
Abstract
Purpose
This study is a systematic review of the literature which seeks to evaluate auditory and quality of life (QOL) outcomes of cochlear implantation in patients with Usher syndrome.
Methods
Systematic review of studies indexed in Medline via PubMed, Ovid EMBASE, Web of Science, CENTRAL and clinicaltrials.gov was performed up to March 9th 2022, conducted in accordance with the PRISMA statement. Patient demographics, comorbidity, details of cochlear implantation, auditory, and QOL outcomes were extracted and summarized.
Results
33 studies reported over 217 cochlear implants in 187 patients with Usher syndrome, comprising subtypes 1 (56 patients), 2 (9 patients), 3 (23 patients), and not specified (99 patients). Auditory outcomes included improved sound detection, speech perception, and speech intelligibility. QOL outcomes were reported for 75 patients, with benefit reported in the majority.
Conclusions
Many patients with Usher syndrome develop improved auditory outcomes after cochlear implantation with early implantation being an important factor.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Usher syndrome, also known as Hallgren syndrome, Usher-Hallgren syndrome, retinitis pigmentosa-dysacusis syndrome, and dystrophia retinae dysacusis syndrome, is an autosomal recessive hereditary ciliopathy characterized by partial or complete sensorineural hearing loss and progressive visual loss due to retinitis pigmentosa [1]. It affects 3–6 persons per 100,000, and in the United States accounts for approximately 50% of all deaf-blindness [2].
Mutations in at least 10 genes thought to account for Usher syndrome are present in both inner ear hair cells and retinal photoreceptors, where they lead to loss of hair cells in the cochlea and progressive loss of rod and cone photoreceptors in the retina [1]. Most patients retain central vision of around 20/40 until age 40 [1]. Peripheral visual losses impair the use of sign language to mitigate communication challenges in this population. Notably, auditory nerve function, gross anatomy and intellectual ability are not typically affected, making these patients good candidates for cochlear implantation.
It is important that patients and their families can make an informed decision based on the best available evidence on cochlear implantation outcomes. Usher syndrome affects several aspects of a patient’s life, many of which cannot be accurately assessed by audiometric tests alone. For instance, the impact of Usher syndrome on a patient’s schooling needs to be considered when clinicians counsel patients and their families about cochlear implants and this knowledge can provide useful insight for teachers educating patients with Usher syndrome also. The use of validated Patient Reported Outcome Measures (PROMs) in measuring cochlear implant outcomes provides further insight into the effects of Usher syndrome on a patient’s quality of life and can also be a useful tool for assessing treatment effectiveness. To our knowledge, there are no systematic reviews in the literature at present that assesses the audiometric outcomes, schooling and PROMs for cochlear implants in patients with Usher syndrome.
Three clinical subtypes of Usher syndrome are recognized, distinguished by severity of hearing loss, age of onset of symptoms, and vestibular involvement (Online Resource 1).
Type 1 is characterized by pre-lingual hearing loss and early onset visual loss and early cochlear implantation in this group has been associated with better auditory outcomes in children with severe or profound congenital hearing loss [3].
Pre-lingual hearing loss in type 2 is mild to severe with preservation of low-frequency hearing, often amenable to amplification by acoustic hearing aids [4].
Type 3 is the least common, accounting for 4% of cases [1]. Progressive sensorineural hearing loss and visual loss develop post-lingually in late childhood, adolescence, or adulthood. Acoustic hearing aids and, more uncommonly, cochlear implants are treatment options in this population, but optimal timing is unclear.
Vestibular function may sometimes be negatively affected by cochlear implantation. However, there is very little evidence of implant-associated vestibular deterioration in Usher syndrome [5].
Objective
In this systematic review, we provide an overview of auditory and PROMs after cochlear implantation in patients with a confirmed diagnosis of Usher syndrome. We aim to provide clinicians with a synthesis of evidence with which to counsel patients and their families on the range of outcomes a patient with Usher syndrome may experience following cochlear implantation.
Methods
Study identification and selection
We conducted a systematic literature search using the databases PubMed, Ovid EMBASE, ISI Web of Science, the Cochrane Register of Controlled Trials and ClinicalTrials.gov from inception to March 9th 2022 using the MeSH and key search terms: cochlear implant*, electric acoustic stimulation, hearing aid, Usher syndrome and derivatives (Online Resource 2). Search results were manually deduplicated. The review was conducted in accordance with the PRISMA statement.
Abstracts were screened for relevance by two independent reviewers (HLC and CML). Two reviewers (two of HLC, CML, DC) independently evaluated full texts for inclusion using predetermined eligibility criteria. Bibliographies of included studies were searched for additional relevant studies. Discrepancies were resolved through consensus or consultation with a third reviewer.
