of material re-distribution by diffusion within the reaction front and of interface reaction to the overall kinetics and qualitatively predicts many features of cellular segregation reactions. In particular, for the case where the mobility of the reaction front is high relative to component diffusion within the reaction front, it predicts a narrow lamellar spacing and a characteristic chemical zoning across individual symplectite phases. By contrast, symplectite phases are chemically homogeneous with equilibrium compositions, and the lamellar spacing is wide, when diffusion within the reaction front is fast relative to the mobility of the front. Hence, the width of a symplectite microstructure, the characteristic spacing between the symplectite phases and chemical zoning of the product phases are intimately connected and bear important kinetic and rate information, which may potentially be useful as a geo-speedometer.
Efforts to adapt kinetic models from materials science for describing symplectites are rare. Ashworth and Chambers (2000) and Degi et al. (2009) applied irreversible thermodynamics to show that for a given symplectite forming reaction and for a given set of P–T–X conditions the rate of reaction front propagation is maximised for a specific spacing of phase alternation in the symplectite, which is selected automatically by the reacting system. Abart et al. (2014) adopted the model of Cahn (1959) for application to myrmekites.
Although instructive and perfectly valid in a qualitative sense, the applicability of these analyses to mineral systems is limited due to the lack of experimental calibration. To the best of our knowledge, no experiments focussing on the kinetics of symplectite formation in silicate systems have been reported in the literature. In this contribution, symplectites produced experimentally at high pressure and temperature in the CaO–MgO–\({\text{SiO}}_2\) model system are presented. The symplectites formed from the breakdown of monticellite with composition \({\text{Ca}}_{0.88}{\text{Mg}}_{1.12}{\text{SiO}}_4\) (Mtc I) along two different pathways. One route involved replacement of Mtc I by an intergrowth of rod-shaped forsterite in a matrix of monticellite (\({\text{CaMgSiO}}_4\), Mtc II), henceforth referred to as symplectite I (Sy I). The second pathway involved replacement of Mtc I by a symplectite comprising forsterite + merwinite, henceforth referred to as symplectite II (Sy II). Both the width of the symplectite replacement microstructures and the characteristic spacing of the symplectite phases were measured for different run durations, temperatures and water contents. In addition, the width, morphology and atomic structure of the reaction front as well as topotaxial orientation relationships between the symplectite phases were examined. Finally, the model of Degi et al. (2009) was extended to account for finite intrinsic interface mobility of the reaction front and applied to extract relations among the relevant thermodynamic and kinetic parameters from our experimental results.
Experimental methods
Experimental procedure
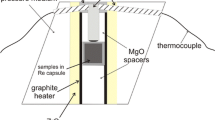
Partial replacement of monticellite by symplectite was produced using large single crystals of synthetic monticellite provided by the Institut für Kristallzüchtung (IKZ, Berlin). The crystals have a composition of \({\text{Ca}}_{0.88}{\text{Mg}}_{1.12}{\text{SiO}}_4\) and are homogenous (Table 1). The crystals were drilled to obtain cylinders of 1.3–2.0 mm length and about 1.5 mm in diameter, which were then dried at \(800\,^{\circ }\hbox {C}\) for 1 h. The cylinders were subsequently cleaned with acetone and water and then placed into platinum capsules of 2 mm diameter, 3 mm length and 0.2 mm wall thickness. In some experiments, tiny flakes of synthetic brucite (\({\text{Mg(OH)}}_2\)) were added to provide different amounts of water making use of the fact that brucite decomposes to periclase (MgO) + water at the applied P and T conditions. The amount of added brucite corresponds to water contents ranging from 0.1 to 0.5 wt% \({\text{H}}_2\text{O}\) of the total charge (Table 1). The platinum capsules were then sealed shut using a plasma welder and placed into a pressure cell consisting of crushable alumina (\({\text{Al}}_2{\text{O}}_3\)). The cell assembly consisted of an outer sleeve of talc and an additional pyrex tube with a graphite furnace. The experiments were carried out in a piston-cylinder apparatus. The temperature was controlled using a Jumo dTRON 16.1 controller with type S thermocouples (Pt/PtRh10). Considering the temperature gradient within the capsule, the uncertainty of the temperature is estimated at \(\pm \,4\,^{\circ }\hbox {C}\) (Massonne and Schreyer 1989). The pressure calibration was done based on a simple force per area calculation related to the piston area at 1 bar, corrected by \(-\,0.4\%\) for lateral piston expansion under pressure. The nominal pressure was then corrected by \(-\,15\%\) to account for frictional loss based on our own laboratory calibration using the quartz–coesite transition. The accuracy of the pressure measurement was estimated to be about ± 0.05 GPa (Frei et al. 2009). The samples were first brought to the final pressure and then heated to the respective temperature along a temperature ramp of about 30 K/min. Run durations were between 10 min and 24 h. The device is water cooled so that the samples could be quenched to below \(200\,^{\circ }\hbox {C}\) within 1 min by switching off the power supply. After quenching, the pressure cells were opened and the Pt-capsules were cleaned from any cell material in an ultrasonic bath. The capsules with content were cut into several sections, ground, and polished using SiC-wafers. Finally, they were embedded in epoxy and polished with \(0.25\,\upmu \hbox {m}\) colloidal silica for several hours.
Analytical techniques
Electron probe microanalysis (EPMA)
The starting material (Mtc I) before and after the run as well as the symplectite phases was analysed using a JEOL JXA-8500F electron microprobe equipped with a field emission gun (FEG). The chemical compositions were obtained by wavelength dispersive analysis. For Mtc I, 15 kV, 20 nA and a spot size of \(5\,\upmu \hbox {m}\) were applied. For the symplectite phases, 15 kV, 15 nA, and a focused beam corresponding to a beam diameter of about 50 nm were used. Well-characterised natural standards were used: albite for Si, anorthite for Ca, and olivine for Mg. The measured Ca-concentrations of forsterite from Sy I were unrealistically high, even with application of a focused beam. This is very likely due to an artefact caused by signal from the monticellite matrix (Mtc II), which was collected unintentionally due to the small size of the forsterite rods with thicknesses of a few hundred nanometers. Analytical TEM using EELS confirmed that forsterite is virtually Ca-free. In addition, high-resolution element distribution maps were produced in WDS mode for Mg, Ca and Si using a focused electron beam at 8 kV and 15 nA with dwell times of 2 ms. Element maps were obtained using beam scan mode with pixel sizes of about \(30 \times 30\, \hbox {nm}^2\).
BSE images were obtained using the same instrument applying a beam current of 10 nA and acceleration voltages of 8–15 kV. Specific section cuts revealed the microstructural properties of the replacement symplectites, including the width of the entire symplectite replacement structure, the characteristic spacing of the symplectite phases, the structure and morphology of the reaction front, and the geometric relations around triple junctions at the front. Reliable and consistent values of the width of the symplectite replacement structures were only obtained for Sy II. In contrast, the width of Sy I is highly variable and no systematics could be detected (see below). For each experiment, the characteristic spacing of the symplectite phases was measured from 20–30 different areas closely behind the reaction front for both symplectite types. Their averaged values along with errors in terms of 1\(\sigma\) are given in Table 1.
