
Abstract
Purpose
Sinonasal lymphoma (SL) is a rare lymphatic neoplasm of the nasal cavities, paranasal sinuses and nasopharynx. Whereas some risk factors for SL subtypes have been identified, their aetiology is unknown. Along with other predisposing factors, the viral association of lymphomas, such as Epstein-Barr virus (EBV) and Burkitt and Hodgkin lymphomas, is well-established. Modern molecular biology techniques have enabled the discovery of novel human viruses, exemplified by the protoparvovirus cutavirus (CuV), associated with cutaneous T-cell lymphoma. These findings, and the anatomical location of the sinonasal tract with its rich microbiome and infectious agents, justify in-depth studies among SL.
Methods
We analysed the presence of 20 viruses of Orthoherpesviridae, Parvoviridae, and Polyomaviridae by qPCR in 24 SL tumours. We performed RNAscope in situ hybridisation (RISH) to localize the viruses. Parvovirus-specific IgG was analysed by enzyme immunoassay and targeted next-generation sequencing (NGS) was applied to detect CuV in plasma.
Results
We detected viral DNA in 15/24 (63%) tumours; nine of EBV, six of human herpesvirus (HHV) -7, four each of HHV-6B and parvovirus B19, two of cytomegalovirus, and one each of CuV and Merkel-cell polyomavirus. We found tumours with up to four viruses per tumour, and localized CuV and EBV DNAs by RISH. Two of the ten plasma samples exhibited CuV IgG, and one plasma sample demonstrated CuV viremia by NGS.
Conclusion
Viruses were frequent findings in SL. The EBV detection rate was high in diffuse large B-cell lymphoma, and co-detections with other viruses were prevalent.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Sinonasal lymphomas (SLs) form a heterogenous entity of lymphomas of a specific anatomical location, the sinonasal tract. The classification of lymphomas follows the 5th edition of the WHO Classification of Haematolymphoid Tumours (WHO- HAEM5). Diffuse large B-cell lymphoma (DLBCL) is the most common subtype followed by extranodal natural killer/T-cell lymphoma (ENK/TL) with geographical variation in prevalence [1, 2].
Microbes cause approximately 15% of all cancers worldwide [3]. The anatomical location of the sinonasal tract is exposed to an array of microbes. Within malignant tumours of the upper respiratory tract, viruses have an evident etiological role, such as the associations of human papillomavirus (HPV) with oropharyngeal squamous cell carcinoma and Epstein-Barr virus (EBV) with lymphatic tumours, such as Burkitt lymphoma, Hodgkin lymphoma, and DLBCL [4,5,6]. Additional risk factors for lymphomas include human immunodeficiency virus (HIV), human herpesvirus-8 (HHV-8), hepatitis B and C viruses, and human T-lymphotropic virus, as well as a family history of lymphomas, immunological disorders, ionizing radiation, agricultural pesticides, and increased body-mass index in young adults [2, 7].
In addition to viruses of Papillomaviridae and Orthoherpesviridae, oncogenic viruses are found within Polyomaviridae (Merkel cell polyomavirus, MCPyV, the cause of Merkel cell carcinoma), and Hepadnaviridae (hepatitis B virus in liver cancer) [8, 9]. Oncolytic viruses, in turn, are found within Parvoviridae [10].
Many new parvoviruses have been detected in the recent decade, and their behaviour and role are under stringent investigation [11]. Cutavirus (CuV), discovered in 2016, has been associated with cutaneous T-cell lymphoma and its precursor parapsoriasis, which makes it interesting in the context of SL [12,13,14,15,16,17]. In addition to CuV, the Protoparvovirus genus includes human bufavirus (BuV) and tusavirus (TuV), and the Bocaparvovirus genus includes the human bocaviruses 1–4 (HBoV1-4) [11, 18, 19].
Other herpesviruses, such as HHV-6 and -7 are widely distributed especially in the gastrointestinal tract and its organs, as well as in the salivary glands [20, 21]. The latent viruses harboured by monocytes, macrophages and CD4+ T lymphocytes, have a unique feature of being able to modify the host-cell immune response, possibly facilitating the effects of other agents or viruses on these cells [20,21,22].
Whereas many herpes-, parvo-, and polyomaviruses are ubiquitous within the general population, the persistence and presence in various tissues and tumours vary [11, 23, 24]. Given the interesting anatomical locus with its rich microbiome, we present a study focusing on SL with the aim to screen for a broad range of DNA viruses not studied before in this context, including “new” viruses detected in the recent decade.
Materials and methods
Ethics
The research ethics committee at the Helsinki University Hospital approved the study (§31/07.03.2019) and a research permission was granted (HUS/332/2019).
Patients and clinical specimens
This was a retrospective cross-sectional study. The patients were treated at the Departments of Otorhinolaryngology—Head and Neck Surgery and Oncology, Helsinki University Hospital, Helsinki, Finland. We recorded the clinical information from hospital charts (Table 1).
