
Abstract
This review article presents select recent studies that form the basis for the development of esmethadone into a potential new drug. Esmethadone is a promising member of the pharmacological class of uncompetitive N-methyl-D-aspartate receptor (NMDAR) antagonists that have shown efficacy for major depressive disorder (MDD) and other diseases and disorders, such as Alzheimer’s dementia and pseudobulbar affect. The other drugs in the novel class of NMDAR antagonists with therapeutic uses that are discussed for comparative purposes in this review are esketamine, ketamine, dextromethorphan, and memantine. We present in silico, in vitro, in vivo, and clinical data for esmethadone and other uncompetitive NMDAR antagonists that may advance our understanding of the role of these receptors in neural plasticity in health and disease. The efficacy of NMDAR antagonists as rapid antidepressants may advance our understanding of the neurobiology of MDD and other neuropsychiatric diseases and disorders.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The contemporary understanding of major depressive disorder (MDD) neurobiology is progressively disengaging from the classic serotonergic hypothesis [1]. Accordingly, the risk–benefit ratio of available antidepressants, which mostly target monoaminergic neurotransmissions, has been increasingly questioned [2]. More recent hypotheses implicate impairments of neural plasticity in the pathogenesis of MDD [3,4,5] through the dysregulation of glutamatergic signaling via N-methyl-D-aspartate receptors (NMDARs) [6, 7]. Individuals with MDD suffer not only from depressed mood but also from cognitive deficits, and animal models of depressive-like behavior display learning deficits that have also been related to the impairment of neural plasticity [8, 9]. In the prefrontal cortex and hippocampus, impairment in neural plasticity has been associated with chronic inescapable stress and other models of depressive-like behavior [10, 11]. Interestingly, patients with MDD have also been shown to have reduced hippocampal volume [12, 13]. While MDD is still primarily considered a mood disorder, the impairment of cognition and motivation may be primary for understanding the neurobiology of this disorder. Furthermore, cognitive deficits in MDD are central in determining the prominent functional loss and disability seen in patients.
In experimental models of depressive-like behavior, reduced synaptic spine volume and impaired spinogenesis are reversed by NMDAR antagonists [14,15,16]. Specifically, Fogaça et al. [16] demonstrated that a single dose of esmethadone increased levels of the synaptic proteins PSD95, Synapsin 1, and GluA1 in the medial prefrontal cortex (mPFC) but not in the hippocampus. In addition, Li et al. [14] reported that ketamine produces a rapid (2-h) and sustained (72-h) increase in synaptic protein levels in the mPFC and increases levels of Synapsin 1 in whole rat hippocampus. The reversal of depressive-like behavior by uncompetitive NMDAR antagonists in experimental animal models appears to be due to the restoration of synaptic proteins through a brain-derived neurotrophic factor (BDNF)-dependent mechanism [14,15,16].
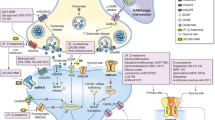
Uncompetitive NMDAR antagonists are a relatively recently described class of molecules with potential clinical applications as rapid antidepressants. One hypothesis for the mechanism of action of uncompetitive NMDAR antagonists in the treatment of depression is shown in Fig. 1 [17]. The “disinhibition hypothesis” is an alternative hypothesis that suggests that ketamine preferentially blocks NMDARs on GABAergic inhibitory interneurons, leading to a decrease of overall inhibition. This, in turn, disinhibits excitatory neurons and enhances excitatory synaptic transmission in the mPFC [18]. Other hypotheses are centered around different receptor systems, including the opioid receptor system and the sigma-1 receptor [19, 20]. While the mechanism of action of uncompetitive NMDAR antagonists for the treatment of depression needs to be further clarified and may differ among different drugs, several uncompetitive NMDAR antagonists have shown promise as antidepressant agents. The rapid antidepressant effects of ketamine have been replicated with esketamine, which has been approved for treatment-resistant depression [21]. The dextromethorphan–bupropion combination has shown efficacy for MDD in phase 2 and phase 3 trials [22, 23] and has been recently approved for the treatment of MDD. NMDAR antagonists have been FDA-approved for the treatment of other diseases and disorders. Memantine is approved for Alzheimer’s disease, and the combination drug dextromethorphan–quinidine is approved for the treatment of pseudobulbar affect. Esmethadone increased circulating BDNF levels in healthy subjects of a phase 1 clinical study [24] and improved subjective cognitive symptoms in patients with MDD in a phase 2 clinical study [25, 26]. Esmethadone (REL-1017) showed rapid, robust, and sustained antidepressant effects in a phase 2 trial conducted in patients with inadequate response to standard antidepressants [25]. Phase 3 studies are underway.