Focused ion beam and scanning electron microscopy (FIB–SEM)
The 3D geometries of some symplectite microstructures were reconstructed from BSE images taken on serial sections with a spacing of about 50 nm using an FEI Quanta 3D FEG Dual Beam instrument at the GFZ Potsdam. Serial sectioning was performed by progressive ion milling using a focused ion beam (FIB) combined with electron imaging with a scanning electron microscope (SEM). Before ion milling, a thin Pt layer was deposited on the area of interest to avoid charging. A \(\hbox {Ga}^+\)-ion beam operated at 30 kV and 7 nA was used to excavate a block of \(20 \times 20 \times 8 \upmu \,\hbox {m}\). The sample was subsequently tilted to \(52^{\circ }\) towards the ion beam, so that ion milling occurs perpendicular to the sample surface. After additional cleaning steps, using ion beam currents of 0.5–1 nA to remove amorphous remnants of the milling process, the system automatically removes 50 nm of the material and records a BSE image of the exposed surface after each milling step using the “auto slice and view” routine provided in the software package by FEI. The successive BSE images were taken using an acceleration voltage of 10 kV and a beam current of 32 pA to avoid charging of the uncoated surfaces. The BSE images were processed using background correction and filtering tools to enhance image quality and contrast, and the different phases we extracted by applying suitable threshold values. The obtained stack of 200–300 BSE images was then converted into a three-dimensional visualisation using the software Avizo Fire 7 from VSG.
Transmission electron microscopy (TEM)
About \(15 \times 10 \times 0.15 \upmu \,\hbox {m}\) sized electron-transparent, site- and orientation-specific foils sampling the replacement front as well as the symplectic intergrowth were extracted from polished thin sections using the FIB device at GFZ Potsdam (FEI FIB 200 TEM). For a detailed description of FIB sample preparation, see Wirth (2004). TEM was carried out on an FEI Tecnai G2 F20 X-Twin transmission electron microscope equipped with a Gatan imaging filter (GIF Tridiem) for acquisition of energy-filtered images, an EDAY X-ray analyser with an ultrathin window, and a Fischione high-angle annular dark field detector (HAADF). The system operates at 200 kV with an FEG electron source. The microstructures were characterised using bright-field and dark-field images acquired as energy-filtered images with a 10 eV window to the zero-loss peak. In addition, energy dispersive X-ray analysis (EDX) in scanning transmission mode (STEM) was applied. The crystallographic orientations between the reactant and product phases were investigated by combining high-resolution imaging with electron diffraction. Electron energy-loss spectroscopy (EELS) analysis was used to check for compositional gradients at high resolution within the symplectite phases and across the phase boundaries.
Results
Symplectite forming reactions
A summary of 21 experimental runs is presented in Table 2. Experimental conditions varied from 1000 to \(1200\,^{\circ }\hbox {C}\), from 1.0 to 1.4 GPa, and from nominally water-free conditions to water contents of 0.5 wt% \({\text{H}}_2{\text{O}}\) of the total charge (solids + water). Run durations of 10 min to 24 h were applied (Table 2).
In all experiments, the homogeneous synthetic monticellite (Mtc I, \({\text{Ca}}_{0.88}{\text{Mg}}_{1.12}{\text{SiO}}_4\)) reacted to form fine-grained aggregates with symplectic microstructure. Representative BSE images are shown in Fig. 2a–f. All stages from incipient reaction up to complete conversion were sampled. The reaction always started at crystal surfaces and along cracks and proceeded into the reactant monticellite by the propagation of a sharp reaction front. Two types of symplectites were observed: on the one hand, Mtc I was replaced by a symplectite consisting of forsterite rods (Fo) within a monticellite (Mtc II) matrix (Sy I). Both the forsterite and the monticellite of Sy I have end-member compositions, \({\text{Mg}}_2{\text{SiO}}_4\) and \({\text{CaMgSiO}}_4\), respectively. The underlying reaction may be written as:
Referring to the nomenclature of Cahn (1959), this is classified as a cellular precipitation reaction. On the other hand, Mtc I was replaced by a regular, lamellar intergrowth of merwinite (Mw) and forsterite (Sy II). Again, the merwinite and the forsterite in the symplectite have end-member compositions, \({\text{Ca}}_3{\text{MgSi}}_2\text{O}_8\) and \({\text{Mg}}_2{\text{SiO}}_4\), respectively. The pertinent reaction
is classified as a eutectoidal decomposition reaction according to Cahn (1959). In several places, Sy I was replaced by a merwinite–forsterite intergrowth, which, in terms of phase assemblage, is similar to Sy II. This indicates that Sy I is metastable with respect to the merwinite–forsterite assemblage. In contrast to the regular lamellar intergrowth of Sy II, a chaotic microstructure lacking shape preferred orientation is formed, when the merwinite–forsterite assemblage replaces Sy I (see Fig. 2a).

Back-scattered electron images of various symplectite microstructures: Mtc I \({\text{Mtc}}_{88}{\text{Fo}}_{12}\), reactant monticellite, Mtc \({\text{Mtc}}_{100}\), monticellite of Sy I, Fo forsterite, Mw merwinite, Sy I Fo - Mtc intergrowth, Sy II Fo - Mw intergrowth; a Run No. PR Sy26 (\(1000\,^{\circ }\hbox {C}\), 0.1 wt% \({\text{H}}_2\text{O}\), 0.17 h). Sy I and Sy II microstructures growing from a crack into Mtc I on both sides. Towards the left Sy II (Mw: light grey and Fo: dark grey) lamallae with regular spacing and oriented approximately perpendicular to the reaction front directly replace Mtc I, the arrows indicate the width of the replacement structure. Towards the right an irregular Mw (light grey)–Fo (dark grey) intergrowth replaces previously formed Sy I (Fo: dark grey, Mtc: light grey). The reaction front, where Sy I replaces Mtc I is rather uneven (upper right). b Run No. PR Sy10 (\(1100^{\circ}\hbox {C}\), 0.5 wt% \({\text{H}}_2\text{O}\), 5h). Sy I (Fo: dark grey, Mtc: light grey) replacing Mtc I. The lamellar spacing at the reaction front is quite regular. Sy I is consumed by an irregular Mw–Fo intergrowth from behind. c Blow-up of b Sharp reaction front between Sy I and Mtc I. The characteristic spacing of Fo and Mtc lamellae is about 1250 nm. d Run No. PR Sy7 (\(1000\,^{\circ }\hbox {C}\), nominally dry, 24h). Sy II microstructures growing from a crack towards both sides with the Mw (light grey) + Fo (dark grey) lamallae oriented approximately perpendicular to the reaction front. The arrows indicate the width of the replacement structure. e Run No. PR Sy10 (\(1100\,^{\circ }\hbox {C}\), 0.5 wt% \({\text{H}}_2\text{O}\), 5h); Sy II with lamellae of Fo (dark grey) and Mw (light grey) lamellae in about equal modal proportions directly replaces Mtc I. Directly at the reaction front the lamellae are oriented approximately perpendicular to the reaction front, whereas behind the front the microstructure is convoluted, indicating successive re-crystallisation. f Blow-up of 3d. About equally-spaced Fo + Mw lamellae at the reaction front. The characteristic spacing is 250 nm on average. The reaction front shows some topography in that Mw protrudes deeper into the Mtc I by a few nm than Fo
In nominally dry experiments, only Sy II is produced (Table 2, Fig. 2d). When water is present, the formation of the metastable assemblage of Mtc II + Fo of Sy I is observed (Fig. 2a–c). In this case, the reaction front propagating into the reactant monticellite has highly irregular shape. Sy II with regularly spaced Mw + Fo lamellae may, however, also occur in water-bearing runs (Fig. 2a, d, f), when it directly replaces the reactant monticellite without forming the transient intermediate Sy I. It can thus be clearly seen from the microstructure of the merwinite–forsterite intergrowth, whether it formed directly from Mtc I via a one-step process, or whether it formed via a two-step process that involves the formation of Sy I as a transient intermediate stage.
