
Abstract
Purpose
To assess the impact of concomitant emphysema on outcomes in patients with idiopathic pulmonary fibrosis (IPF).
Methods
The IPF-PRO Registry is a US registry of patients with IPF. The presence of combined pulmonary fibrosis and emphysema (CPFE) at enrollment was determined by investigators’ review of an HRCT scan. Associations between emphysema and clinical outcomes were analyzed using Cox proportional hazards models.
Results
Of 934 patients, 119 (12.7%) had CPFE. Compared with patients with IPF alone, patients with CPFE were older (median 72 vs 70 years); higher proportions were current/former smokers (88.2% vs 63.7%), used oxygen with activity (49.6% vs 31.9%) or at rest (30.8% vs 18.4%), had congestive heart failure (13.6% vs 4.8%) and had prior respiratory hospitalization (25.0% vs 16.7%); they had higher FVC (median 71.8 vs 69.4% predicted) and lower DLco (median 35.3 vs 43.6% predicted). In patients with CPFE and IPF alone, respectively, at 1 year, rates of death or lung transplant were 17.5% (95% CI: 11.7, 25.8) and 11.2% (9.2, 13.6) and rates of hospitalization were 21.6% (14.6, 29.6) and 20.6% (17.9, 23.5). There were no significant associations between emphysema and any outcome after adjustment for baseline variables. No baseline variable predicted outcomes better in IPF alone than in CPFE.
Conclusion
Approximately 13% of patients in the IPF-PRO Registry had CPFE. Physiologic characteristics and comorbidities of patients with CPFE differed from those of patients with IPF alone, but the presence of emphysema did not drive outcomes after adjustment for baseline covariates.
Trial registration
ClinicalTrials.gov, NCT01915511; registered August 5, 2013.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Idiopathic pulmonary fibrosis (IPF) is a chronic interstitial lung disease associated with progressive decline in lung function, debilitating symptoms, and high mortality [1]. Emphysema is a common comorbidity in patients with IPF [2], but the clinical course of combined pulmonary fibrosis and emphysema (CPFE) continues to be debated. In a case series of 61 patients with CPFE published in 2005, almost half of the patients had pulmonary hypertension (PH) by echocardiogram and median survival was 6.1 years [3]. Subsequent studies have reported mortality in patients with CPFE similar to that in patients with IPF alone [4,5,6] or worse than in patients with IPF alone [7,8,9,10]. Comparing studies is challenging due to the different definitions used for CPFE, variation in the algorithms used to obtain the chest CT scans, and case ascertainment biases.
How emphysema in patients with IPF affects disease progression and survival and how patients with CPFE should be followed remain poorly understood. Compared with patients with IPF alone, patients with CPFE typically have higher forced vital capacity (FVC) but lower diffusing capacity of the lungs for carbon monoxide (DLco) [4]. Different studies have suggested that the measure of lung function that provides the best indicator of prognosis in patients with CPFE may be DLco [11] or forced expiratory volume in 1 s (FEV1) [12]. We used data from the IPF-PRO Registry, a multicenter US registry of patients with IPF, to compare the clinical characteristics and outcomes of patients with CPFE and IPF alone and to determine whether baseline measures, such as pulmonary function tests, correlate differently with outcomes in patients with CPFE versus IPF alone.
Methods
The design of the IPF-PRO Registry has been published [13]. This registry enrolled patients with IPF that was diagnosed or confirmed at the enrolling center in the past 6 months. Patients who were on a lung transplant list or participating in a randomized clinical trial could not be enrolled, but patients could be listed for transplant or enter clinical trials after enrollment. Patients in the registry are followed prospectively while receiving usual care, with follow-up visits scheduled approximately every 6 months. The study was approved by the Duke University Institutional Review Board (Pro00046131). The protocol was approved by the relevant Institutional Review Boards and/or local Independent Ethics Committees prior to patient enrollment at each site listed in the Acknowledgments. All patients provided informed consent.