We collected FFPE tumours representing 24 SL patients diagnosed during 1987–2018 from the Helsinki Biobank (permission no §73/15.05.2019, HUS/118/2019). The Helsinki Biobank provided plasma samples from seven SL patients with available FFPE tumour samples and additionally from three SL patients with no tumour samples available.
We analysed all tumours by qPCR for viral DNA, and the high-load ones by RNAscope in situ hybridisation (RISH) for cell tropism. We collected plasma samples to investigate the serological status of parvoviruses B19V, BuV, CuV and TuV, and one underwent targeted NGS.
DNA extraction
All tumour biopsies were collected in a PCR-sterile manner from FFPE tissue blocks as 2-mm punch biopsies in sterile 1.5 ml microcentrifuge tubes. The correct localization was evaluated microscopically from the representative tissue slide by the first author, and whenever in doubt, confirmed by the senior pathologist. If enough material were available, two distinct biopsies from the tumour were taken. Prior to puncturing, new HE slices were cut from paraffin blocks to ensure the punching to hit tumour tissue. DNA was extracted with QIAamp DNA FFPE Tissue Kit (Qiagen, Heiden, Germany), according to the manufacturer’s protocol, with slight modifications: the paraffin treatment with xylene was done twice and 40 µl of proteinase K was used. We eluted the DNA preps in 60 µl AE buffer and stored them at -20°C. From one plasma sample (patient L31), we extracted DNA with QIAamp DNA blood Mini Kit (Qiagen), according to the manufacturer’s protocol. The DNA yields and human cell quantity were evaluated by comparing viral loads with that of the human reference gene RNase P by qPCR [25].
Virus DNA detection by qPCR
The viruses analysed with qPCR included the herpesviruses (herpes simplex virus-1 and -2, varicella zoster virus, EBV, cytomegalovirus (CMV), HHV-6A, HHV-6B, HHV-7, and HHV-8), the polyomaviruses (BK polyomavirus (BKPyV), JC polyomavirus (JCPyV), and MCPyV), and the parvoviruses (B19V, BuV, CuV, TuV, and HBoV1–4). Details are listed in Table 2 [14, 25,26,27,28]. All qPCR reactions contained either Maxima probe qPCR Master Mix (Thermo Fischer Scientific, Pittsburgh, PA, USA) or TaqPath ProAmp master mix Thermo Fisher), as described [29]. We performed all real-time qPCR assays with AriaMx Realtime PCR System (Agilent Technologies, Santa Clara, CA, USA).
We included molecular biology grade water in all qPCR reactions as a non-template control and used ten-fold diluted plasmids (101–106), containing each viral target regions as qPCR standards and as positive controls.
Virus DNA detection by NGS
We processed one plasma sample of patient L31, in whose tissue sample we had detected high copy numbers of CuV DNA, by targeted NGS, given the unknown nature of this rather newly discovered virus. The viruses targeted with the NGS are listed in Table 2. We fragmented the DNA mechanically and prepared the libraries with unique double indexes and performed in solution capture-targeted enrichment of the viral DNAs [29, 30]. The enriched library was sequenced, at the FIMM genomics unit of the University of Helsinki, with Novaseq 6000 (S1, PE151 kit; Illumina). The pipeline was validated as described in Pratas et al. [31] and has been tested on a wide range of materials, including human tissues, bone, serum/plasma, and formalin-fixed samples [24]. A blank extraction control was carried downstream alongside the sample throughout the entire process, spanning library preparation, enrichment, and sequencing.
NGS data analysis
The data were analysed with TRACESPipeLite, a fully automatic program [30,31,32]. When in low breadth coverage (< 15%), we manually inspected and confirmed the individual reads by BLAST. The viral reads of cutavirus were manually verified using BLAST and confirmed to match exclusively cutavirus.
In situ hybridisation
We chose five tumours for RISH; (Advanced Cell Diagnostics (ACD), Newark, CA), each representing the highest virus DNA load (3.45 × 102–2.08 × 106 cpm) of either CuV, EBV, HHV-6B, HHV-7 or B19V, and five additional virus DNA-negative SL tumours. We performed RISH for the CuV PCR-positive tumour of patient L31, the EBV PCR-positive tumour of patient L28, the HHV-6B and -7 PCR-positive tumour of patient L35, and the B19V PCR-positive tumour of patient L36. We applied RISH on 5-µm thick FFPE sections on SuperFrost glass slides with RNAscope 2.0 HD Red Chromogenic Reagent Kit (ACD) with probes targeting the regions BILF-1 (catalogue Number 44781) of EBV, U4 (870,801) of HHV-7; U36 (521,401) of HHV-6B, NS (869,391) and VP1 (872,961) of CuV and NS1 (496,871) of B19V, according to the manufacturer’s protocol. The human housekeeping gene PPIB and the bacterial gene DapB (ACD) served as positive and negative technical controls, respectively.