Proposed mechanism of kinase involvement in uncompetitive NMDAR antagonist-mediated rapid antidepressant effects. A In the normal phenotype, physiological NR1-2D homeostatic tonic Ca2+ influx appropriately regulates calmodulin-dependent protein kinase III (CaMKIII) phosphorylation of eukaryotic elongation factor 2 (eEF2), which results in adequate homeostatic maintenance and availability of synaptic proteins required for action potential (AP)-mediated neural plasticity. B In the depressive phenotype, increased Ca2+ influx through NR1-2D channels upregulates CaMKIII-eEF2 activity, leading to the halting of synaptic protein production and availability, impairing AP-mediated neural plasticity. C Resolution of the depressive phenotype is possible through the action of uncompetitive NMDAR antagonists, such as REL-1017, which block excessive tonic Ca2+ currents. This blockade may restore homeostatic maintenance and availability of synaptic proteins, re-enabling physiological AP-mediated synaptic plasticity
Esmethadone (REL-1017)
Esmethadone (d-methadone; dextromethadone; REL-1017) is the opioid inactive (S)-enantiomer of racemic methadone and is a novel uncompetitive NMDAR antagonist [27, 28]. Esmethadone is a promising, once-daily, oral, rapid antidepressant candidate [25]. If phase 3 results reproduce the robust and sustained efficacy seen in phase 2, esmethadone could potentially become the first-in-class agent among emerging second-generation (post-ketamine), oral, uncompetitive NMDAR antagonists with rapid antidepressant effects. This work reviews the current knowledge on the pharmacology of esmethadone and its ongoing development for the treatment of MDD.
Interactions of esmethadone with the NMDAR in silico and in vitro
The interactions of esmethadone with the NMDAR have been recently characterized in silico (Fig. 2) [28]. The in vitro activity of esmethadone has been compared with other uncompetitive NMDAR antagonists (Tables 1, 2 and 3) [28]. Furthermore, the known influence of physiological magnesium on NMDAR subtype preference by uncompetitive NMDAR antagonists [29] has also been characterized for esmethadone (Table 4) [28].

This rendering shows the interactions of uncompetitive NMDAR antagonists with the NR1-2D subtype in silico [28]. The structure of NR1-2D was obtained by electron microscopy (panel A, Protein Data Bank [PDB] code 6WHT). The black box highlights the drug-binding site. Structures of the complexes between esmethadone (light blue), arketamine (magenta), and esketamine (purple) with NR1-2D in the open conformation model (PDB code 6WHT) and the closed conformation model (PDB code 6WHS) can be seen in panels (B–D) and (E–G) [28]
The pharmacological interactions of esmethadone with human heterodimeric NMDARs described by Bettini and colleagues highlighted low NMDAR receptor affinity, NR1-2D subtype preference, ketamine-like trapping in the channel pore, and a propensity for undocking from the NMDAR in the open conformation. Importantly, the unique characteristics of esmethadone’s interaction with NMDARs, along with its lower potency compared to ketamine [28], may explain the lack of dissociative effects seen in clinical trials [25, 30]. Similarly, the ketamine enantiomer arketamine may be effective as an antidepressant with fewer dissociative effects because of its lower NMDAR affinity as compared to the ketamine enantiomer esketamine [31]. Other NMDAR antagonists, such as memantine and lanicemine, may lack consistent antidepressant effects in patients with MDD because of their low trapping [32] as compared to the higher trapping shown by ketamine and esmethadone. Additional in vitro experiments showed that esmethadone reduces Ca2+ influx induced by L-glutamate at very low concentrations, as well as Ca2+ influx due to quinolinic acid (QA) and gentamicin stimulation. Therefore, esmethadone may protect cells from the excessive calcium entry via NMDARs that are hyperactivated by very low concentrations of glutamate and by endogenous (e.g., QA) and exogenous (e.g., gentamicin) molecules [33].
Two clinical studies designed to assess the human abuse potential and performed in recreational drug users showed no meaningful abuse potential for esmethadone in this patient population [34, 35]. In these studies, dextromethadone was compared to oxycodone, ketamine, and dextromethorphan. Dextromethorphan is an over-the-counter antitussive drug and NMDAR uncompetitive antagonist with affinity for the NMDAR that is approximately threefold higher than esmethadone [28]. The primary metabolite of dextromethorphan, dextrorphan, also has NMDAR affinity [36], in contrast with 2-ethylidene-1,5-dimethyl-3,3-diphenylpyrrolidine (EDDP), the primary metabolite of esmethadone, which is inactive. These differences in potency and metabolism may explain the higher drug liking score of 300 mg oral dextromethorphan compared to 150 mg oral esmethadone in recreational drug users, as reported by Shram and colleagues [34].
Notably, cortical neurons of rats exposed to high doses of esmethadone did not show evidence of Olney’s lesions or other neuropathological changes [37], in contrast with other uncompetitive NMDAR antagonists known to produce Olney’s lesions [38,39,40,41]. This lack of evidence for potential neurotoxicity may be related to the relatively lower affinity of esmethadone binding at NMDARs, as demonstrated in radioligand binding assays, fluorometric imaging plate reader assays, and automated and manual patch assays [27, 28].
Lack of opioid activity by esmethadone: in vitro, animal, and human evidence
Since the introduction of methadone in the US in 1946 [42] and because of the structural similarity with levomethadone (the opioid-active mu agonist levo-enantiomer), many studies have examined the interactions of the dextro-enantiomer esmethadone with opioid receptors and its potential for eliciting opioid agonist effects in animal models and humans. Receptor affinity studies using esmethadone in rat models show 20-fold lower affinity for mu opiate receptors compared to the opioid-active enantiomer, levomethadone [43]. We performed two radioligand binding assays at human opioid receptors using esmethadone, levomethadone, and EDDP (Relmada studies performed by Eurofins: TW04-0009163 and TW04-0009695, submitted to FDA under IND 133345). In these studies, esmethadone exhibited a 27- to 40-fold lower affinity for human mu opioid receptors as compared to levomethadone (IC50 610/410 nM and IC50 14.6/14.7 nM for esmethadone and levomethadone, respectively). The major metabolite of esmethadone, EDDP, had no meaningful opioid affinity (Relmada studies submitted to FDA under IND 133345).