A total of 1002 patients at 46 sites were enrolled between June 2014 and October 2018. Patients who did not have a high-resolution computed tomography (HRCT) scan, FEV1 value and FVC value at enrollment, or had no data recorded after enrollment, were excluded from these analyses. A patient was considered to have CPFE if “clinically significant emphysema” was present on an HRCT scan in the opinion of the investigator, assessed at the time of enrollment into the registry. In the presentation of patient characteristics at enrollment, continuous variables are presented as median (25th percentile, 75th percentile) and categorical variables as number and per cent of patients. Continuous variables were compared between patients with CPFE versus IPF alone using Wilcoxon rank sum tests. Categorical variables were compared using Pearson's chi-squared tests.
Clinical outcomes studied over follow-up were as follows: death; lung transplant; hospitalization (all-cause); death or lung transplant; and death, lung transplant, or hospitalization (all-cause). Cumulative incidence curves (for lung transplant and hospitalization, with death as a competing risk) and Kaplan–Meier event curves (for other outcomes) were estimated using all available follow-up data. Event rates at 1 year were estimated. Associations between the presence of emphysema and each outcome were assessed using univariable and multivariable Cox proportional hazards models. For lung transplant and hospitalization, Fine and Gray models were used, with death considered as a competing risk. In addition, associations between the presence of emphysema based on a broader definition (clinically significant emphysema on HRCT and/or FEV1/FVC < 0.7 at enrollment) and each outcome were assessed using the same modeling approach. The univariable model included the presence of emphysema as the only covariate. Multivariable models included emphysema and variables identified as being associated with the outcome in the entire registry cohort (Supplementary Table 1).
Associations between the GAP (gender-age-lung physiology) score [14] and the composite physiologic index (CPI) [15] and outcomes were assessed using multivariable models. The association between baseline variables and disease severity metrics and outcomes in patients with CPFE versus IPF alone were assessed using interaction tests. A significant interaction indicated that the relationship between the baseline variable and the outcome was different depending on whether the patient had emphysema.
When performing the modeling, the linearity assumption was assessed for all continuous covariates using restricted cubic splines, and transformations such as piecewise linear splines were used to accommodate for violations where necessary. The proportional hazards assumption was checked for CPFE in all models using the Kolmogorov-type supremum test, and no violations were found. Missing data were handled using multiple imputation: the missing data were filled in five times to generate five complete data sets using the Full Conditional Specification method; each complete data set was analyzed using standard statistical analyses; the results from the five complete datasets were averaged to generate the final inferential results.
The proportion of patients with new-onset PH (as reported by the investigator) during follow-up was assessed descriptively.
Results
Patients
Of 934 patients in the analysis cohort, 119 (12.7%) had CPFE at enrollment based on the presence of clinically significant emphysema on an HRCT scan, in the opinion of the investigator. Compared with patients with IPF alone, patients with CPFE were older (median age 72 vs 70 years) and higher proportions were current/former smokers (88.2% vs 63.7%), used oxygen with activity (49.6% vs 31.9%), used oxygen at rest (30.8% vs 18.4%), had congestive heart failure (13.6% vs 4.8%), and had a prior respiratory hospitalization (25.0% vs 16.7%) (Table 1). Patients with CPFE had a higher median FVC (71.8 vs 69.4% predicted) and lower median DLco (35.3 vs 43.6% predicted) than patients with IPF alone. The distribution of pulmonary function tests, GAP scores, and CPI at enrollment are presented in Supplementary Figs. 1 and 2. Patients with CPFE had worse St. George’s Respiratory Questionnaire (SGRQ) activity scores (median 66.2 vs 54.5) and better Cough and Sputum Assessment Questionnaire (CASA-Q) cough impact (median 87.5 vs 75.0) and symptoms (median 66.7 vs 58.3) domain scores than patients with IPF alone (Table 2). When CPFE was defined using a broader definition of clinically significant emphysema on HRCT and/or FEV1/FVC < 0.7, 149 patients (16.0%) had CPFE at enrollment.