Immunohistochemistry (IHC) combined with RISH
To identify the host cells of CuV in the tumour of patient L31, we combined RNAscope in-situ hybridization (RISH) with fluorescent multiplex immunohistochemistry (mIHC) on 5-mm FFPE tissue sections, with multiple cellular markers on the same tissue section. The CuV probe (Probe-V-CuV-gp3-gp4-gp5 [NC_039050.1 (nt 2023-3562)], targets the sense DNA strand and mRNA of the VP gene region [15]. Probes targeting the human housekeeping gene PPIB and the bacterial gene DapB (ACD) served as positive and negative technical controls, respectively.
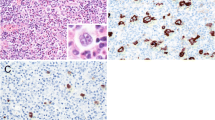
RISH (RNAscope 2.5 HD reagent kit-BROWN; [PN 322310; ACD, Newark, USA]) was employed according to the manufacturer’s protocol with slight modification. After addition of the AMP6 reagent, Alexa Fluor™ 488 Tyramide Reagent (Thermo Fisher [cat: B40953]) was applied to the tissue sections in a 1:100 dilution for 5 min, to make the RISH signal fluorescent. This was then followed by mIHC, as previously described (Blom et al., 2017), with some modifications. Briefly, primary antibodies raised in different species were applied pairwise in sequential staining rounds and detected by AlexaFluor647 and AlexaFluor750 fluorochrome-conjugated secondary antibodies. The following primary biomarker antibodies (1:200): helper T cells, (Rabbit-anti-CD4; Abcam; ab133616), cytotoxic T cells (Mouse-anti-CD8; Dako; M7103), macrophages (Mouse-anti-CD163; Thermo; MS-340) and B cells (Rabbit-anti-CD163; Abcam ab188571). DAPI was applied as a nuclear stain. After each round of staining the tissues were scanned on a Zeiss Axioscan.Z1 slide scanner. Before the next round of staining, coverslips were soaked off in wash buffer and the tissues were incubated in a bleaching solution (TBS/24 mM NaOH/4.5% H2O2) for 1 h at room temperature. Then the slides were heated in 10 mM Tris–HCl/1 mM EDTA, pH 9, for 20 min at 99°C to inactivate the primary antibodies of the previous staining round. Representative CuV RISH-mIHC- and RISH only-treated SL biopsy tissues are shown in Fig. 1.

Virus RNAscope in situ hybridisation and cell-marker immunohistochemistry on FFPE tissues of two SL patients. RNAscope 2.5 Assay-RED (a–h) or Assay-BROWN (i) in combination with the following CD markers in IHC (j–l): CD4, CD8, CD20, and CD163, as below. Counterstained with hematoxylin. Red (a–h), brown (i), and green (j–l) punctuated dots represent positive RISH signals. a CuV NS1 RISH for CuV PCR-positive SL section. b CuV NS1 RISH, same section as a., another area magnified. c A consecutive section, same histological area as in a., stained with CuV VP1. d. EBV BILF-1 RISH for qPCR EBV-positive SL section. e. CuV NS1 RISH for CuV PCR-negative SL section. f Human PPIB mRNA probe (positive control). g. Bacterial dapB probe (negative control). h EBV BILF-1 RISH for EBV PCR-negative SL section. i CuV NS1 RISH Brown for CuV PCR-positive SL section. j A consecutive section, same histological area as in (i), stained for CD4 + (red) and CD8 (light blue) T cells after CuV VP1 RISH (green punctuated dots). k Same section from different area as in (j.), stained for CD4 + (red) and CD 8 + (light blue) T cells after CuV VP1 RISH (green punctuated dots). l Same section and histological area as in (k), with a consecutive staining for CD20 + (red) B cells and CD163 (light blue) macrophages. Light grey stain shows nuclear DAPI
Parvovirus IgG enzyme immunoassay (EIA)
Seven of the 24 patients included in this study, had plasma samples stored at the Helsinki Biobank. Plasma samples but no FFPE tumour blocks were available from three additional patients with SL. We tested all the 10 plasma samples by in-house IgG EIAs, using biotinylated VP2 virus-like particles (VLP) as antigen for BuV, CuV and TuV, as well as for B19V, as previously described [33, 34]. The initial results for protoparvovirus IgGs were confirmed with a competition assay [33]. Optical densities (ODs) were measured at 450 nm (Multiskan EX; Thermo Fischer Scientific).
Results
Patient characteristics
Clinical information was available for 17 patients and unavailable for seven patients (Table 1). Overall, 50% of the patients were female. All the tumours were known to be situated in the sinonasal area, but the exact location of the tumour was not available for eight patients. In addition to SL DLBCL-NOS (NOS—not otherwise specified), patient L18 had a systemic T-cell lymphoma. Pathological reports were available for all patients, but in some cases the diagnosis did not follow the latest lymphoma classification due to old age of samples. The histological SL subtypes represented DLBCL-NOS (n = 16, 67%), mantle-cell lymphoma (MCL, n = 2, 8%), and smaller entities (Table 1). Immunostainings for EBV and ALK (anaplastic lymphoma kinase positive) were reported for some samples: samples L19 and L21 were EBNA2 and LMP1 negative, L25 was ALK unknown, L28 was positive and L34 was negative for EBER by chromogenic in situ hybridisation. It is noteworthy that there were no lymphomas of NK-cell subtype within this cohort.