Animal studies show a lack of meaningful opioid effects and lack signs of withdrawal after abrupt discontinuation of esmethadone [44,45,46]. Furthermore, the results of these earlier preclinical studies were replicated in recent studies that showed esmethadone does not cause reinforcing effects, physical dependence, or withdrawal in rats [47]. These preclinical studies are corroborated by early human studies indicating no meaningful abuse potential from esmethadone [42, 48, 49] and by more recent clinical studies employing state-of-the-art methodology [34, 35, 47]. Taken together, preclinical and clinical studies confirm this 2019 Drug Enforcement Administration statement: “The d-isomer lacks significant respiratory depressant action and addiction liability, but possesses antitussive activity” [50]. The lack of opioid activity of esmethadone, in contrast with the opioid activity of levomethadone, is in line with the known stereoselectivity of opioid agonist activity for opioid enantiomers: esmethadone, dextromethorphan, and dextro-morphine are all inactive at opioid receptors, in contrast with the opioid agonist drugs levomethadone, levomethorphan, and levo-morphine [42, 43, 45, 51, 52]. Finally, the successful substitution of racemic methadone with half the dose of levomethadone in over 1500 patients with opioid use disorder indirectly supports the lack of opioid activity of esmethadone [53].
While the scientific evidence for esmethadone’s lack of meaningful opioid agonist activity is conclusive, the layman’s assumption may still be one of similarity of opioid effects to racemic methadone and levomethadone. This erroneous assumption may need additional educational efforts from the scientific community and from treating physicians to dispel addiction concerns that are unsupported by scientific data and that may interfere with its potential use as an antidepressant.
Antidepressant-like activity of esmethadone: preclinical studies
Esmethadone has rapid antidepressant-like activity in the rat forced swim test [54], an established model of depressive-like behavior predictive of antidepressant effects in humans. Aside from reversing depressive-like behavior in preclinical paradigms of depression, esmethadone, similarly to ketamine, may also reverse neuronal dysfunctions associated with depressive-like behavior by increasing synaptic spine volume and restoring spinogenesis [14, 16]. Remarkably, the reversal of depressive-like behavior by esmethadone and other NMDAR antagonists appears to rely on the restoration of synaptic proteins via a BDNF-dependent mechanism [14, 15, 55]. Figure 1 shows a current molecular hypothesis for the rapid relief of depressive behaviors and associated symptoms by esmethadone and other uncompetitive NMDAR antagonists [17]. While NMDAR antagonism is thought to be the mechanism of action of the antidepressant effects of uncompetitive NMDAR channel blockers, activity at other receptor systems, including opioid receptors [19] and sigma receptors [20], is also hypothesized. Esmethadone, aside from its uncompetitive NMDAR antagonist activity, shows affinity for other receptors (Table 5), which may also be implicated in its potential therapeutic effects.
Clinical studies assessing safety, tolerability, and efficacy of esmethadone in MDD
The safety, tolerability, and efficacy of esmethadone was assessed in two phase 1 trials and one phase 2 trial (Table 6). A single ascending dose (SAD) clinical trial demonstrated safety and tolerability of esmethadone in single doses of up to 150 mg. The 150 mg dose was deemed the maximum tolerated dose (MTD) based on the insurgence of nausea and vomiting. No patient experienced opioid-like euphoria or ketamine-like dissociative symptoms [30]. The lack of esmethadone-induced opioid-like euphoria and lack of ketamine-like dissociation at MTD was also confirmed in two studies designed to assess human abuse potential [34, 35]. The safety and tolerability of esmethadone administered daily at doses of 25 mg, 50 mg, and 75 mg for 10 days were then tested in a multiple ascending dose (MAD) trial [30]. In these subjects, there was no evidence of withdrawal after abrupt discontinuation of the 10-day course of esmethadone.
In these SAD and MAD studies [30], esmethadone exhibited linear pharmacokinetics with dose proportionality for most single-dose and multiple-dose parameters. Single doses up to 150 mg and daily doses up to 75 mg for 10 days were well tolerated with mostly mild treatment-emergent adverse events and no severe or serious adverse events. There was no evidence of respiratory depression, dissociative and psychotomimetic effects, or withdrawal signs and symptoms upon abrupt discontinuation. In regard to the effects of esmethadone on the QTc interval, an overall dose–response effect was observed, with higher doses resulting in larger QTcF (QT interval corrected using the Fridericia formula) changes from baseline. Importantly, none of the changes was considered clinically significant. Similar effects of the QTcF were observed in the phase 2 study [25]. No detectable conversion of esmethadone to levomethadone occurred in vivo.
Two randomized, double-blind, active- and placebo-controlled crossover studies were designed to evaluate the abuse potential of esmethadone compared with oxycodone (oxycodone study) or ketamine (ketamine study) in healthy recreational drug users. Three doses of esmethadone were evaluated in each study: 25 mg (the proposed therapeutic daily dose for MDD treatment), 75 mg (loading dose), and 150 mg (MTD). Positive controls were 40 mg oral oxycodone in the oxycodone study and 0.5 mg/kg intravenous ketamine infused over 40 min in the ketamine study. The ketamine study included 300 mg oral dextromethorphan as an exploratory comparator. The primary endpoint was the maximum effect (Emax) for drug liking, assessed using a bipolar 100-point visual analog scale (VAS). In the oxycodone study and the ketamine study, 47 and 51 participants completed all treatment arms, respectively. In both studies, esmethadone doses ranging from therapeutic (25 mg) to six times therapeutic (150 mg) had a statistically significant and clinically meaningful (p < 0.001) lower drug liking VAS Emax compared with positive controls. Results were consistent for all secondary endpoints, including measurements of overall drug liking and willingness to take the drug again, in both studies. Moreover, in the ketamine study, drug liking VAS Emax scores for esmethadone at all tested doses were significantly lower versus dextromethorphan (p < 0.05) (exploratory endpoint). In conclusion, these studies indicated no meaningful abuse potential for esmethadone.