Symplectite microstructures
Symplectite formation starts at the crystal surfaces or along cracks in the reactant monticellite and proceeds via a sharp reaction front. The reaction fronts and the symplectite microstructures are devoid of porosity on the micrometer- down to the tens of nanometer-scale, which can be resolved by scanning electron microscopy. Whereas the front of Sy I has a rather irregular shape, it is comparatively smooth for Sy II. For small degrees of conversion, the replacement structures formed by Sy II have fairly uniform thicknesses. Experiments at high water contents of 0.5 wt% and with long run durations result in thick, irregularly shaped replacement structures showing an internal “garben microstructure” with highly variable grain size and lacking shape-preferred orientation. The garben microstructure is affected by repeated stages of recrystallisation and discontinuous coarsening (Fig. 2e).
Three-dimensional reconstructions of both the Sy I and the Sy II microstructures from the same sample (PR Sy10), both directly replacing the reactant monticellite are displayed in Fig. 3. The symplectite microstructures were sampled close to, but not including the reaction front. Sy I consists of only weakly interconnected, about 200 nm wide forsterite rods, which are aligned in a subparallel manner and embedded in a matrix of Mtc II. Sy II consists of a lamellar intergrowth of forsterite and merwinite with approximately equal modal proportions. The lamellae are highly interconnected.

Three-dimensional reconstructions of the symplectite microstructures as obtained from BSE imaging of successive FIB cuts. Run No. PR Sy10 (\(1100\,^{\circ }\hbox {C}\), 0.5 wt% \({\text{H}}_2\text{O}\), 5h). Sections represent parts of the microstructures taken close to but not including the reaction front. The lateral resolution of the images is about 50–100 nm, the step size for the material sputtered from the surface before the acquisition of each image was 50 nm. a Sy I, consisting of isolated Fo rods (green) in a matrix of \({\text{Mtc}}_{100}\) (not shown); b Sy II, lamellar intergrowth of forsterite (green) and merwinite (not shown)
Phase compositions
EPMA analyses of the reactant monticellite \({\text{Mtc}}_{88}{\text{Fo}}_{12}\) (Mtc I) and of the symplectite phases are given in Table 1. A large number of point analyses from various Sy I and Sy II samples indicate that the precursor is chemically homogeneous. This also holds close to the reaction front. Mtc II of Sy I is chemically homogeneous and with \({\text{Ca}}_{1.01}{\text{Mg}}_{0.99}{\text{SiO}}_4\) (Table 1) has close to end-member composition. An Mg-distribution map for Sy I (PR Sy10; \(1100\,^{\circ }\hbox {C}\), 0.5 % \({\text{H}}_2\text{O}\), 5h) is shown in Fig. 4. It is interesting to note that despite continuous miscibility between Mtc I and Mtc II and their structural similarity, the boundary between the two is sharp on the scale of the element distribution map (Fig. 4) and on the BSE images (Fig. 2c).

Mg-distribution map of a Sy I domain (PR Sy10; \(1100\,^{\circ }\hbox {C}\), 0.5 wt% \({\text{H}}_2\text{O}\), 5h) close to the reaction front displaying forsterite rods (red: high Mg-concentrations) in a homogeneous matrix of \({\text{Mtc}}_{100}\). The reaction front to the \({\text{Mtc}}_{88}{\text{Fo}}_{12}\) precursor (right) is sharp within the spatial resolution of the FEG-EPMA analysis. The width of the forsterite rods is about 200 nm
Similarly, in all Sy II samples merwinite is homogeneous and, within analytical error, has end-member composition (\({\text{Ca}}_{3.01}{\text{Mg}}_{0.99}{\text{Si}}_2\text{O}_8\); Table 1). Again, there are neither compositional variations detectable close to the reaction front, nor close to the interface with the forsterite lamellae in the Mw–Fo intergrowth. Forsterite rods of Sy I, and Fo lamellae of Sy II are too small for reliable analysis by EPMA (Table 1). In both cases, ATEM combined with EELS confirms that Fo also has close to end-member composition.
Symplectite nanostructures
TEM micrographs (sample PR Sy10) taken in various modes are displayed in Fig. 5a–f, revealing the textural relationships between the precursor Mtc I and the symplectite phases Mtc II + Fo of Sy I and Mw + Fo of Sy II.

TEM images displaying the textural relations between the reactant Mtc I and the symplectite phases Mtc + Fo of Sy I, and Mw + Fo of Sy II, respectively. All images stem from run No. PR Sy 10, \(1100\,^{\circ }\hbox {C}\), 0.5 wt% \({\text{H}}_2\text{O}\), 5h; see Fig. 2e). a Bright-field image of TEM foil no. 3699 cut perpendicular to the reaction front displaying replacement by a lamellar Mw–Fo intergrowth. Note the triple junctions between Mw, Fo and Mtc I. Ahead of the reaction front, the reactant Mtc I is free of strain contrasts, and also within the symplectite strain contrast is absent. b Lattice fringe image showing the boundary (arrows) between Mtc I and a single Fo lamella of Sy II. No specific crystallographic orientation relationship exists between the two. c Detail of the reaction front between Fo and Mtc I. The lattice planes of the Fo directly fit that of the reactant Mtc I. There is no amorphous phase or formerly fluid-filled space in between. d Dark-field image of the TEM foil no. 3696 cut perpendicular to the reaction front (not visible). The image shows the Sy I intergrowth of one Fo rod surrounded by \(\hbox {Mtc}_{100}\) (see Fig. 2c). There are regular misfit dislocation arrays along the two interfaces (arrows) indicating crystallographic orientation relationships. In addition, strain contrasts are present in Mtc. e Lattice fringe image of the interface between Sy I phases, Fo and Mtc. The (010) planes of both phases coincide. f FFT-diffraction pattern from e. (010) of Fo and Mtc show identical orientation
Where Mtc I is directly replaced by Mw + Fo (Sy II) the reaction front changes its orientation in a discontinuous fashion at the triple junctions among the three phases, whereby Mw bulges into Mtc I (convex towards Mtc I) and Fo shows a concave interface towards Mtc I (Fig. 5a). There is no indication of strain contrasts in the reactant monticellite directly ahead of the reaction front, nor at the interfaces between the symplectite phases. The structural width of the Sy II reaction front between the reactant Mtc I and the Fo in the symplectite is smaller than the largest visible lattice spacing of about 0.83 nm, and there is no detectable distortion of the crystal lattice of the precursor or the replacement phases (Fig. 5b, c). There is no specific crystal orientation relationship between the reactant Mtc I and the newly formed forsterite in Sy II. Most importantly, there is no amorphous phase or open space detectable at the reaction front. Moreover, all phases have uniform compositions. Sy I shares many of the nanometre-scale features with Sy II. The reaction front is equally narrow and all phases have uniform compositions. By contrast, the symplectite phases Mtc II and Fo show a specific crystal orientation relationship with \(\text (010)_{{\text{Fo}}} \parallel (010)_{Mtc}\) (Fig. 5d–f). There are arrays of regularly spaced misfit dislocation along their interfaces, and strain contrasts are observed in the Mtc II matrix (Fig. 5d). The Mtc II–Fo interfaces may be classified as semi-coherent, where the regularly spaced dislocations compensate the misfit between the Mtc II and Fo lattices (Fig. 5d). In essence, Sy I is composed of a few large single crystals of Mtc II with the forsterite rods embedded with similar crystallographic orientation. There are no precipitates of forsterite in the interior of the reactant Mtc I. Instead, the reaction starts at the surfaces or along cracks and proceeds via a sharp, irregularly shaped reaction front.
Influence of water content, temperature, and run duration on symplectite microstructures
Figure 6 displays the dependence of the characteristic spacing \(\lambda\) of the phase alternation in Sy I and Sy II on bulk water content for 11 experiments performed at \(1000\,^{\circ }\hbox {C}\) and 1.2 GPa. The characteristic spacing is independent of run duration. In Sy I, it varies from 332 nm at 0.1 wt% to 429 nm at 0.5 wt% bulk water content. In Sy II, it ranges from 223 to 290 nm within the range from nominally dry to water contents of 0.2 wt% \({\text{H}}_2\text{O}\) of the total charge. Given the measurement error, the dependence of the characteristic spacing on the water content is weak for Sy I, and it is insignificant for Sy II.