The cohort included many patients with compromised immunity before the manifestation of SL (Table 1). Reasons such as malnutrition or tobacco smoking, were not reported in the patient charts.
Presence of virus DNA by qPCR
We detected virus DNA in 15/24 (63%) tumours with viral loads ranging between 4.4 and 2.1 × 106 copies per one million cells (cpm) (Table 3). Nine tumours were positive for EBV DNA (38%; 3.6 × 101–2.1 × 106 cpm). The DLBCL-NOS subtype harboured EBV DNA in 6/16 (38%) of the tumours. Both MCLs were EBV-DNA positive as well as the single anaplastic tumour.
Six tumours were positive for HHV-7 DNA (25%; 2.9 × 101–4.8 × 102 cpm), detected in almost all subtypes present in our study: DLBCL-NOS, MCL, and anaplastic lymphoma. We found B19V DNA and HHV-6B DNA in four tumours each (17%; 2.9 × 101–2.1 × 103 cpm; 4.4 × 101–3.5 × 102 cpm, respectively): B19V DNA in DLBCL-NOS, and HHV-6B in MCL, and DLBCL-NOS. We found CMV DNA in two tumours (8%; 1.6 × 102–8.7 × 102 cpm) representing MCL, and DLBCL-NOS, respectively; and detected CuV DNA (3.1 × 105 cpm) and MCPyV DNA (2.1 × 102 cpm) in one tumour each (4%): both DLBCL-NOS. The human single-copy RNase-P qPCR served for quantification of human cells with a variation of 2.3 × 103–1 × 106 (mean: 2.7 × 104) cells/ul DNA extract. All positive findings were reproducible.
Two or more different viral DNAs were co-detected in 8/24 (33%) tumours with up to four different viruses per tumour (Table 3). EBV was co-detected with other viruses in 78% of EBV-positive tumour samples, most often with HHV-7. HHV-6B, -7, and CMV were never the only viral findings in tumours but were present in combinations with other viruses; with EBV in 22%, 44%, and 100%; with B19V in 17%, 25%, and 0%; and with MCPyV in 0%, 25%, and 0% of HHV-6B, HHV-7 or CMV-positive SL samples, respectively.
Among the five patients that were immunocompromised before the onset of the lymphoma, we detected virus DNA in four. Two out of the three patients with HIV were EBV-DNA positive. A slight emphasis was noted in the overall virus DNA prevalence (63% vs 80%) and the co-detections (1.6 virus co-detections/tumour vs 1.8 co-detections/tumour), between our groups of healthy, and the previously immunocompromised individuals, respectively.
While the mean age of the patients at the time of SL diagnosis was 66 years (range 34–86), the mean age at diagnosis for the virus DNA-positive and -negative groups was 62 years (SD ± 14, range 34–84) and 73 years (SD ± 12, range 46–86), respectively. However, the age difference was not statistically significant (p = 0.06).
Presence of CuV DNA in blood plasma by NGS
As the relevance of CuV persistence is unknown and interesting, we analysed the plasma sample from the patient with CuV PCR-positive SL (SL31) with targeted NGS to investigate a possible viremia (Table 3). The plasma sample was NGS positive for CuV (seven unique reads). We could, however, not repeat the result of plasma CuV positivity with qPCR.
Location of CuV, EBV, HHV-6B and HHV-7 in tissues
To investigate EBV, we chose a high copy-number DLBCL-NOS tumour (patient L28, AIDS positive, Tables 1, 3). The EBV probe BILF-1 demonstrated scattered signals on a broad area of the tumour (Fig. 1d). In the CuV-positive tumour (patient L31), we detected repeatedly intense positive signals by both CuV probes VP1 and NS1, matching with each other on the layout, with signals in single spots or small clusters spread out unevenly throughout the tumour (Fig. 1 a-c).
When combining RISH with IHC on the same slide, we could localize CuV in CD4+ T cells, however, with fewer recognizable positive CuV spots when compared to RISH alone (Fig. 1a–c and i–l). We did not detect CD8+ , B cells or macrophages overlapping or in direct contact with CuV signals.
To detect HHV-6B and -7, and B19V, we chose tumour samples with the highest, however still modest, virus loads (2.07 × 102 cpm; patient L35 for both HHVs, 2.07 × 103 cpm for B19V), without solid findings. PCR-negative tumour controls remained all negative.