The safety, tolerability, and efficacy of esmethadone were tested in a phase 2 study [25]. This study aimed to examine the effects of esmethadone in patients with MDD with inadequate response to standard antidepressants during the course of a major depressive episode. This was a randomized, double-blind, placebo-controlled trial, comprising three arms, designed to assess the safety, tolerability, pharmacokinetics, and efficacy of two dosages of esmethadone (25 mg or 50 mg orally once a day) administered for 7 days and conducted in ten centers across the United States. Patients were randomly assigned in a 1:1:1 ratio to placebo (N = 22), 25 mg/day esmethadone (N = 19), or 50 mg/day esmethadone (N = 21). All patients were maintained on their stable dose of standard antidepressant. Safety scales included the four-item Positive Symptom Rating Scale for psychotomimetic symptoms, the Clinician-Administered Dissociative States Scale for dissociative symptoms, the Clinical Opiate Withdrawal Scale for withdrawal signs and symptoms, and the Columbia Suicide Severity Rating Scale for suicidality. Efficacy was evaluated based on changes in the Montgomery–Åsberg Depression Rating Scale (MADRS) score. All 62 randomly assigned patients were included in the full analysis set population. Patients experienced only mild to moderate transient adverse events, and there was no evidence of dissociative, psychotomimetic, or opioid effects or withdrawal signs and symptoms, confirming the safety and tolerability results of phase 1 studies [30]. Clinically meaningful and statistically significant improvement in MADRS score started on day 4 with both esmethadone doses and was sustained through day 7 (last dose) and day 14 (7 days after the last dose), with effect sizes from 0.7 to 1.0. This trial confirmed the very favorable safety, tolerability, and pharmacokinetic profiles of esmethadone and indicated that esmethadone had rapid and sustained antidepressant effects compared with placebo in patients with inadequate responses to antidepressant treatments.
Table 7 lists publications from phase 1 and phase 2 sub-analyses.
Uncompetitive NMDAR antagonists: pharmacokinetics, tolerability, and safety considerations
Among the upcoming pharmacological class of NMDAR antagonists that may work as rapid antidepressants in patients, esmethadone stands out because of its very favorable tolerability and safety profile. The efficacy and safety of esmethadone may be determined by its selectivity for tonically hyperactive NR1-2D subtypes at doses therapeutic for MDD [28]. In addition, esmethadone has an ideal pharmacokinetic profile that allows once-daily oral administration [25, 30]. Ketamine and its enantiomers can only be administered intravenously or intranasally due to variable oral absorption. In addition, the safety window for ketamine and esketamine may be too narrow: at dosages in current use for the treatment of depression, approximately 70% of patients experience dissociative symptoms [56]. The combination drug dextromethorphan–bupropion is better tolerated than ketamine and esketamine [22] but carries the combined side effects of two different drugs with the burdens of polypharmacy, which may be especially relevant when this combination drug is under consideration for patients who are already taking other drugs.
Furthermore, ketamine and dextromethorphan have been reported to cause Olney’s lesions in rats. While the significance of this neuropathological animal finding is unknown, it cannot be discounted. Up to recently, the therapeutic uses of ketamine (for anesthesia) and dextromethorphan (for cough suppression) have been intermittent. Their current use for the treatment of MDD is likely to be chronic. The safety of the chronic uses of ketamine and dextromethorphan will need to be confirmed in post-marketing analyses. In contrast, esmethadone does not cause Olney’s lesions in rats [33], suggesting that its long-term use may be safer compared to NMDAR antagonists that have been found to cause these lesions. The safety of esmethadone is also indirectly supported by over 70 years of chronic racemic methadone use in millions of patients with pain and opioid use disorder. Most of these patients are exposed to esmethadone serum levels greater than those seen in patients treated with the dose proposed for MDD. The average methadone dose for opioid use disorder is approximately 75 mg daily, and 50% of this dose is esmethadone. The esmethadone exposure in these patients with opioid use disorder and pain is, therefore, higher than the exposure of patients with MDD treated with 25 mg esmethadone. No long-term detrimental neurological consequences have been described in patients treated chronically with racemic methadone.
In conclusion, due to the favorable pharmacological features described above, if ongoing phase 3 studies confirm the promising phase 2 results, esmethadone may potentially become the best-in-class agent for safety, tolerability, and efficacy among uncompetitive NMDAR antagonists with rapid antidepressant effects.
Data availability
Not applicable.
Code availability
Not applicable.