Characteristic spacing \(\lambda\) (nm) of phase alternation in Sy I (open) and Sy II (black circles) for 13 experiments plotted versus \({\text{H}}_2\text{O}\) content at \(1000^{\circ}\hbox {C}\) and 1.2 GPa except for two experiments at 1.1 and 1.4 GPa (all values from Table 2). There is no Sy II with regular lamellar microstructure formed at 0.5 wt% \({\text{H}}_2\text{O}\) (PR Sy 34)
By contrast, the characteristic spacing is strongly temperature dependent for both symplectite types. It varies from about 400–1200 nm in the temperature range from \(1000\,^{\circ }\hbox {C}\) to \(1100^o\hbox {C}\) for Sy I, and from 250 nm to 700 nm between 1000 and \(1200\,^{\circ }\) for Sy II. The data are shown in Fig. 7 and include water contents of 0.1–0.5 wt% water for Sy I, and between nominally dry and 0.2 wt% water for Sy II (see Table 2).

Characteristic spacing \(\lambda\) (nm) of Sy I (open) and Sy II (black circles) lamellae plotted versus T ranging from 1000 to \(1200\,^{\circ }\hbox {C}\). All data are from Table 2. The pressure was 1.2 GPa except for Sy II from PR Sy 40, where P = 1.0 GPa. The data include water contents between nominally dry and 0.2 wt% \({\text{H}}_2\text{O}\) for Sy II lamellae; and between 0.1 and 0.5 wt% \({\text{H}}_2\text{O}\) for Sy I lamellae. There are no data for Sy I at \(1200\,^{\circ }\hbox {C}\) (Pr Sy35) because the reactant Mtc I is entirely consumed. For Sy I lamellae \(\lambda\) increases about linearly and steeply from 400 nm at \(1000\,^{\circ }\hbox {C}\) to about 1200 nm at \(1100\,^{\circ }\hbox {C}\); for Sy II lamellae there is a linear increase from 250 nm at \(1000\,^{\circ }\hbox {C}\) to about 700 nm at \(1200\,^{\circ }\hbox {C}\)
The rate of reaction front propagation was determined from time-series experiments. Due to the irregular shape of the reaction front, highly variable widths of the symplectite microstructures were observed for Sy I. When present, the Sy I domains are always much broader than the Sy II domains indicating that Sy I reaction fronts propagate at substantially higher rates than Sy II reaction fronts.
In contrast, Sy II replacement microstructures that formed directly from Mtc I show relatively constant widths (Table 2; Fig. 8). At \(1000\,^{\circ }\hbox {C}\), 0.1 wt% \({\text{H}}_2\text{O}\), the rim width increases from 4 to 17 to \(32\,\upmu \hbox {m}\) with increasing run durations of 10, 20, and 40 min (see Fig. 8). Due to considerable scatter of the data, it is not possible to decide whether the reaction front propagated at a constant rate (linear rate law) or followed a \(\sqrt{t}\) dependence. Irrespective of the growth behaviour, extrapolations of a linear or a parabolic rate law to zero thickness of the symplectite replacement structure yield \(t>0\) indicating retarded growth probably due to sluggish nucleation. An experiment performed at \(1200\,^{\circ }\hbox {C}\) and 0.1 wt% \({\text{H}}_2\text{O}\), 20 min produced a rim width of \(21\,\upmu \hbox {m}\), only \(4\,\upmu \hbox {m}\) wider than that at \(1000\,^{\circ }\hbox {C}\) at otherwise identical conditions, indicating that a temperature increase only moderately enhances the propagation rate of the replacement front. This is supported by two experiments performed at 1100 and \(1200\,^{\circ }\hbox {C}\) at nominally dry conditions for a run duration of 30 min (Fig. 8), where the rim widths are 31 and \(36\,\upmu \hbox {m}\), respectively. This is about consistent with the wet experiment at \(1200\,^{\circ }\hbox {C}\) (20 min), and also with the wet experiments at \(1000\,^{\circ }\hbox {C}\) (10, 20, and 40 min) and suggests that there is no significant difference in the propagation rate of Sy II microstructures between nominally dry experiments and experiments with 0.1 wt% \({\text{H}}_2\text{O}\) added. Another experiment at \(1000\,^{\circ }\hbox {C}\) at nominally dry conditions (PR Sy 7; not shown in Fig. 8, see also Fig. 2) resulted in a rim width of only \(2.5\,\upmu \hbox {m}\) after 24 h. This is interpreted to be due to nucleation delay at low temperature.

Widths of Sy II microstructures emanating from the surface or fractures inside the precursor for experiments at short run durations versus run duration. Black squares: \(1000\,^{\circ }\hbox {C}\); 1.2 GPa; 0.1 wt% \({\text{H}}_2\text{O}\); and 10, 20, and 40 min duration. Open square: \(1200\,^{\circ }\hbox {C}\); 1.2 GPa; 0.1 wt% \({\text{H}}_2\text{O}\), 20 min. Half filled circle: \(1100\,^{\circ }\hbox {C}\); 1.2 GPa, dry, 30 min. Open circle: \(1200\,^{\circ }\hbox {C}\), 1.2 GPa, dry, 30 min. The time series of the three experiments at \(1000\,^{\circ }\hbox {C}\) at identical conditions indicate a linear increase of the widths with time. All values are from Table 2
Discussion
Our experimental results show that at low water contents of up to about 0.2 wt% \({\text{H}}_2\text{O}\) of the total charge, Mtc I (\({\text{Mtc}}_{88}{\text{Fo}}_{12}\)) that is subjected to P-T conditions outside its stability field breaks down to a fine-grained symplectic intergrowth via two pathways. Mtc I may either be directly replaced by the stable assemblage Mw + Fo (Sy II), or, alternatively, the reaction may proceed via formation of the transient intermediate assemblage Mtc II + Fo (Sy I), before the stable merwinite + forsterite assemblage takes over. Both reaction types proceed via the propagation of sharp reaction fronts and both the reactant and the product phases exhibit uniform compositions. The characteristic spacing of phase alternation in the symplectite microstructures is strongly temperature dependent, but rather insensitive to the water content. At water contents of 0.5 wt% \({\text{H}}_2\text{O}\) of the total charge the overall reaction rate is drastically increased and Mtc I is replaced by a garben microstructure composed of the stable assemblage merwinite + forsterite.
In natural symplectites, the characteristic spacing of phase alternation may vary from the nanometer to the micrometer scale, and the question arises which parameters control the characteristic spacing of symplectite phases. In the following, we adopt the thermodynamic model for sympelctite formation that was used earlier by Ashworth and Chambers (2000), Degi et al. (2009), and Abart et al. (2012) to analyse the microstructures obtained from our synthesis experiments. Thereby, only the regular intergrowth of Sy II, which formed directly from Mtc I, is considered. In principle, the Sy I microstructure with its regular rod-matrix geometry would also be amenable to a thermodynamic analysis. Due to the highly variable thickness of the Sy I microstructures produced in our experiments, this microstructure is, however, unsuitable for extracting quantitative relations and is not further addressed. The microstructure of the Mw–Fo assemblage replacing Sy I is bare of any geometrical regularities and cannot be analysed with the thermodynamic model that follows. It was assumed in the previous studies that material re-distribution by diffusion within the reaction front is the only rate-limiting process during symplectite formation. We allow for the interface reaction proceeding at the reaction front as an additional potentially rate-limiting process. The thermodynamic extremal principle (Onsager 1931; Ziegler 1961; Svoboda and Turek 1991; Svoboda et al. 2005; Fischer et al. 2014) is employed to derive the corresponding evolution equation. Finally, our experimental results are discussed in the light of the extended thermodynamic model.