Pulmonary Hypertension
Among patients with available data, 12 of 117 patients (10.3%) with CPFE and 52 of 810 patients (6.4%) with IPF alone had a history of PH at enrollment (p = 0.13). New-onset PH was reported during follow-up in 4 of 105 patients (3.8%) with CPFE and 43 of 758 (5.7%) patients with IPF alone at enrollment.
Associations Between CPFE and Outcomes
For all the outcomes studied, estimates for event rates at 1 year were numerically higher in patients with CPFE versus IPF alone at enrollment (Table 3; Fig. 1; Supplementary Figs. 3–6). However, there were no statistically significant associations between the presence of emphysema at enrollment and any outcome evaluated in univariable or multivariable models (Fig. 2). Similarly, there were no significant associations between the presence of emphysema at enrollment and any outcome evaluated when the broader definition of emphysema was used (Supplementary Table 2 and Supplementary Fig. 7).

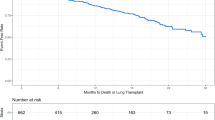
Kaplan–Meier plot of time to death, lung transplant, or hospitalization by presence of emphysema at enrollment

Associations between emphysema at enrollment and clinical outcomes.aAdjustment variables included in the models were as follows (all at enrollment): age, BMI, FVC % predicted, DLco % predicted, oxygen use at rest, and history of coronary artery disease or heart failure for time to death. Age, BMI, FEV1 % predicted, FVC % predicted, DLco % predicted, oxygen use at rest, oxygen use with activity, and prior diagnosis of IPF (before referral to enrolling center) for time to lung transplant. BMI, FEV1 % predicted, and oxygen at rest for time to hospitalization. Age, BMI, FEV1 % predicted, FVC % predicted, DLco % predicted, oxygen use at rest, oxygen use with activity, history of coronary artery disease or heart failure, and prior diagnosis of IPF (before referral to enrolling center) for time to death or lung transplant and for time to death, lung transplant or hospitalization
Associations Between Baseline Variables and Outcomes in Patients with CPFE Versus IPF Alone
There were no significant interactions between baseline variables and CPFE status for the outcomes of death, or death or lung transplant (data not shown). There were no significant interactions between baseline FVC, FEV1, or DLco and CPFE status for any of the outcomes. For the outcome of hospitalization, evidence of an interaction was found between CPFE status and oxygen use at rest (interaction p-value = 0.026) and age (interaction p-value = 0.032). In patients with CPFE, the risk of hospitalization was higher among those who were using oxygen at rest than in those who were not (HR 2.43 [95% CI: 1.39, 4.26]). Oxygen use at rest was not associated with a higher risk of hospitalization among patients with IPF alone (HR 1.16 [95% CI: 0.84, 1.60]). In patients with CPFE, among patients aged > 75 years, the risk of hospitalization was lower for older patients (HR 0.81 [95% CI: 0.67, 0.99] per 1-year increase above 75 years). Among patients with IPF alone, in the same age group, age was not associated with risk of hospitalization (HR 1.03 [95% CI: 0.98, 1.09] per 1-year increase above 75 years). Among patients aged ≤ 75 years, age was not associated with risk of hospitalization in either group (CPFE: HR 1.03 [95% CI: 0.97, 1.10] per 1-year increase up to 75 years; IPF alone: HR 1.00 [95% CI: 0.98, 1.02] per 1-year increase up to 75 years).
Associations Between GAP Score and CPI and Outcomes in Patients with CPFE Versus IPF Alone
Overall, the risk of death, or death or lung transplant, was higher among patients with a higher GAP score. Although there was no significant interaction between GAP score and CPFE for any outcome, the risk of hospitalization was numerically higher in patients who had a higher GAP score in patients with CPFE but not in patients with IPF alone (Table 4).
Overall, the risk of death, lung transplant, death or lung transplant, or death, lung transplant or hospitalization was higher with increasing CPI when CPI was > 45. Although there was no significant interaction between CPI and CPFE for any outcome, the risk of hospitalization, or death, lung transplant, or hospitalization, was numerically higher in patients with a higher CPI in patients with CPFE but not in patients with IPF alone (Table 4).