References
Moncrieff J, Cooper RE, Stockmann T, Amendola S, Hengartner MP, Horowitz MA (2022) The serotonin theory of depression: a systematic umbrella review of the evidence. Mol Psychiatry https://doi.org/10.1038/s41380-022-01661-0
Jakobsen JC, Gluud C, Kirsch I (2020) Should antidepressants be used for major depressive disorder? BMJ Evid Based Med 25:130–130. https://doi.org/10.1136/bmjebm-2019-111238
Mathews DC, Henter ID, Zarate CA (2012) Targeting the glutamatergic system to treat major depressive disorder: rationale and progress to date. Drugs 72:1313–1333. https://doi.org/10.2165/11633130-000000000-00000
Boku S, Nakagawa S, Toda H, Hishimoto A (2018) Neural basis of major depressive disorder: beyond monoamine hypothesis. Psychiatry Clin Neurosci 72:3–12. https://doi.org/10.1111/pcn.12604
Henter ID, de Sousa RT, Zarate CA Jr (2018) Glutamatergic modulators in depression. Harv Rev Psychiatry 26:307–319. https://doi.org/10.1097/HRP.0000000000000183
Nicoll RA (2017) A brief history of long-term potentiation. Neuron 93:281–290. https://doi.org/10.1016/j.neuron.2016.12.015
Hansen KB, Yi F, Perszyk RE, Furukawa H, Wollmuth LP, Gibb AJ, Traynelis SF (2018) Structure, function, and allosteric modulation of NMDA receptors. J Gen Physiol 150:1081–1105. https://doi.org/10.1085/jgp.201812032
Mahati K, Bhagya V, Christofer T, Sneha A, Shankaranarayana Rao BS (2016) Enriched environment ameliorates depression-induced cognitive deficits and restores abnormal hippocampal synaptic plasticity. Neurobiol Learn Mem 134:379–391. https://doi.org/10.1016/j.nlm.2016.08.017
Bora E, Harrison BJ, Yücel M, Pantelis C (2013) Cognitive impairment in euthymic major depressive disorder: a meta-analysis. Psychol Med 43:2017–2026. https://doi.org/10.1017/S0033291712002085
Kim JJ, Diamond DM (2002) The stressed hippocampus, synaptic plasticity and lost memories. Nat Rev Neurosci 3:453–462. https://doi.org/10.1038/nrn849
Moda-Sava RN, Murdock MH, Parekh PK, Fetcho RN, Huang BS, Huynh TN, Witztum J, Shaver DC, Rosenthal DL, Alway EJ, Lopez K, Meng Y, Nellissen L, Grosenick L, Milner TA, Deisseroth K, Bito H, Kasai H, Liston C (2019) Sustained rescue of prefrontal circuit dysfunction by antidepressant-induced spine formation. Science 364:eaat8078. https://doi.org/10.1126/science.aat8078
Videbech P, Ravnkilde B (2004) Hippocampal volume and depression: a meta-analysis of MRI studies. Am J Psychiatry 161:1957–1966. https://doi.org/10.1176/appi.ajp.161.11.1957
Neumeister A, Wood S, Bonne O, Nugent AC, Luckenbaugh DA, Young T, Bain EE, Charney DS, Drevets WC (2005) Reduced hippocampal volume in unmedicated, remitted patients with major depression versus control subjects. Biol Psychiatry 57:935–937. https://doi.org/10.1016/j.biopsych.2005.01.016
Li N, Lee B, Liu RJ, Banasr M, Dwyer JM, Iwata M, Li XY, Aghajanian G, Duman RS (2010) mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science 329:959–964. https://doi.org/10.1126/science.1190287
Autry AE, Adachi M, Nosyreva E, Na ES, Los MF, Cheng P-f, Kavalali ET, Monteggia LM (2011) NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature 475:91–95. https://doi.org/10.1038/nature10130
Fogaça MV, Fukumoto K, Franklin T, Liu R-J, Duman CH, Vitolo OV, Duman RS (2019) N-Methyl-D-aspartate receptor antagonist d-methadone produces rapid, mTORC1-dependent antidepressant effects. Neuropsychopharmacology 44:2230–2238. https://doi.org/10.1038/s41386-019-0501-x
Stahl SM, De Martin S, Mattarei A, Bettini E, Pani L, Guidetti C, Folli F, de Somer M, Traversa S, Inturrisi CE (2022) Esmethadone (REL-1017) and other uncompetitive NMDAR channel blockers may improve mood disorders via modulation of synaptic kinase-mediated signaling. Int J Mol Sci 23:12196. https://doi.org/10.3390/ijms232012196
Duman RS, Sanacora G, Krystal JH (2019) Altered connectivity in depression: GABA and glutamate neurotransmitter deficits and reversal by novel treatments. Neuron 102:75–90. https://doi.org/10.1016/j.neuron.2019.03.013
Bonaventura J, Lam S, Carlton M, Boehm MA, Gomez JL, Solis O, Sanchez-Soto M, Morris PJ, Fredriksson I, Thomas CJ, Sibley DR, Shaham Y, Zarate CA Jr, Michaelides M (2021) Pharmacological and behavioral divergence of ketamine enantiomers: implications for abuse liability. Mol Psychiatry 26:6704–6722. https://doi.org/10.1038/s41380-021-01093-2
Keam SJ (2022) Dextromethorphan/Bupropion: first approval. CNS Drugs 36:1229–1238. https://doi.org/10.1007/s40263-022-00968-4
Popova V, Daly EJ, Trivedi M, Cooper K, Lane R, Lim P, Mazzucco C, Hough D, Thase ME, Shelton RC, Molero P, Vieta E, Bajbouj M, Manji H, Drevets WC, Singh JB (2019) Efficacy and safety of flexibly dosed Esketamine nasal spray combined with a newly initiated oral antidepressant in treatment-resistant depression: a randomized double-blind active-controlled study. Am J Psychiatry 176:428–438. https://doi.org/10.1176/appi.ajp.2019.19020172