Thermodynamic model for symplectite formation by a eutectoidal reaction
Consider a binary system with the components A and B and with phases \(\alpha\), \(\beta\), \(\gamma\) (see Fig. 9a), where \(\gamma\) is metastable with respect to the assemblage \(\alpha + \beta\), so that there is a thermodynamic force driving the reaction
Let the product assemblage \(\alpha + \beta\) take the form of a symplectite consisting of a lamellar intergrowth, which replaces the reactant phase \(\gamma\) at a sharp reaction front such as shown in Fig. 9b. Symplectite formation requires material re-distribution by diffusion within the reaction front and interface reactions at the reaction front to proceed in parallel. Both diffusion and interface reactions are dissipative processes and my potentially be rate limiting. They are both accounted for in the following thermodynamic model.
Let the g-X curves of all phases be sharply convex downward (see Fig. 9a), so that their compositions are essentially fixed with \(X_{\alpha }< X_{\gamma } < X_{\beta }\). The stoichiometric reaction equation thus reads
where \(n_{\alpha }\), \(n_{\beta }\), \(n_{\gamma }\), are the composition vectors, and \(\nu _{\alpha }\), \(\nu _{\beta }\), are the stoichiometric coefficients, and \(\nu _{\gamma }\) was set to unity. Let the reaction front have a thickness \(\delta\) and let it move with velocity u relative to a material point in phase \(\gamma\). The lamellae of the symplectitic intergrowth are oriented perpendicular to the reaction front and alternate at a regular spacing \(\lambda\) (see Fig. 9b). The modal proportions of \(\alpha\) and \(\beta\) in the symplectite are
where \(\Omega _{\alpha }\) and \(\Omega _{\beta }\) are the molar volumes of \(\alpha\) and \(\beta\), respectively. Let us assume that any volume change associated with reaction (3) is accommodated by transformation strain in the direction perpendicular to the reaction front. Then, the interface velocity v with respect to a material point in the symplectite is related to the interface velocity u with respect to a material point in phase \(\gamma\) through
The free energy change \(\Delta G\) associated with reaction reads
where \(\Delta _r\bar{g} = \frac{\Delta _rg}{\Omega _{\gamma }}\) is the free energy change of reaction per unit volume of the reactant phase \(\gamma\), and \(\sigma\) is the interfacial energy per unit area of \(\alpha\)-\(\beta\) interface, and \(\Delta G\) refers to a unit volume of the reactant phase \(\gamma\).

a schematic molar Gibbs energy diagram for the binary system A–B with phases \(\alpha\), \(\beta\), \(\gamma\) with sharp downwards convex g-X surfaces so that the compositions of the pases are essentially fixed; b geometry of transforming system; c component fluxes into and out of the reaction interface associated with interface motion and diffusive flux along the reaction interface in the direction perpendicular to the lamellae in the \(\alpha\)–\(\beta\) aggregate (y-direction), \(m_{\alpha }\) is the modal proportion of phase \(\alpha\) in the symplectite
Material re-distribution within the reaction front We assume that growth has reached a steady state, and the component concentrations within and their variation along the reaction front are considered stationary. Any composition difference between incoming and outgoing material must thus be compensated for by corresponding diffusive fluxes within the reaction front in the direction perpendicular to the \(\alpha\)– \(\beta\) lamellae, which corresponds to the y-direction in Fig. 9b, c. Conservation of mass requires
where \(J_i\) is the diffusive flux of component i in y-direction within the reaction front, and \(S_i^{\alpha }\) and \(S_i^{\beta }\) may be regarded as source/sink terms for component i with
Integration of Eq. (6) over the lamella width yields
where \(0 \le y \le \frac{\lambda }{2} m_{\alpha }\) corresponds to the section of the reaction front, where \(\gamma\) is in contact with \(\alpha\), and \(\frac{\lambda }{2} m_{\alpha } \le y \le \frac{\lambda }{2}\) corresponds to the section of the reaction front, where \(\gamma\) is in contact with \(\beta\).
Dissipation We assume linear kinetics so that the constitutive law for diffusion reads
where \(F_i = -\nabla \mu _i\) is the thermodynamic driving force for diffusion with \(\mu _i\) being the chemical potential of component i, and \(L_i\) is the phenomenological coefficient of diffusion of component i, which is related to the self diffusion coefficient of component i. For a thermodynamically ideal system, the relation is (Lasaga 1998; Petrishcheva and Abart 2017)
where \(c_i\) is the molar concentration, \(D_i\) is the self diffusion coefficient, R is the gas constant, and T is the absolute temperature. Noting that the dissipation per unit volume due to diffusion of component i is \(q_i = J_i F_i\) (DeGroot and Mazur 1984) the dissipation due to diffusion during interface motion reads
where the integral is taken over the volume in which diffusion takes place. In the case at hand, this is the volume of the reaction interface that is given by the interface thickness \(\delta\) in x-direction, half the lamella spacing in y-direction and unit length in z-direction. Inserting for \(J_i\) from Eq. (7) and multiplying by \(\frac{2}{\lambda }\) so that \(Q_{\text {diff}}\) refers to unit area of the reaction front yields
It will be convenient to rewrite Eq. (9) as:
where
and \(\frac{M_{\text {diff}}}{\lambda ^2}\) may be interpreted as an interface mobility related to the necessary diffusion within the reaction front (Abart et al. 2012).
In addition to the material re-distribution within the reaction front, propagation of the symplectite reaction front also requires that an interface reaction proceeds. The breaking of bonds in the reactant phase \(\gamma\), the transfer of matter across the propagating reaction front and the making of new bonds in the symplectite phases are all dissipative processes. As a consequence, the mobility of the reaction front is finite, an effect that we refer to as the intrinsic interface mobility. Again assuming linear kinetics the constitutive law for interface motion may be written as:
where \(M_{\text {if}}\) is the intrinsic interface mobility, and \(F_{\text {if}}\) is the thermodynamic driving force for interface motion with \(F_{\text {if}} = \sum _i \Delta \mu _i\), in which \(\Delta \mu _i\) is the jump in chemical potential of component i across the interface. Noting that the dissipation due to motion of an interface with finite intrinsic interface mobility is \(q_{\text {if}} = u F_{\text {if}}\) (DeGroot and Mazur 1984), the dissipation due to the motion of an interface with finite intrinsic interface mobility \(M_{\text {if}}\) and with velocity u reads
where \(Q_{\text {if}}\) refers to unit area of the moving interface. Combining the effects from the re-distribution of components by diffusion within the reaction front and its finite intrinsic mobility, the total dissipation due to interface motion is
Gibbs energy The rate of Gibbs energy change associated with the transformation of the homogeneous precursor phase \(\gamma\) into a symplectic lamellar intergrowth of \(\alpha\) and \(\beta\) is given by
where \(\Delta G\) is given by Eqs. (5) and (13) refers to a unit area of the reaction front.
Evolution equation The evolution of the system is described in terms of the single kinetic parameter u, and the Thermodynamic Extremal Principle reduces to the constraint \(Q_{\text {tot}} = -\dot{G}\), which yields
where \(M_{\text {tot}} =1/\left( \frac{\lambda ^2}{M_{\text {diff}}} + \frac{1}{M_{\text {if}}} \right)\) may be regarded as the bulk interface mobility, which accounts for both the diffusion within the reaction front and the interface reaction. Note that Eq. (14) corresponds to a linear rate law indicating that for a given thermodynamic driving force and a given bulk interface mobility, a symplectite reaction front propagates at a constant velocity, which is directly proportional to the bulk interface mobility, and to the thermodynamic driving force.