Parvovirus enzyme immunoassay
To search for signs of immune system activation, and to survey the seroprevalence of the ‘new’ protoparvoviruses, we performed protoparvovirus and B19V EIAs for all the 10 plasma samples available. Two out of the 10 plasma samples were positive for CuV-specific IgG (absorbance values 1.5 and 2.1). One of the CuV IgG-positive plasma samples belonged to patient L31, who was positive for CuV DNA both in FFPE tumour (qPCR) and in plasma (NGS). The other CuV IgG-positive plasma belonged to a SL patient with no FFPE-tumour sample available. One plasma sample was BuV2-IgG positive. The IgG prevalence of B19V was 60% (6/10).
Discussion
We explored the presence of herpes-, parvo- and polyomaviruses in SL. Our results revealed multiple viruses present in SL, often simultaneously, potentially related to the anatomical location and sensitive methods. We explored the entity further by NGS, RISH, and parvovirus EIAs for in-depth analysis.
In epidemiological studies, the mean age of SL patients is 63–68 years [1, 2], corresponding to that of our patient cohort, with a tendency towards earlier onset of disease in the virus-positive patients. No previous comprehensive SL virus-screening studies exist, but the virus-related tendency of younger age at disease onset is evident in other virus-associated tumours, such as HPV-positive oropharyngeal carcinoma and carcinoma ex pleomorphic adenoma of the salivary glands [35, 36].
We detected viral DNA in 63% of the 24 SL tumours. EBV DNA was the most prevalent virus finding (38%), followed by HHV-7 and -6B. These viruses are ubiquitous, and the primary infection occurs usually in early childhood or adolescence. EBV resides primarily in the B lymphocytes of the lymphatic system, the prevalence on the mucous membranes of the nose and nasopharynx varies between 0 and 10% [23, 37]. The EBV prevalence of 38% (6/16) in the DLBCL-NOS subtype observed in this study is highly above the range of the previously reported DLBCL EBV prevalence of less than 5% in the Western countries and of 4–14% worldwide [38, 39]. The high rate of EBV in our study could relate to the anatomical impact of the sinonasal tract compared to a systemic disease and the high sensitivity of PCR.
The incidence rate of EBV positive DLBCL in patients with HIV-infection reaches up to 20–60% [38, 39]. HIV infection, especially at the AIDS stage, may predispose to malignant transformation of B cells into DLBCL, with EBV and HCV as important etiological factors [39, 40]. Our study supports these results. We noticed an accentuated EBV involvement in our previously immunocompromised patients with HIV, HCV, or with additional primary malignancies, with an EBV-DNA positivity of 67% in their SL tumours. We were able to detect EBV throughout the tumour with RISH targeting BILF-1, a probe targeting a constitutively expressed G-protein-coupled receptor essential for EBV-mediated immunosuppression and oncogenesis [41], throughout the tumour with RISH (Fig. 1d).
In our cohort both sinonasal MCLs harboured EBV DNA. This unexpected finding contrasts with those of Carvalho et al. [42], where EBV ISH was negative in all 20 MCLs.. The discrepancy may be explained by the anatomical location, low virus loads (1.8 × 102–9.2 × 102 cpm) coupled with high sensitivity of our qPCR, and the biological diversity of MCL [42, 43]. The association between EBV and MCL remains unresolved and would benefit from larger mantle cell SL series with a high-sensitive approach.
As for the other members of the Orthoherpesviridae, HHV-7 DNA was detected in 25%, HHV-6B DNA in 17% and CMV DNA in 8% of the SL tumours. This is in line with the results of Hernandez-Losa et al. in 2005 with 15%, 27%, 4%, of HHV-7, -6B and CMV, respectively, in fresh-frozen tissue samples of any lymphoma subtypes [44]. Within healthy individuals, predominately HHV-7, and to a lesser extent HHV-6, were nearly always present in oral, and in approximately 50% of nasal swab samples [23]. Unfortunately, RISH targeting the U38 gene of HHV-6B, the product of which is associated with active replication, and the U4 gene of HHV-7 (unknown function), was not consistent probably due to low viral DNA loads or lack of active replication (Table 3).
Like other herpesviruses, HHV-6B and -7 establish life-long persistence and especially HHV-6B can reactivate in immunocompromised patients [21]. Proteins coded by HHV-6 and -7 have immunomodulatory functions by acting on HLA class I expression and chemokine receptors and thereby may weaken virus-specific immune responses by creating a microenvironment that favours viral persistence [21]. In addition, HHV-6 might facilitate and enhance the infections of HIV, HCMV, and EBV [21, 44, 45]. It is noteworthy, that EBV was almost exclusively detected in combination with other viruses, usually HHV-6B or -7 (Table 3). Two out of three patients with HIV were positive for HHV-6B or -7 as well. Therefore, the role of other viruses, such as HHV-6B and -7, could have an assisting role in disease development, but this would require larger studies for verification.