Tabuteau H, Jones A, Anderson A, Jacobson M, Iosifescu DV (2022) Effect of AXS-05 (Dextromethorphan-Bupropion) in major depressive disorder: a randomized double-blind controlled trial. Am J Psychiatry 179:490–499. https://doi.org/10.1176/appi.ajp.21080800
Iosifescu DV, Jones A, O’Gorman C, Streicher C, Feliz S, Fava M, Tabuteau H (2022) Efficacy and safety of AXS-05 (Dextromethorphan-Bupropion) in patients with major depressive disorder: a phase 3 randomized clinical trial (GEMINI). J Clin Psychiatry 83:21m14345. https://doi.org/10.4088/JCP.21m14345
De Martin S, Gabbia D, Folli F, Bifari F, Fiorina P, Ferri N, Stahl S, Inturrisi CE, Pappagallo M, Traversa S, Manfredi PL (2021) REL-1017 (Esmethadone) increases circulating BDNF levels in healthy subjects of a phase 1 clinical study. Front Pharmacol 12:671859. https://doi.org/10.3389/fphar.2021.671859
Fava M, Stahl S, Pani L, De Martin S, Pappagallo M, Guidetti C, Alimonti A, Bettini E, Mangano RM, Wessel T, de Somer M, Caron J, Vitolo OV, DiGuglielmo GR, Gilbert A, Mehta H, Kearney M, Mattarei A, Gentilucci M, Folli F, Traversa S, Inturrisi CE, Manfredi PL (2022) REL-1017 (Esmethadone) as adjunctive treatment in patients with major depressive disorder: a phase 2a randomized double-blind trial. Am J Psychiatry 179:122–131. https://doi.org/10.1176/appi.ajp.2021.21020197
Guidetti C, Fava M, Pani L, Pappagallo M, Serra G, DeMartin S, Mattarei A, Manfredi PL (2022) A phase 2a double-blind randomized trial of REL-1017 (Esmethadone) in patients with MDD: analysis of subscales from the symptoms of depression questionnaire. CNS Spectr 27:235. https://doi.org/10.1017/S1092852922000359
Gorman AL, Elliott KJ, Inturrisi CE (1997) The d- and l-isomers of methadone bind to the non-competitive site on the N-methyl-D-aspartate (NMDA) receptor in rat forebrain and spinal cord. Neurosci Lett 223:5–8. https://doi.org/10.1016/s0304-3940(97)13391-2
Bettini E, Stahl SM, De Martin S, Mattarei A, Sgrignani J, Carignani C, Nola S, Locatelli P, Pappagallo M, Inturrisi C, Bifari F, Cavalli A, Alimonti A, Pani L, Fava M, Traversa S, Folli F, Manfredi PL (2022) Pharmacological comparative characterization of REL-1017 (Esmethadone-HCl) and other NMDAR channel blockers in human heterodimeric N-methyl-D-aspartate receptors. Pharmaceuticals (Basel) 15:997. https://doi.org/10.3390/ph15080997
Kotermanski SE, Johnson JW (2009) Mg2+ imparts NMDA receptor subtype selectivity to the Alzheimer’s drug memantine. J Neurosci 29:2774–2779. https://doi.org/10.1523/JNEUROSCI.3703-08.2009
Bernstein G, Davis K, Mills C, Wang L, McDonnell M, Oldenhof J, Inturrisi C, Manfredi PL, Vitolo OV (2019) Characterization of the safety and pharmacokinetic profile of D-methadone, a novel N-methyl-D-aspartate receptor antagonist in healthy, opioid-naive subjects: results of two phase 1 studies. J Clin Psychopharmacol 39:226–237. https://doi.org/10.1097/JCP.0000000000001035
Zhang JC, Yao W, Hashimoto K (2022) Arketamine, a new rapid-acting antidepressant: a historical review and future directions. Neuropharmacology 218:109219. https://doi.org/10.1016/j.neuropharm.2022.109219
Sanacora G, Smith MA, Pathak S, Su HL, Boeijinga PH, McCarthy DJ, Quirk MC (2014) Lanicemine: a low-trapping NMDA channel blocker produces sustained antidepressant efficacy with minimal psychotomimetic adverse effects. Mol Psychiatry 19:978–985. https://doi.org/10.1038/mp.2013.130
Bettini E, De Martin S, Mattarei A, Pappagallo M, Stahl SM, Bifari F, Inturrisi CE, Folli F, Traversa S, Manfredi PL (2022) The N-methyl-D-aspartate receptor blocker REL-1017 (Esmethadone) reduces calcium influx induced by glutamate, quinolinic acid, and gentamicin. Pharmaceuticals (Basel) 15:882. https://doi.org/10.3390/ph15070882
Shram M, Henningfield J, Apseloff G, Gorodetzky C, De Martin S, Vocci F, Sapienza F, Kosten T, Huston J, Buchhalter A, Ashworth J, Lanier R, Folli F, Traversa S, Inturrisi CE, Manfredi P, Pappagallo M (2022) No meaningful abuse potential in recreational ketamine users of REL-1017 (Esmethadone Hydrochloride), a new NMDAR antagonist and potential rapid-acting antidepressant. Paper presented at the American Society of Clinical Psychopharmacology (ASCP) Annual Meeting. Scottsdale, AZ
Shram M, Henningfield J, Apseloff G, Gorodetzky C, De Martin S, Vocci F, Sapienza F, Kosten T, Huston J, Buchhalter A, Ashworth J, Lanier R, Folli F, Traversa S, Inturrisi CE, Manfredi PL, Marco P (2022) No meaningful abuse potential in recreational opioid users of REL-1017 (Esmethadone Hydrochloride), a new NMDAR antagonist and potential rapid-acting antidepressant. Paper presented at the American Society of Clinical Psychopharmacology (ASCP) Annual Meeting. Scottsdale, AZ
Jaffe DB, Marks SS, Greenberg DA (1989) Antagonist drug selectivity for radioligand binding sites on voltage-gated and N-methyl-D-aspartate receptor-gated Ca2+ channels. Neurosci Lett 105:227–232. https://doi.org/10.1016/0304-3940(89)90042-6