The explicit relations among reaction front velocity and the relevant thermodynamic and kinetic parameters can be seen, when Eq. (14) is expanded in the form
From Eq. (15), an interesting interdependence of the reaction front velocity u and the characteristic lamellar spacing \(\lambda\) emerges. At small values of \(\lambda\), the reaction front velocity decreases with decreasing \(\lambda\) due to the reduction of the thermodynamic driving force resulting from the addition of the interface energy associated with the newly formed \(\alpha\)–\(\beta\) interfaces. When
the numerator in Eq. (15) and thus the thermodynamic driving force vanishes, and the symplectite forming reaction stops. At large values of \(\lambda\), the reaction front velocity decreases with increasing \(\lambda\) due to the fact that diffusion becomes successively more inefficient with increasing diffusion distance. For a given set of kinetic parameters (\(M_{\text {if}}\) and \(M_{\text {diff}}\)) and interfacial energy (\(\sigma\)), the velocity of the reaction front is maximised at a specific lamellar spacing \(\lambda _{\text {max}}\) (see Fig. 10a). It is supposed that \(\lambda _{\text {max}}\) is automatically selected by the system as it maximises the rate of free energy dissipation. At a given degree of reaction overstepping (\(\Delta _r\bar{g}\)) and a fixed value of \(M_{\text {diff}}\), \(\lambda _{\text {max}}\) depends on the intrinsic interface mobility \(M_{\text {if}}\) and on interface energy \(\sigma\) (see Fig. 10b). It increases with increasing \(\sigma\) and with decreasing \(M_{\text {if}}\). If the intrinsic interface mobility \(M_{\text {if}}\) is high, all the driving force is available for chemical segregation by diffusion within the reaction front, and diffusion is rate limiting. In the diffusion-controlled regime, the evolving microstructure minimizes diffusion distances, and \(\lambda _{\text {max}}\) is relatively small. If, in contrast, the intrinsic interface mobility \(M_{\text {if}}\) is low, and interface reaction is rate limiting, diffusion can be effective over larger distances, and the resulting \(\lambda _{\text {max}}\) is relatively large ensuring minimum possible formation of new interfaces in the symplectite. The characteristic lamellar spacing of a symplectite is thus controlled by the relative contributions of the interface reaction and of the diffusion within the reaction front to the dissipation of free energy during motion of the reaction front.

a Reaction front velocity u as a function of characteristic spacing \(\lambda\) for different values of \(M_{\text {if}}\), small circles indicate \(u_{\text {max}}\); b \(\lambda _{\text {max}}\) as a function of \(M_{\text {if}}\) for different values of \(\sigma\)
Application to symplectite formation in the CaO–MgO–SiO2 system
In the following, we apply the model (15) for analysing the formation of Sy II according to reaction (2). The Gibbs energies and molar volumes of the pure phases were taken from the thermodynamic data of Holland and Powell (1998), and the mixing model of Warner and Luth (1973) was used for calculating the Gibbs energy of the monticellite–forsterite solid solution (see Table 3). Model (15) gives a relation among the velocity of the symplectite reaction front, the characteristic spacing of the lamellae constituting the symplectite, the thermodynamic driving force for reaction, the excess free energy associated with merwinite–forsterite interfaces in the symplectite, the intrinsic mobility of the symplectite reaction front, and its mobility due to chemical segregation by diffusion within the reaction front. Out of these parameters, the characteristic spacing \(\lambda\) and the velocity u of the reaction front are directly accessible through experiment. The thermodynamic driving force for reaction is calculated from the thermodynamic data. The excess free energy \(\sigma\) associated with the interfaces between the symplectite phases, the intrinsic mobility \(M_{\text {if}}\) of the symplectite reaction front and its mobility due to diffusion \(M_{\text {diff}}\) is not known. Figure 11 illustrates the relations among the model parameters as expressed by Eq. (15). It shows contours for different values of \(\sigma\) (solid lines) and \(M_{\text {if}}\) (dashed lines) in a u versus \(\lambda\) coordinate frame. The left diagram corresponds to temperatures of \(1000\,^{\circ }\hbox {C}\), and the right diagram shows the relations for \(1200\,^{\circ }\hbox {C}\). Both diagrams were drawn for a pressure of 1.2 GPa and \(M_{\text {diff}} = 10^{- 19} {\text{m}}^6 {\text{J}}^{-1} s^{-1}\). The heavy open circle represents the values of u and \(\lambda\) that were determined from experiment.
The solid lines indicate that for a given interfacial energy \(\sigma\) the reaction-front velocity drops drastically with decreasing intrinsic interface mobility, while the characteristic spacing \(\lambda\) increases. The increase of \(\lambda\) with decreasing intrinsic interface mobility is rather modest at relatively high intrinsic interface mobility but it becomes more pronounced towards low intrinsic interface mobility. It is seen in Fig. 11a that the characteristic lamellar spacing and the reaction front velocity obtained in the experiment at \(1000\,^{\circ }\hbox {C}\) are well reproduced for values of \(M_{\text {if}}\) = \(10^{- 15} {\text{m}}^4 {\text{J}}^{-1} {\text{s}}^{-1}\), \(M_{\text {diff}} = 10^{- 19} \text{m}^6 \text{J}^{-1} \text{s}^{-1}\) and interfacial energies in the range of \(0.1\,\hbox {J/m}^2\) \(\le \sigma \le 0.5\,\hbox {J/m}^2\). In contrast, an unrealistically high interfacial energy and intrinsic interface mobility would be required to reproduce the \(\lambda\)- and u-values obtained from the experiment at \(1200\,^{\circ }\hbox {C}\) when \(M_{\text {diff}}\) is kept at \(10^{- 19} \text{m}^6 \text{J}^{-1} {\text{s}}^{-1}\) (see Fig. 11b). Diffusion is a thermally activated process, and it may be expected that \(M_{\text {diff}}\) is temperature dependent. Figure 12 shows isopleths for constant \(M_{\text {diff}}\) in the u versus \(\lambda\) coordinate frame, while \(\sigma\) was kept constant at \(0.1 \,\hbox {J/m}^2\). It is seen that the experimentally observed \(\lambda\)- and u-values can be reproduced, when \(M_{\text {diff}}\) is increased by a factor of about 100 relative to the value of \(M_{\text {diff}}\) that is suitable for the experiments at \(1000\,^{\circ }\hbox {C}\). Interestingly, \(M_{\text {if}}\) does not appear to show large differences between the 1000 and the \(1200\,^{\circ }\hbox {C}\) case, whereas \(M_{\text {diff}}\) does suggesting that \(M_{\text {diff}}\) is substantially more temperature dependent than \(M_{\text {if}}\).

Reaction front velocity u as a function of the characteristic lamellar spacing \(\lambda\); the solid lines refer to constant interfacial energy \(\sigma\) between the symplectite phases, and the dashed lines refer to constant intrinsic mobility \(M_{\text {if}}\) of the reaction front a relations at \(1000\,^{\circ }\hbox {C}\), 1.2 GPa, b \(1200\,^{\circ }\hbox {C}\), 1.2 GPa; both diagrams were constructed for \(M_{\text {diff}} = 10^{- 19}\text{m}^6 \text{J}^{-1}\text{s}^{-1}\); the open circles indicate the u-\(\lambda\) relations observed in experiment

Reaction front velocity u as a function of characteristic lamellar spacing \(\lambda\); the solid lines refer to constant mobility due to diffusion \(M_{\text {diff}}\), and the dashed lines refer to constant intrinsic mobility \(M_{\text {if}}\) of the reaction front; the diagram is drawn for a temperature of \(1200\,^{\circ }\hbox {C}\) and a pressure of 1.2 GPa and a constant interfacial energy \(\sigma = 0.1{\text{Jm}}^{-2}\) between the symplectite phases; the open circle indicates the u-\(\lambda\) relations observed in experiment
The systematic increase of the lamellar spacing in Sy II with increasing temperature is thus best explained by different activation energies of interface reaction and of diffusion within the reaction front. The observed increase in lamellar spacing with increasing temperature implies that the activation energy of interface reaction is lower than the activation energy of diffusion within the reaction front. Comparatively low activation energy for interface reaction compared to diffusion was reported from layer growth experiments in the \({\text{MgO}}\)–\({\text{Al}}_2{\text{O}}_3\) system (Götze et al. 2014). With regard to symplectites, this implies that symplectite formation tends to be interface-reaction controlled at high temperature and experiences a transition to diffusion-controlled growth towards lower temperatures. This inference is supported by the observation of a systematic decrease of the characteristic spacing of phases in several generations of myrmekite that grew at successively lower temperatures in a cooling batholith (Abart et al. 2014). For the last myrmekite generation, which was formed at the lowest temperature, the latter authors reported a characteristic composition zoning of the plagioclase matrix, which they ascribed to the successively more sluggish diffusion in the myrmekite replacement front with decreasing temperature.