CuV is a rather recently discovered protoparvovirus [12], present in a relevant proportion of skin biopsies of cutaneous T-cell lymphoma (CTCL) 8.5–38% [13,14,15]. Therefore, we explored one DLBCL patient with a high CuV load (3.06 × 105 cpm) in the FFPE tumour. We detected CuV DNA both in the SL tumour by qPCR (Tables 1, 3) and in the plasma by NGS, showing CuV DNAemia/viremia, previously reported only in one highly immunocompromised patient [46]. We did, however, not detect CuV DNA in the plasma by our qPCR, probably due to fragmentation or mismatches in the critical primer-or probe-binding areas. RISH was positive with both CuV probes NS1 and VP1 (Fig. 1a–c). We combined chromogenic RISH with fluorescent IHC, and CuV positivity was detected in CD4+ T cells, but not in B cells or CD8+ T cells. While no definitive conclusions can be made, our findings support the idea of CuV being linked to T cells in accordance with the finding of CuV detected in CTCL [13, 14]. Further, we detected CuV IgG in 2/10 (20%) of our SL plasma samples, which is interesting when compared to the low seroprevalence in the general population of below 6%, but similar to the higher seroprevalences of 9.5% and 33% in CTCL patients [14, 17, 33]. However, our sample size is too small to make firm conclusions about the prevalence.
There are some limitations to this study. Although we have been able to demonstrate the efficient use of FFPE tumour samples in virus, DNA screening [15, 29, 36], the harsh formalin fixation process harms the DNA and may interfere with the discovery of viruses. Small tumour sample sizes and low virus loads may further affect the results. To detect virus activity, RNA studies from fresh-frozen samples would be essential. In addition, the low volume and heterogeneity of our tumours are evident, and since both diagnostics and classification of lymphomas evolve continuously, the exact subtyping according to the latest classification was not possible in this study. In addition, clinical detailed data were lacking for older patients, which could affect the results and their interpretation, especially when discussing the characteristics of virus prevalence within certain lymphoma subtypes. Neither does our cohort represent a ‘typical’ setting of lymphomas, since there were no NK-T-cell lymphomas present.
Conclusion
SLs constitute a rare entity of various lymphomas in a distinct anatomical site with proximity to a massive microbial load. The association of both DLBCL and MCL with EBV were highlighted in our cohort. The role of CMV, HHV-6 and -7, with recurring findings in SL, may provoke a disease-friendly microenvironment. The high viral loads in qPCR of a newcomer in virus research, CuV, is an intriguing finding of this study, with positive viral signals in RISH, CuV-DNA positivity in plasma, and the putative high CuV IgG seroprevalence among SL patients compared to the general population. Larger sample series focusing on aspects of virus activity would be an essential next step.
The mere presence of a virus is not proof of a causal association with disease development. However, persisting viruses might still be able to affect the cell [47]. The behaviour of the recently discovered viruses is unknown, and therefore, all insights are beneficial for future research.
Data availability
The datasets generated during and/or analyzed during the current study are not publicly available due to the sample size and rarity of these tumors. Data are available from the corresponding author on reasonable request.
References
Dubal PM, Vazquez DRA, Patel TD, Baredes S, Eloy JA (2015) A comparative population-based analysis of sinonasal diffuse large B-cell and extranodal NK/T-cell lymphomas. Laryngoscope 125:1077–1083
Eide JG et al (2023) Primary sinonasal lymphoma: a multi-institutional experience of clinical presentation, treatment, and outcomes. Int Forum Allergy Rhinol 13:1492–1502 (in eng)
Plummer M, de Martel C, Vignat J, Ferlay J, Bray F, Franceschi S (2016) Global burden of cancers attributable to infections in 2012: a synthetic analysis. Lancet Glob Health 4(9):e609–e616. https://doi.org/10.1016/S2214-109X(16)30143-7. (in eng)
Syrjänen K, Syrjänen S, Lamberg M, Pyrhönen S, Nuutinen J (1983) Morphological and immunohistochemical evidence suggesting human papillomavirus (HPV) involvement in oral squamous cell carcinogenesis. Int J Oral Surg 12(Generic):418–424. https://doi.org/10.1016/S0300-9785(83)80033-7. (in eng)
Wolf H, ZurHausen WHH, Becker V (1973) EB viral genomes in epithelial nasopharyngeal carcinoma cells. Nature 244(138):245–247 (in eng)