Bifari F, Pappagallo M, Bleavins M, Traversa S, Folli F, Manfredi PL (2022) REL-1017 (Esmethadone), a novel NMDAR blocker for the treatment of MDD is not neurotoxic in Sprague-Dawley rats. Front Pharmacol 13:863959. https://doi.org/10.3389/fphar.2022.863959
Morris PJ, Burke RD, Sharma AK, Lynch DC, Lemke-Boutcher LE, Mathew S, Elayan I, Rao DB, Gould TD, Zarate CA Jr, Zanos P, Moaddel R, Thomas CJ (2021) A comparison of the pharmacokinetics and NMDAR antagonism-associated neurotoxicity of ketamine, (2R,6R)-hydroxynorketamine and MK-801. Neurotoxicol Teratol 87:106993. https://doi.org/10.1016/j.ntt.2021.106993
Olney JW, Labruyere J, Price MT (1989) Pathological changes induced in cerebrocortical neurons by phencyclidine and related drugs. Science 244:1360–1362. https://doi.org/10.1126/science.2660263
Olney JW, Labruyere J, Wang G, Wozniak DF, Price MT, Sesma MA (1991) NMDA antagonist neurotoxicity: mechanism and prevention. Science 254:1515–1518. https://doi.org/10.1126/science.1835799
Wozniak D, Dikranian K, Ishimaru M, Nardi A, Corso T, Tenkova T, Olney J, Fix A (1998) Disseminated corticolimbic neuronal degeneration induced in rat brain by MK-801: potential relevance to Alzheimer’s disease. Neurobiol Dis 5:305–322. https://doi.org/10.1006/nbdi.1998.0206
Eddy NB, Halbach H, Braenden OJ (1957) Synthetic substances with morphine-like effect: clinical experience; potency, side-effects, addiction liability. Bull World Health Organ 17:569–863
Codd EE, Shank RP, Schupsky JJ, Raffa RB (1995) Serotonin and norepinephrine uptake inhibiting activity of centrally acting analgesics: structural determinants and role in antinociception. J Pharmacol Exp Ther 274:1263–1270
Ramabadran K, Bansinath M (1986) A critical analysis of the experimental evaluation of nociceptive reactions in animals. Pharm Res 3:263–270. https://doi.org/10.1023/A:1016355200944
Bolan EA, Tallarida RJ, Pasternak GW (2002) Synergy between μ opioid ligands: evidence for functional interactions among μ opioid receptor subtypes. J Pharmacol Exp Ther 303:557–562. https://doi.org/10.1124/jpet.102.035881
Lemberg K, Kontinen VK, Viljakka K, Kylänlahti I, Yli-Kauhaluoma J, Kalso E (2006) Morphine, oxycodone, methadone and its enantiomers in different models of nociception in the rat. Anesth Analg 102:1768–1774. https://doi.org/10.1213/01.ane.0000205751.88422.41
Henningfield J, Gauvin D, Bifari F, Fant R, Shram M, Buchhalter A, Ashworth J, Lanier R, Pappagallo M, Inturrisi C (2022) REL-1017 (Esmethadone; d-Methadone) does not cause reinforcing effect, physical dependence and withdrawal signs in Sprague Dawley rats. Sci Rep 12:1–15. https://doi.org/10.1038/s41598-022-15055-3
Isbell H, Wilker A, Eisenman AJ, Daingerfield M, Frank K (1948) Liability of addiction to 6-dimethylamino-4-4-diphenyl-3-heptanone (Methadon, Amidone or 10820) in man: experimental addiction to methadon. Arch Intern Med 82:362–392. https://doi.org/10.1001/archinte.1948.00020040042004
Fraser H, Isbell H (1962) Human pharmacology and addictiveness of certain dextroisomers of synthetic analgesics. Bull Narc 14:25–35
Drug Enforcement Administration (2019) Methadone. Drug Enforcement Administration. https://www.deadiversion.usdoj.gov/drug_chem_info/methadone/methadone.pdf. Accessed 11 Nov 2022
Pasternak GW, Pan Y-X (2013) Mu opioids and their receptors: evolution of a concept. Pharmacol Rev 65:1257–1317. https://doi.org/10.1124/pr.112.007138
Pappagallo M, Inturrisi CE, Manfredi PL (2022) Comment on “Novel glutamatergic modulators for the treatment of mood disorders: current status.” CNS Drugs 36:203–204. https://doi.org/10.1007/s40263-021-00891-0
Soyka M, Zingg C (2009) Feasability and safety of transfer from racemic methadone to (R)-methadone in primary care: clinical results from an open study. World J Biol Psychiatry 10:217–224. https://doi.org/10.1080/15622970802416057
Hanania T, Manfredi P, Inturrisi C, Vitolo OV (2020) The N-methyl-D-aspartate receptor antagonist d-methadone acutely improves depressive-like behavior in the forced swim test performance of rats. Exp Clin Psychopharmacol 28:196–201. https://doi.org/10.1037/pha0000310
Fogaça MV, Duman RS (2019) Cortical GABAergic dysfunction in stress and depression: new insights for therapeutic interventions. Front Cell Neurosci 13:87. https://doi.org/10.3389/fncel.2019.00087
Molero P, Ramos-Quiroga JA, Martin-Santos R, Calvo-Sánchez E, Gutiérrez-Rojas L, Meana JJ (2018) Antidepressant efficacy and tolerability of Ketamine and Esketamine: a critical review. CNS Drugs 32:411–420. https://doi.org/10.1007/s40263-018-0519-3
Acknowledgements
Editorial support for the current manuscript was performed by Máté Fischer, Sarah A. Laredo, Julia Dey, and Lauren Hummel of Metis Medical Media, supported by Relmada Therapeutics, Inc., according to Good Publication Practices (GPP3). All opinions, conclusions, and data interpretation lie with the authors.