The transition from interface-reaction controlled to diffusion-controlled propagation of a symplectite reaction front with decreasing temperature and the associated decrease in the spacing of phase alternation is probably a general feature of symplectites. For a given type of symplectite, the characteristic spacing of phase alternation can thus be used as a thermometer. The characteristic spacing of symplectite phases cannot, however, be used as a universal temperature indicator, as the transition from interface-reaction controlled to diffusion controlled symplectite formation very likely occurs at different P–T conditions for different symplectites.
The highly organised lamellar or rod-shaped microstructures of Sy I and Sy II ensure maximum reaction rate at given pressure–temperature conditions. In both cases, the microstructure emerges from the competition between minimising transport distances during chemical segregation within the reaction front, on the one hand, and minimising the area of newly formed interfaces, on the other hand. This coupling is modulated by the finite rate of interface reaction that is necessary for the reaction front to propagate. Both interface reaction and chemical segregation within the reaction front substantially contribute to the total energy dissipation, and the well-organised microstructures ensure that the two processes are coupled in such a way that the rate of energy dissipation is maximised. In essence, both Sy I and Sy II form by similar mechanisms. The main difference between the two lies in the geometry of phase alternation (rod-matrix versus lamellar geometry) and in the extent of chemical mass transfer that needs to occur at the reaction front. In Sy I, one of the product phases (Mtc II) has nearly the same composition as the reactant Mtc I, whereas both the merwinite and the forsterite of Sy II differ substantially in composition from the precursor Mtc I. At given component mobilities, this leads to systematically larger characteristic spacing of the phase alternation in Sy I as compared to Sy II.
The coupling between chemical mass transfer and interface reaction is substantially different for the less well organised “garben type” microstructure that is formed at high water contents of 5 wt% of the total charge. Formation of this microstructure requires chemical mass transfer over larger distances than the symplectic microstructure type. The “garben type” microstructure thus indicates that chemical mass transfer was substantially enhanced at high water contents so that there was no need for an evolution of a microstructure that minimizes transport distances. It appears to be a general trend that with increasing component mobility such as induced by the presence of water, less well organised microstructures form (Spruzeniece et al. 2017). The above developed thermodynamic model for symplectite formation is based on a regular lamellar geometry of the symplectite. It may well be extended to a matrix-rod geometry of the symplectite microstructure such as the one of Sy I, but it is not suitable for describing a “garben type” microstructure.
Conclusions
In piston cylinder experiments performed at temperatures ranging from 1000 to \(1200\,^{\circ }\hbox {C}\), at pressures in the range of 1.0–1.4 GPa and applying water contents of 0.0–0.5 wt% of the total charge for run durations of 10 min to 24 h homogeneous monticellite crystals with the composition \({\text{Ca}}_{0.88}{\text{Mg}}_{1.12}{\text{SiO}}_4\) reacted to form the stable assemblage merwinite + forsterite. At water contents below about 0.2 wt% of the total charge, the reaction products take the form of a symplectite comprising a lamellar merwinite–forsterite intergrowth, which is the only reaction product at nominally dry conditions. If water is added by applying small amounts of brucite (\({\text{Mg(OH)}}_2\)) in the starting assembly, a second type of symplectite comprising forsterite rods immersed in a matrix of monticellite with end-member composition (\({\text{CaMgSiO}}_4\)) may form as a transient intermediate product. On the long run, this monticellite–forsterite symplectite is replaced by the stable assemblage merwinite + forsterite. At water contents of \(\ge 0.5\) wt% of the total charge, the overall reaction rate is greatly enhanced, and a “garben type” microstructure comprising merwinite and forsterite is produced. When formed directly from the reactant monticellite, the merwinite–forsterite symplectite is characterised by a well-defined characteristic spacing of the forsterite and merwinite lamellae, which is largely independent of pressure and water content, but shows a pronounced increase with increasing temperature. Moreover, the merwinite–forsterite symplectite forms domains with relatively constant thickness replacing the reactant monticellite along its surfaces. The reaction front velocity as determined from the thickness of the symplectite domains and run duration shows only a weak increase with increasing temperature and with increasing water contents in the range of nominally dry to about 0.5 wt% of the total charge.
A thermodynamic model accounting for interface reaction and for chemical segregation by diffusion within the reaction front as the two potentially rate-limiting processes was developed based on the Thermodynamic Extremum Principle. The model delivers a relation among two thermodynamic variables, namely the thermodynamic driving force for reaction and the free energy associated with the interfaces between the newly formed symplectite phases, two kinetic parameters given by the intrinsic mobility of the reaction front and its mobility due to chemical segregation by diffusion, and two parameters that are directly amenable to observation, namely the characteristic lamellar spacing and the reaction front velocity. Application of this model to the symplectite synthesis experiments in the CaO–MgO–\({\text{SiO}}_2\) system reveals that the pronounced increase of lamellar spacing with increasing temperature is due to a relatively high activation energy for diffusion within the reaction front as compared to the activation energy for interface reaction. This inference is in line with observations on natural systems, where the characteristic spacing of symplectite phases also appears to increase with increasing temperature. Surprisingly, the water content does not appear to have a measurable effect on the characteristic lamellar spacing and on the overall conversion rate. Nevertheless, the availability of water plays a crucial role, because symplectite formation only occurs in a relatively narrow window of low water contents, \(\le 5\) wt% of the total charge in our experiments. Fundamentally different, spatially less organised “garben-type” microstructures are generated at higher water contents. The pronounced temperature dependence of the characteristic spacing of phase alternation in a symplectite can potentially be used as a geo-thermometer. For symplectites to be used as geo-speedometers, the two potentially rate limiting processes, the interface reaction and the diffusion within a moving reaction front need to be calibrated independently. With our synthesis experiments, we could shed light on the interplay between the two processes and give some indication of the numerical values of the respective kinetic parameters, a quantitative calibration of the two processes is, however, beyond reach.