Discussion
The presence of emphysema in patients with IPF is often viewed as a unique phenotype, with worse prognosis and a high likelihood of concomitant PH. We evaluated this impression by analyzing a large real-world prospective multicenter dataset of patients with IPF. Of 934 subjects, 119 were deemed to have significant emphysema, yielding a prevalence of CPFE of 13%. Using a broader definition that included the FEV1/FVC ratio, 16% of the registry population had CPFE. We found that the presence of emphysema did not independently confer risk for poor outcomes, after adjustment for a comprehensive set of baseline variables.
The prevalence of CPFE in our population was lower than in several prior studies [2, 5, 7,8,9, 16], but not all prior studies [4]. A lower prevalence of smoking, the requirement for emphysema to be regarded as “clinically significant”, and recruitment of a broader population including patients with less severe disease are possible explanations for the relatively low prevalence of CPFE in our study. Similar to prior reports, in our study patients with CPFE tended to have more severe disease at baseline, as evidenced by a greater proportion using supplemental oxygen and worse pulmonary function test parameters, compared with patients without emphysema. Additionally, patients with emphysema were older, had more comorbidities, and had a higher likelihood of prior respiratory hospitalization compared with those without emphysema.
Several single-center reports have suggested worse outcomes in patients with emphysema [7,8,9,10] but in most of these studies, the impact of CPFE on mortality did not persist after multivariable adjustment. In other studies, the presence of emphysema was not associated with mortality [2, 4, 5]. A meta-analysis including 13 studies found no significant difference in mortality between patients with IPF alone and CPFE at 1, 3, or 5 years [6]. It is possible that findings of increased mortality in patients with CPFE compared with those with IPF alone have been driven largely by confounding variables, including age, tobacco exposure, smoking-related comorbidities, and degree of fibrosis [7, 10]. Our study provides further evidence that CPFE is not an independent risk factor for poor outcomes in patients with IPF. There were no significant interactions between baseline variables and CPFE status for any outcome studied: these variables had the same predictive utility in patients with CPFE as IPF alone.
Patients with CPFE had worse quality of life, as measured by the SGRQ activity score, than those with IPF alone. The impact on physical activity associated with emphysema may in part be indicative of greater physiologic impairment. However, the effect of cough on health-related quality of life, as assessed using the CASA-Q cough domains, was less in patients with CPFE, with between-group medians approximating the minimal important difference for patients with chronic obstructive pulmonary disease [17]. The reasons for the lower impact of cough in the patients with CPFE are unclear, but the more frequent use of inhaler therapy and systemic corticosteroids may be partly responsible.
Although FVC is often relatively preserved in patients with CPFE compared with IPF alone, we found no evidence that baseline FVC or DLco were less useful prognostic markers in the CPFE group. It is possible that changes in FVC or DLco may be differentially prognostic in the CPFE population, but we did not evaluate this. Similarly, disease severity metrics, including GAP score and CPI, did not predict the likelihood of death or lung transplant differently in patients with CPFE than IPF alone. There were non-significant trends for a greater likelihood of hospitalization with worse GAP or CPI values in patients with CPFE than IPF alone (interaction p-values 0.07 and 0.06, respectively). Although not conclusive, these data suggest the possibility that CPFE should be assessed as a covariate when using hospitalization as an outcome predicted by GAP or CPI score. Given the similar point estimates for risk, it is possible that worsening physiology, rather than age or sex, account for the increased risk of hospitalization in those with worse scores.
An association between emphysema and PH has been observed in patients with IPF [3, 7, 18]. While we found that PH was numerically more frequent in patients with CPFE than in patients with IPF alone, the prevalence of recognized PH in our CPFE population was only 12%. This could be due to under-diagnosis, but other factors may account for the discrepant findings. For example, because of the prevailing belief that CPFE and PH are strongly associated, practicing clinicians may test more aggressively for PH in patients with CPFE, which may have led to higher estimates of its prevalence in prior reports. In combination with pulmonary fibrosis, emphysema markedly reduces DLco, which may also lead to earlier evaluation for PH [5]. Additionally, the combined effects of parenchymal destruction from pulmonary fibrosis and emphysema result in defects in a higher percentage of the lung. When the percentage of total lung volume is taken into account, the presence of emphysema per se is not associated with PH in patients with IPF [19].