Funding
This research was sponsored by Relmada Therapeutics, Inc., Coral Gables, FL, USA. Maurizio Fava—Dr. Fava is employed by or has received compensation from companies or institutions that received funding from Relmada Therapeutics. Stephen M. Stahl—Dr. Stahl has received compensation from Relmada Therapeutics as a consultant. Sara De Martin—Dr. De Martin has received grant support from MGGM LLC and consultation fees from Neuroarbor LLC and is employed by or has received compensation from companies or institutions that received funding from Relmada Therapeutics. Andrea Mattarei—Dr. Mattarei has received grant support from MGGM LLC and consultation fees from Neuroarbor LLC and is employed by or has received compensation from companies or institutions that received funding from Relmada Therapeutics. Ezio Bettini—Dr. Bettini is employed by or has received compensation from companies or institutions that received funding from Relmada Therapeutics. Stefano Comai—Dr. Comai has received grant support from MGGM LLC and is employed by or has received compensation from companies or institutions receiving funding from Relmada Therapeutics. Andrea Alimonti—Dr. Alimonti is employed by or has received compensation from companies or institutions that received funding from Relmada Therapeutics, has received research funding from MGGM LLC, and is inventor on patent n. WO2020181194A1. Francesco Bifari—No disclosures. Luca Pani—Dr. Pani has received compensation from Relmada Therapeutics as a consultant. Franco Folli—Dr. Folli has received consultant fees from Relmada Therapeutics. Clotilde Guidetti—Dr. Guidetti has received grant support from Relmada Therapeutics and personal fees from MGGM LLC and is employed by or has received compensation from companies or institutions that received funding from Relmada Therapeutics. Alberto Furlan—Dr. Furlan is employed by or has received compensation from companies or institutions that received funding from Relmada Therapeutics. Jacopo Sgrignani—Dr. Sgrignani is employed by the Institute for Research in Biomedicine (IRB) in Bellinzona, Switzerland, which has received funding from Relmada Therapeutics. Patrizia Locatelli—Dr. Locatelli is employed by the Institute for Research in Biomedicine (IRB) in Bellinzona, Switzerland, which has received funding from Relmada Therapeutics. Andrea Cavalli—Dr. Cavalli is associated with the Swiss Institute of Bioinformatics (SIB) and The Italian Institute for Technology (IIT), both of which receive funding from Relmada Therapeutics. Cedric O’Gorman—Dr. O’Gorman is employed by Relmada Therapeutics. Sergio Traversa—Dr. Traversa is employed by Relmada Therapeutics. Charles E. Inturrisi—Dr. Inturrisi has received compensation from Relmada Therapeutics as a consultant and is an inventor on patents related to esmethadone. Marco Pappagallo—Dr. Pappagallo has received compensation from Relmada Therapeutics as a consultant. Paolo L. Manfredi—Dr. Manfredi has received compensation from Relmada Therapeutics as a consultant and is an inventor on patents related to esmethadone.
Author information
Authors and Affiliations
Contributions
Conceptualization: MF, SMS, SDM, AA, CG, ST, CEI, MP and PLM; methodology: MF, SMS, EB, CG, JS, PL, AC, AF and PLM; software: JS, PL, AC, MF and FF; validation: FF; formal analysis: FF; investigation: EB, JS and PLM; resources: ST; data curation: FF, CG and PLM; writing—original draft preparation: MF, SMS, SDM, CG and PLM; writing—review and editing: MF, SMS, SDM, AM, SC, LP, CG, FF, CO, ST, MP, AA and PLM; visualization: FF, JS, AF, PL and PLM; supervision: FF and PLM; project administration: ST and PLM; funding acquisition: ST. All the authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
Relmada Therapeutics and its consultants and employees contributed to all aspects of this manuscript, including all described experimental work.
Ethical approval
Not applicable.
Consent for publication
Not applicable.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fava, M., Stahl, S.M., De Martin, S. et al. Esmethadone-HCl (REL-1017): a promising rapid antidepressant. Eur Arch Psychiatry Clin Neurosci 273, 1463–1476 (2023). https://doi.org/10.1007/s00406-023-01571-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00406-023-01571-4