References
Abart R, Schmid R, Harlov D (2001) Metasomatic coronas around hornblendite xenoliths in granulite facies marble, Ivrea zone, N-Italy: constraints on component mobility. Contrib Miner Pet 141:473–492
Abart R, Petrishcheva E, Joachim B (2012) Thermodynamic model for growth of reaction rims with lamellar microstructure. Am Miner 97:231–240
Abart R, Heuser D, Habler G (2014) Mechanisms of myrmekite formation: case study form the Weinsberg granite, Maldanubian zone, Upper Austria. Contrib Miner Pet 168:2–14
Altherr R, Kalt A (1996) Metamorphic evolution of ultrahigh-pressure garnet peridotites from the Variscan Vosges Mts. (France). Chem Geol 134(1–3):27–47
Ashworth J, Chambers A (2000) Symplectic reaction in olivine and the controls of intergrowth spacing in symplectites. J Pet 41:285–304
Cahn J (1959) The kinetics of cellular segregation reactions. Acta Metall 7:18–28
Carlswell D, Moller C, OBrien P (1989) Origin of sapphirine–plagioclase symplectites in metabasites from Mitterbachgraben, Dunkelsteinerwald granulite complex, Lower Austria. Eur J Miner 1(3):455–466 ISSN 0935-1221
Christian JW (2002) The theory of transformations in metals and alloys. Elsevier, Amsterdam
Degi J, Abart R, Török K, Bal i E, Wirth R, Rhede D (2009) Symplectite formation during decompression induced garnet breakdown in lower crustal mafic granulite xenoliths: mechanisms and rates. Contrib Miner Pet 159:293–314
DeGroot R, Mazur S (1984) Non equilibrium thermodynamics. Dover, England
Fischer F, Svoboda J, Petryk M (2014) Thermodynamic extremal principles for irreversible processes in materials science. Acta Mater 67:1–20
Foley SF, Andronikov AV, Jacob DE, Melzer S (2006) Evidence from Antarctic mantle peridotite xenoliths for changes in mineralogy, geochemistry and geothermal gradients beneath a developing rift. Geochim Cosmochim Acta 70(12):3096–3120
Frei D, F G, Liebscher A, Wunder B, Klemme S, Blundy J (2009) Trace element partitioning between orthopyroxene and anhydrous silicate melt on the lherzolite solidus from 1.1 to 3.2 gpa and 1230 \(^o\)c to 1535 \(^o\)c in the model system \(\text{ Na }_2O-CaO-MgO-Al_2O_3-SiO_2\). Contrib Miner Pet 157:473–490
Gaidies F, Milke R, Heinrich W, Abart R (2017) Mineral reaction kinetics: microstructures, textures and chemical compositions, vol 16. EMU notes in mineralogy, chapter metamorphic mineral reactions: porphyroblast, corona and symplectite growth. European Mineralogical Union and Mineralogical Society of Great Britain and Ireland, London, pp 469–540
Gittins J, Fawcett J, Brooks C, Rucklidge J (1980) Intergrowths of nepheline–potassium feldspar and kalsilite–potassium feldspar: a re-examination of the ‘pseudo-leucite problem’. Contrib Miner Pet 73:119–126
Godard G, Martin S (2000) Petrogenesis of kelyphites in garnet peridotites: a case study from the Ulten zone, Italian alps. J Geodyn 30:117–145
Götze L, Abart R, Milke R, Schorr S, Zizak I, Dohmen R, Wirth R (2014) Growth of magnesio-aluminate spinel in thin film geometry—in-situ monitoring using synchrotron X-ray diffraction and thermodynamic model. Phys Chem Min 41:681–693
Harley S, Buick I (1992) Wollastonite–scapolite assemblages as indicators of granulite pressure temperature fluid histories—the Rauer group, East Antarctica. J Pet 33(3):693–728 ISSN 0022-3530
Harley S, Santosh M (1995) Wollastonite at Nuliyam, Kerala, southern India—a reassessment of \(CO_2\)-infiltration and charnockite formation at a classic locality. Contrib Miner Pet 120:83–94
Holland T, Powell R (1998) An internally consistent thermodynamic data set for phases of petrological interest. J Metamorph Geol 16(3):309–343
Keankeo W, Taylor W, FitzGerald J (2000) Clinoferrosilite-bearing kelyphite: a breakdown product of xenolithic garnet, Delegate breccia pipes, New South Wales, Australia. Miner Mag 64(3):469–479
Lasaga A (1998) Kinetic theory in the earth sciences. Princeton University Press, Princeton
Massonne H, Schreyer W (1989) Stability field of the high-pressure assemblage talc + phengite and 2 new phengite barometers. Eur J Miner 1(3):391–410
Messiga B, Bettini E (1990) Reactions behavior during kelyphite and symplectite formation—a case-study of mafic granulites and eclogites from the Bohemian massif. Eur J Miner 2(1):125–144
Mukhopadhyay B (1991) Garnet breakdown in some deep seated garnetiferous xenoliths from the central Sierra-Nevada—petrologic and tectonic implications. Lithos 27(1):59–78
Obata M (2011) Kelyphite and symplectite: textural and mineralogical diversities and universality, and a new dynamic view of their structural formation, chapter 5. New frontiers in tectonic research. in Tech, Croatia, sharkov, ev. edn, pp 93–122
Obata M, Ozawa K, Naemura K, Miyake A (2013) Isochemical breakdown of garnet in orogenic garnet peridotite and its implication to reaction kinetics. Miner Pet 107:881–895
Onsager L (1931) Reciprocal relations in irreversible processes I. Phys Rev 37(4):405–426
Oswald P, Bechhoefer J, Libchaber A (1987) Instabilities of a moving nematic isotropic interface. Phys Rev Lett 58(22):2318–2321
Petrishcheva E, Abart R (2017) Mineral reaction kinetics: microstructures, textures and chemical compositions, vol 16 of EMU Notes in Mineralog, chapter diffusion: some mathematical foundations and applications in mineralogy. European Mineralogical Union and Mineralogical Society of Great Britain and Ireland, London, pp 255–294
Phillips ER (1974) Myrmekite—one hundred years later. Lithos 7:181–194
Spacek P, Ackerman L, Habler G, Abart R, Ulrych J (2013) Garnet breakdown, symplectite formation and melting in basanite-hosted peridotite xenoliths from Zinst (Bavaria, Bohemian Massif). J Pet 54(8):1691–1723
Spruzeniece L, Piazolo S, Daczko NR, Kilburn MR, Putnis A (2017) Symplectite formation in the presence of a reactive fluid: insights from hydrothermal experiments. J Metamorph Geol 35:281–299
Spry A (1969) Metamorphic textures. Pergamon Press, Oxford
Steinbach I, Apel M (2007) The influence of lattice strain on pearlite formation in Fe–C. Acta Mater 55:4817–4822
Svoboda J, Turek I (1991) On diffusion-controlled evolution of closed solid-state thermodynamic systems at constant temperature and pressure. Philos Mag Part B 64:749–759
Svoboda J, Turek I, Fischer F (2005) Application of the thermodynamic extremal principle to modeling of thermodynamic processes in material sciences. Philos Mag 85:3699–3707
Vaks V (2004) Kinetics of phase separation and ordering in alloys. Phys Rep 391:157–242
Vernon R (2004) A practical guide to rock microstructure. Cambridge University Press, Cambridge
Warner R, Luth W (1973) 2-phase data for join monticellite (CaMgSiO4)–forsterite \((Mg_2SiO_4)\)—experimental results and numerical-analysis. Am Miner 58(11–1):998–1008
Wirth R (2004) Focused ion beam (FIB): a novel technology for advanced application of micro- and nanoanalysis in geosciences and applied mineralogy. Eur J Miner 16(6):863–876. https://doi.org/10.1127/0935-1221/2004/0016-0863 ISSN 0935-1221
Zener C (1958) Pearlite. Trans Am Inst Min Metall Engrs 167:550
Ziegler H (1961) Two extremal principles in irreversible thermodynamics. Ing. Arch 30:410–416
Acknowledgements
Open access funding provided by University of Vienna. This project was funded by the DFG Project AB 314/2-1 in the framework of the FOR 741 research unit. We thank Liene Spruzeniece and Ralf Milke for their thorough reviews and for their valuable hints that helped to improve the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Chris Ballhaus.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Remmert, P., Heinrich, W., Wunder, B. et al. Synthesis of monticellite–forsterite and merwinite–forsterite symplectites in the CaO–MgO–SiO2 model system: influence of temperature and water content on microstructure evolution. Contrib Mineral Petrol 173, 5 (2018). https://doi.org/10.1007/s00410-017-1429-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00410-017-1429-y