Vockerodt M et al (2015) The Epstein-Barr virus and the pathogenesis of lymphoma. J Pathol 235(2):312–322. https://doi.org/10.1002/path.4459
Sehn LH, Salles GA-O (2021) Diffuse large B-cell lymphoma. N Engl J Med 384(9):842–858 (in eng)
Feng H, Shuda M, Chang Y, Moore PS (2008) Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science (New York, N.Y.) 319(5866):1096–1100. https://doi.org/10.1126/science.1152586. (in eng)
Beasley RP, Hwang LY, Lin CC, Chien CS (1981) Hepatocellular carcinoma and hepatitis B virus. A prospective study of 22 707 men in Taiwan. Lancet 2(8256):1129–1133 (in eng)
Nüesch JP, Lacroix J, Marchini A, Rommelaere J (2012) "Molecular pathways: rodent parvoviruses–mechanisms of oncolysis and prospects for clinical cancer treatment. Clin Cancer Res 18(13):3516–3523. https://doi.org/10.1158/1078-0432.CCR-11-2325. (in eng)
Söderlund-Venermo M (2019) Emerging human parvoviruses: the rocky road to fame. Annu Rev Virol 6(1):71–91. https://doi.org/10.1146/annurev-virology-092818-015803. (in eng)
Phan TG et al (2016) A new protoparvovirus in human fecal samples and cutaneous T cell lymphomas (mycosis fungoides). 496: 299–305
Kreuter A et al (2018) Cutavirus infection in primary cutaneous B- and T-cell lymphoma. JAMA Dermatol 154(8):965–967. https://doi.org/10.1001/jamadermatol.2018.1628. (in eng)
Väisänen E et al (2019) Cutavirus DNA in malignant and nonmalignant skin of cutaneous T-cell lymphoma and organ transplant patients but not of healthy adults. (68(11):1904–1910, (in eng)
Mohanraj U, Konttinen T, Salava A, Väkevä L, Ranki A, Söderlund-Venermo M (2023) Significant association of cutavirus with parapsoriasis en plaques: high prevalence both in skin swab and biopsy samples. Clin Infect Dis 7(7):987–990. https://doi.org/10.1093/cid/ciad320. (in eng)
Hashida Y, Nakajima K, Higuchi T, Nakai K, Daibata M (2023) Involvement of cutavirus in a subset of patients with cutaneous T-cell lymphoma with an unfavorable outcome. J Clin Virol 165:105523 (in eng)
Mohanraj U, Väkevä L, Ranki A, Söderlund-Venermo M (2024) Prevalence, tropism, and activity of cutavirus in circulating blood lymphocytes, stool, and skin biopsy specimens of patients with cutaneous T-cell lymphoma and parapsoriasis en plaques. J Med Virol 86:11024–11030
Phan TG et al (2012) Acute diarrhea in West African children: diverse enteric viruses and a novel parvovirus genus. J Virol 86(20):11024–11030 (in eng)
Qiu J, Söderlund-Venermo M, Young NS (2017) Human parvoviruses. Clin Microbiol Rev 30(1):43–113 (in eng)
Yamanishi K, Mori Y, Pellett PE (2013) Herpesviridae > Chapter 64 - Human Herpesviruses 6 and 7. In: Knipe DM, Howley PM (eds) Fields Virology, vol. 2, 6th edn. Wolters Kluwer/Lippincott Williams & Wilkins, Philadelphia, pp 2058–2079
Agut H, Bonnafous P, Gautheret-Dejean A (2016) Human Herpesviruses 6A, 6B, and 7. Microbiol Spectrum. https://doi.org/10.1128/microbiolspec.DMIH2-0007-2015. (in eng)
Eliassen E et al (2018) Human herpesvirus 6 and malignancy: a review. Front Oncol 8:512. https://doi.org/10.3389/fonc.2018.00512. (in eng)
Wylie KM, Mihindukulasuriya KA, Zhou Y, Sodergren E, Storch GA, Weinstock GM (2014) Metagenomic analysis of double-stranded DNA viruses in healthy adults. BMC Biol 12:71 (in eng)
Pyöriä L, Pratas D, Toppinen M, Hedman K, Sajantila A, Perdomo M (2023) Unmasking the tissue-resident eukaryotic DNA virome in humans. Nucleic Acids Res 51(7):3223–3239 (in eng)
Toppinen M et al (2015) A new quantitative PCR for human parvovirus B19 genotypes. J Virol Methods 218:40–45 (in eng)
Pyöriä L et al (2020) HERQ-9 is a new multiplex PCR for differentiation and quantification of all nine human herpesviruses. mSphere 5(3):e00265-e320. https://doi.org/10.1128/mSphere.00265-20. (in eng)
Goh S, Lindau C, Tiveljung-Lindell A, Allander T (2009) Merkel cell polyomavirus in respiratory tract secretions. Emerg Infect Dis 15(3):489–491 (in eng)
Kantola K et al (2010) Real-time quantitative PCR detection of four human bocaviruses. J Clin Microbiol 48(11):4044–4050. https://doi.org/10.1128/JCM.00686-10. (in eng)
Jauhiainen M et al (2023) Herpesviruses, polyomaviruses, parvoviruses, papillomaviruses, and anelloviruses in vestibular schwannoma. J Neurovirol 29(2):226–231 (in eng)
Toppinen M et al (2020) The landscape of persistent human DNA viruses in femoral bone. Forensic Sci Int Genet 48:102353 (in eng)
Pratas D, Toppinen M, Pyöriä L, Hedman K, Sajantila A, Perdomo MF (2020) A hybrid pipeline for reconstruction and analysis of viral genomes at multi-organ level. GigaScience 9:1-11. https://doi.org/10.1093/gigascience/giaa086. (in eng)