Inclusion and exclusion criteria
PICO eligibility criteria were used:
-
Population: adults or children with a confirmed diagnosis of Usher syndrome.
-
Intervention: cochlear implant, unilateral or bilateral.
-
Comparison: any or none.
-
Outcome: post-implantation (1) objective measurements of sound detection, speech recognition, speech intelligibility, and/or (2) PROM scores including quality of life measures.
Exclusion criteria: (1) animal study, (2) pharmacological model, (3) genetic diagnosis of Usher syndrome without associated phenotype, (4) mixed groups where data from patients with Usher syndrome cannot be extracted, (5) opinion, editorial or review article, (6) non-English language, (7) full text unavailable.
Studies from the same institution were assessed using treatment dates and authorship to determine the likelihood of overlapping datasets and those deemed to be at high risk are grouped in this review.
Data extraction and synthesis
Two reviewers independently extracted data from included studies using standardized Microsoft Excel (Redmond, WA) spreadsheets piloted during our preliminary literature search, that were compared to ensure accuracy. Inconsistencies were resolved through consensus. Mean and range data for patient age, outcome data and time to last follow-up were recorded by preference where available. Where figure resolution permitted accurate extraction, graphical data were used.
Quality assessment
Two reviewers independently scored included studies for evidence quality using the OCEBM 2011 Levels of Evidence 2.1 grading system [6]. Each study was assessed using the 2012 risk of bias checklist for quality assessment of non-randomized studies (adapted for case series where required) [7].
The protocol for this systematic review was registered prospectively in the PROSPERO database and can be found at https://www.crd.york.ac.uk/PROSPERO/display_record.php?RecordID=185102.
Results
Thirty-three publications met our inclusion and exclusion criteria after full-text screening (Fig. 1), representing the experiences of 187 patients with Usher syndrome and cochlear implant(s). Diagnoses included Usher type 1 (56 patients), Usher type 2 (9 patients), Usher type 3 (23 patients), and Usher of non-specified type (99 patients). 120 unilateral and 29 bilateral CI insertions were reported; five studies did not specify whether CI insertion was unilateral or bilateral (Table 1).

Preferred reporting items for systematic reviews and meta-analyses flow diagram. From: Page MJ, McKenzie JE, Bossuyt PM, Boutron I, Hoffmann TC, Mulrow CD, et al. The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. BMJ 2021;372:n71. https://doi.org/10.1136/bmj.n71. For more information, visit: http://www.prisma-statement.org/
Twenty-seven studies were published in otolaryngology journals, four in ophthalmology journals, and two in general medical journals. Included studies represent diverse contributions from around the world. Studies were conducted in the USA (seven), The Netherlands (five), UK (four), France (three), Saudi Arabia (three), Australia (two), Sweden (two), Brazil (one), Colombia (one), Finland (one), Germany (two), India (one), Japan (one), Spain (one), and Turkey (one). The majority (63%) were retrospective single-center case reports, case series or cohort studies. There were no randomized studies, as might be expected for severe to profound deafness. Four studies from The Netherlands were deemed likely to report from an overlapping dataset and are therefore reported together in this systematic review.
Auditory outcomes
Sound detection
Audiometry data was reported by 16 studies, four pre-implantation [8,9,10,11], four post-implantation [12,13,14,15], and eight both pre- and post-implantation [8, 16,17,18,19,20,21,22] (Online Resource 3). Where reported, pre-operative PTA4 (pure tone average 0.5–4 kHz) was above 90 dB HL across all studies, and all patients exhibited improved sound detection post-operatively.
Five studies [16,17,18,19, 22] of Usher type 1 representing twenty-eight patients reported both pre- and post-implant auditory thresholds. Mean pre-implant PTA4 was 113 dB HL. Mean post-implant aided PTA4 was 37.3 dB HL (range 10–50 dB HL).
One study [20] of Usher type 2 representing eight patients reported a mean pre-implant PTA4 of 98 dB HL, and mean post-implant aided PTA4 of 34 dB SPL.
One study [21] of Usher type 3 representing nineteen patients reported a mean pre-implant PTA4 of 110 dB HL, and mean post-implant aided PTA4 of 34 dB HL.
Speech perception
Speech perception data was reported by 30 studies [8,9,10, 12, 13, 15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39], using a wide variety of assessment tools (Online Resource 3). Most patients demonstrated improved speech perception, either closed or open set recognition. At least 19 patients achieved no demonstrable improvement in speech perception at follow-up: all were diagnosed with Usher type 1 or were pre-lingually deaf [22, 23, 29, 30, 35], two had comorbid cognitive conditions unrelated to Usher syndrome [35], and one originated from a country that did not speak the language spoken by rehabilitation center staff [29]. Of those without additional comorbidities or language barriers, eight were adults [22, 30], four were adolescents [22, 30], and four were children implanted after the age of 5 years [22, 23, 30]. Four patients became non-users of their device [22, 23]. Hinderink et al. [18] found no statistically significant differences in cochlear implant outcomes between Usher type 1 and non-syndromic pre-lingually deaf patients—the sample size of the Usher (four patients) and non-Usher group (five patients) in this study was small and the cause of deafness in the non-Usher group was limited to three patients with meningitis, one with Mondini dysplasia and one patient with a hereditary cause.