Our study has limitations. Case definition was based solely on the investigator’s appraisal of “clinically significant” emphysema on the baseline HRCT scan. Several prior studies [4, 5, 7, 8, 16] have used quantitative assessments of emphysema. The lower prevalence of emphysema in our study compared with most prior studies suggests that we included only more severe/apparent emphysema, which may have resulted in a higher chance of finding a statistical association with worse outcomes. Instead, we found no independent association between emphysema and important outcomes. We acknowledge that the broader definition that we used for CPFE (i.e., clinically significant emphysema on HRCT and/or FEV1/FVC < 0.7) may have been confounded by conditions other than emphysema that cause airway obstruction (e.g., asthma). It is possible that we underestimated the prevalence of PH since there was no pre-specified assessment for PH. Even if a significant proportion of PH cases were missed, the prevalence of PH in our study is substantially lower than prior reports and it is likely that the majority of unrecognized PH would be classified as mild.
Conclusion
Our data challenge the prevailing conception of CPFE; they suggest that confounding variables such as age or complications of smoking may account for the high mortality associated with CPFE in prior reports. Whether CPFE is a unique biologic entity or simply represents the additive effect of two superimposed diseases requires further study.
A plain language summary of this article is available at www.usscicomms.com/respiratory/kim/IPF-PROCPFE.
Data Availability
The datasets analyzed during the current study are not publicly available, but are available from the corresponding author on reasonable request.
Code availability
Not applicable.
References
Raghu G, Remy-Jardin M, Myers JL et al (2018) Diagnosis of idiopathic pulmonary fibrosis. An official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med 198(5):e44–e68
Doubková M, Švancara J, Svoboda M et al (2018) EMPIRE Registry, Czech part: impact of demographics, pulmonary function and HRCT on survival and clinical course in idiopathic pulmonary fibrosis. Clin Respir J 12(4):1526–1535
Cottin V, Nunes H, Brillet PY et al (2005) Combined pulmonary fibrosis and emphysema: a distinct underrecognised entity. Eur Respir J 26(4):586–593
Ryerson CJ, Hartman T, Elicker BM et al (2013) Clinical features and outcomes in combined pulmonary fibrosis and emphysema in idiopathic pulmonary fibrosis. Chest 144(1):234–240
Jacob J, Bartholmai BJ, Rajagopalan S et al (2017) Functional and prognostic effects when emphysema complicates idiopathic pulmonary fibrosis. Eur Respir J 50(1):1700379
Jiang CG, Fu Q, Zheng CM (2019) Prognosis of combined pulmonary fibrosis and emphysema: comparison with idiopathic pulmonary fibrosis alone. Ther Adv Respir Dis 13:1753466619888119
Mejía M, Carrillo G, Rojas-Serrano J et al (2009) Idiopathic pulmonary fibrosis and emphysema: decreased survival associated with severe pulmonary arterial hypertension. Chest 136(1):10–15
Sugino K, Ishida F, Kikuchi N et al (2014) Comparison of clinical characteristics and prognostic factors of combined pulmonary fibrosis and emphysema versus idiopathic pulmonary fibrosis alone. Respirology 19(2):239–245
Tokgoz Akyil F, Sevim T, Akman C et al (2016) The predictors of mortality in IPF - does emphysema change the prognosis? Sarcoidosis Vasc Diffuse Lung Dis 33(3):267–274
Zhang L, Zhang C, Dong F et al (2016) Combined pulmonary fibrosis and emphysema: a retrospective analysis of clinical characteristics, treatment and prognosis. BMC Pulm Med 16(1):137
Yoon HY, Kim TH, Seo JB et al (2019) Effects of emphysema on physiological and prognostic characteristics of lung function in idiopathic pulmonary fibrosis. Respirology 24(1):55–62
Schmidt SL, Nambiar AM, Tayob N et al (2011) Pulmonary function measures predict mortality differently in IPF versus combined pulmonary fibrosis and emphysema. Eur Respir J 38(1):176–183