Pratas D et al (2018) Metagenomic composition analysis of an ancient sequenced polar bear jawbone from Svalbard. Genes (Basel) 9(9):445. https://doi.org/10.3390/genes9090445. (in eng)
Väisänen E et al (2018) Global Distribution of Human Protoparvoviruses. Emerg Infect Dis 24(7):1292–1299 (in eng)
Kaikkonen L et al (1999) Acute-phase-specific heptapeptide epitope for diagnosis of parvovirus B19 infection. J Clin Microbiol 37(12):3952–3956 (in eng)
Marur S, D'Souza G, Westra WH, Forastiere AA (2010) HPV-associated head and neck cancer: a virus-related cancer epidemic. Lancet Oncol 11(8):781–789 (in eng)
Jauhiainen MK et al (2021) The presence of herpesviruses in malignant but not in benign or recurrent pleomorphic adenomas. Tumour Biol 43(1):249–259. https://doi.org/10.3233/TUB-211519
Adham M et al (2013) Epstein-Barr virus DNA load in nasopharyngeal brushings and whole blood in nasopharyngeal carcinoma patients before and after treatment. Clin Cancer Res 19(8):2175–2186 (in eng)
Chabay P (2021) Advances in the pathogenesis of EBV-associated diffuse large B cell lymphoma. Cancers 13(11):2717 (in eng)
Shannon-Lowe C, Rickinson AB, Bell AI (2017) Epstein-Barr virus-associated lymphomas. Philos Trans R Soc Lond B Biol Sci 372(1732):20160271. https://doi.org/10.1098/rstb.2016.0271. (PMID:28893938;PMCID:PMC5597738)
Horgan D et al (2022) Tackling mantle cell lymphoma in Europe. LID 10(9):1682. https://doi.org/10.3390/healthcare10091682. (in eng)
Tsutsumi N et al (2021) Structural basis for the constitutive activity and immunomodulatory properties of the Epstein-Barr virus-encoded G protein-coupled receptor BILF1. Immunity 54(7):1405-1416.e7 (in eng)
Carvalho MVR et al (2023) Mantle cell lymphoma involving the oral and maxillofacial region: a study of 20 cases. Oral Surg Oral Med Oral Pathol Oral Radiol 135(1):101–109 (in eng)
Shah BD, Martin P, Sotomayor EM (2012) Mantle cell lymphoma: a clinically heterogeneous disease in need of tailored approaches. Cancer Control 19(3):227–235 (in eng)
Hernández-Losa J et al (2005) Lack of association of polyomavirus and herpesvirus types 6 and 7 in human lymphomas. Cancer 103(2):293–298 (in eng)
Munawwar A, Singh S (2016) Human herpesviruses as copathogens of HIV infection, their role in HIV transmission, and disease progression. J Lab Phys 8(1):5–18 (in eng)
Zanella M et al (2021) Unmasking viral sequences by metagenomic next-generation sequencing in adult human blood samples during steroid-refractory/dependent graft-versus-host disease. Microbiome 9(1):28 (in eng)
Xu M et al (2022) Prevalence, cell tropism, and clinical impact of human parvovirus persistence in adenomatous, cancerous, inflamed, and healthy intestinal mucosa. Front Microbiology 24(13):914181 (in eng)
Acknowledgements
We thank the following persons: Pia Saarinen for assistance in tumour handling, Noora Keski-Säntti for assistance in clinical data collection, and Outi Mielonen, Man Xu, Sally Chesnut, and Lari Pyöriä for valuable guidance and assistance in laboratory work. We thank Annabrita Schoonenberg for assistance with IHC and the FIMM Digital Microscopy and Molecular Pathology Unit supported by Helsinki University and Biocenter Finland.
RISH Images were generated using 3DHISTECH Panoramic 250 FLASH II digital slide scanner at Genome Biology Unit supported by HiLIFE and the Faculty of Medicine, University of Helsinki, and Biocenter Finland.
Funding
Open Access funding provided by University of Helsinki (including Helsinki University Central Hospital). The Helsinki University Hospital research fund (STS), the University of Helsinki doctoral programme (MJ), the Sigrid Jusélius Foundation (MSV), the Life and Health Medical Support Association (MSV), the Finnish-Norwegian Medical Foundation (UM), the Ida Montin Foundation (UM), and the Viral Disease Research Foundation (UM).
Author information
Authors and Affiliations
Contributions
M.J.K, M.S.-V., and S.T.S performed study concept and design. M.J.K and U.M. performed development of methodology and analysis of the data. M.J.K performed acquisition of the data and writing, review, and revision of the paper. S.T.S, A.M., and M.S.-V. supervision of the data. All authors read, revised, and approved the final paper.
Corresponding author
Ethics declarations
Conflict of interest
The authors report no conflict of interest to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jauhiainen, M.K., Mohanraj, U., Perdomo, M.F. et al. Presence of herpesviruses, parvoviruses, and polyomaviruses in sinonasal lymphoma. Eur Arch Otorhinolaryngol 281, 4201–4211 (2024). https://doi.org/10.1007/s00405-024-08702-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00405-024-08702-0