O'Brien EC, Durheim MT, Gamerman V et al (2016) Rationale for and design of the Idiopathic Pulmonary Fibrosis-PRospective Outcomes (IPF-PRO) registry. BMJ Open Respir Res 3(1):e000108
Ley B, Ryerson CJ, Vittinghoff E et al (2012) A multidimensional index and staging system for idiopathic pulmonary fibrosis. Ann Intern Med 156(10):684–691
Wells AU, Desai SR, Rubens MB et al (2003) Idiopathic pulmonary fibrosis: a composite physiologic index derived from disease extent observed by computed tomography. Am J Respir Crit Care Med 167(7):962–969
Cottin V, Hansell DM, Sverzellati N et al (2017) Effect of emphysema extent on serial lung function in patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 196(9):1162–1171
Rebelo P, Oliveira A, Paixão C et al (2020) Minimal clinically important differences for patient-reported outcome measures of cough and sputum in patients with COPD. Int J Chron Obstruct Pulmon Dis 15:201–212
Cottin V, Le Pavec J, Prévot G et al (2010) Pulmonary hypertension in patients with combined pulmonary fibrosis and emphysema syndrome. Eur Respir J 35(1):105–111
Jacob J, Bartholmai BJ, Rajagopalan S et al (2018) Likelihood of pulmonary hypertension in patients with idiopathic pulmonary fibrosis and emphysema. Respirology 23(6):593–599
Raghu G, Collard HR, Egan JJ et al (2011) An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 183(6):788–824
Acknowledgements
We thank the principal investigators and enrolling centers in the IPF-PRO Registry. The authors meet criteria for authorship as recommended by the International Committee of Medical Journal Editors (ICMJE). The authors did not receive payment for development of this article. Writing support was provided by Julie Fleming and Wendy Morris of Fleishman-Hillard, London, UK, which was contracted and funded by Boehringer Ingelheim Pharmaceuticals, Inc. Boehringer Ingelheim was given the opportunity to review the article for medical and scientific accuracy as well as intellectual property considerations.
Funding
The IPF-PRO™ Registry is funded by Boehringer Ingelheim Pharmaceuticals, Inc. (BIPI) and co-ordinated by the Duke Clinical Research Institute (DCRI).
Author information
Authors and Affiliations
Consortia
Contributions
HJK, LDS, MLN, and DAC were involved in the design of the study. HJK, DLH, LDM, and DAC were involved in data acquisition. MLN and ASH were involved in data analysis. All the authors were involved in the interpretation of the data and in the writing and critical review of the manuscript and have approved the final version.
Corresponding author
Ethics declarations
Conflict of interest
HJK has no disclosures. LDS, MLN, ASH are employees of the Duke Clinical Research Institute (DCRI), which receives funding support from Boehringer Ingelheim Pharmaceuticals, Inc, to co-ordinate the IPF-PRO/ILD-PRO Registry. DLH reports research fees from Boehringer Ingelheim. LDM reports personal fees from Boehringer Ingelheim and Genentech. SB is an employee of Boehringer Ingelheim Pharmaceuticals, Inc. TBL was an employee of Boehringer Ingelheim Pharmaceuticals, Inc at the time this research was conducted. DAC reports personal fees from Boehringer Ingelheim and Genentech.
Ethical Approval
The study was approved by the Duke University Institutional Review Board (Pro00046131). The protocol was also approved by the relevant Institutional Review Boards and/or local Independent Ethics Committees prior to patient enrollment at each site.
Consent to Participate
All patients provided informed consent.
Consent for Publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kim, H.J., Snyder, L.D., Neely, M.L. et al. Clinical Outcomes of Patients with Combined Idiopathic Pulmonary Fibrosis and Emphysema in the IPF-PRO Registry. Lung 200, 21–29 (2022). https://doi.org/10.1007/s00408-021-00506-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00408-021-00506-x