All patients with Usher type 2 for whom speech perception was reported demonstrated open-set speech recognition post-implantation [15, 20], with seven patients demonstrating improved accuracy in best-aided conditions [20]. In a single study, there was no significant difference in speech perception between patients with Usher type 2a and non-visually impaired patients with post-lingual hereditary deafness [20].
Both studies (representing 20 patients) that reported speech perception post-implantation in patients with Usher type 3 reported significant improvement in accuracy of open-set speech perception[21, 29].
Speech production
Speech intelligibility was reported by seven studies [9, 12, 13, 24, 27, 29, 36], four of which reported data as per a classification system: Central Institute for the Deaf Speech Intelligibility Evaluation (CID-SPINE) [36], Speech Intelligibility Rating (SIR) [12, 24], or categories of speech production [29] (Online Resource 3).
Two studies [29, 36] reported pre- and post-implantation speech intelligibility. Improvement was reported in 10 of 12 children (83%) without complex sentence production pre-implantation. From spared words (1 child) or no production (9 children), 50% could produce complex sentences at 9–96 months follow-up [29]. No patients experienced a decline in speech intelligibility post-implantation.
Post-implantation SIR scores were reported to improve with time, from 1.06 at 3 months, to 4.3 at 12 months follow up (mean value for 27 children implanted before 6 years old) [24]. Alzhrani et al. [12] reported a mean SIR score of 4.9 (range 4–5) for 9 children (unspecified duration of follow-up).
Schooling
Three studies [17, 18, 22] reported pre-implantation schooling environments for children with Usher type 1: school for the deaf or special school (10 children) [18,19,20,21,22], mainstream school with interpreter (2 children) [17], or mainstream school (interpreter use not specified, 4 children) [22] (Online Resource 3).
Post-implantation, Imtiaz et al. [19] reported that all three of the children with Usher type 1 in their study were in mainstream education, and one of the four patients previously enrolled in a school for the deaf reported by Hinderink et al. [18] was enrolled in mainstream school, sometimes requiring the help of an interpreter (Online Resource 3).
Of those children with Usher type not specified who were of formal education age at follow-up, reported post-implantation schooling included: mainstream school (6 children) [12, 37, 38], mainstream school with sign-language support (1 child) [36], hearing-impaired unit in mainstream school (3 children) [12], school for the deaf (1 child) [36] (Online Resource 3).
In all other studies, educational setting was not reported.
Communication mode
Communication preferences were reported in three studies pre-implantation [18, 26, 27], five studies post-implantation [12, 35, 37, 40], and four studies both pre- and post-implantation [8, 16, 22, 31] (Online Resource 3).
Among patients with Usher type 1, pre-implantation communication preferences included oral (5 patients) [16, 22], combination oral/sign (2 patients) [22], sign (9 patients) [16, 22], total or lip-reading (6 patients) [16, 18], and no language (1 patient) [22]. Post-implantation, communication preferences included auditory-oral (9 patients) [16, 22, 40], combination oral/sign (5 patients) [22, 40], sign (5 patients) [22], total or lip-reading (7 patients) [16]. Of the two studies reporting both pre- and post-implant data, Liu et al. [16] report uptake of total amongst the children who used sign pre-implantation, and Hoshino et al. [22] report uptake of supplementary sign in two children who underwent late implantation: one oral child and one child without language.
Pre- but not post-implantation communication preference is reported for four pre-lingually deaf patients with Usher of non-specified type: two used cueing supplement [26], one used sign [27], and one used total [26].
Post- but not pre-implantation communication preference is reported for 44 pre-lingually deaf children and one child with progressive hearing loss with Usher of non-specified type: 30 used auditory-oral [12, 23, 35, 37], two used combination oral/sign [23], two used sign [23], two used a primarily oral form of total [35], two used a primarily manual form of total [35], and two used augmentative communication [35].
Two adult post-lingually deafened patients with Usher of non-specified type communicated via palm writing with [8] or without [31] Braille alphabet pre-implantation, and by auditory-oral communication at post-implant follow-up, including telephone use in one case [8].
Four patients became non-users of their device (7% of USH1, 2% of total population sampled) [22, 23].
No data regarding communication preference was available for patients with Usher types 2 and 3.
Electroacoustic stimulation
Two patients with Usher syndrome type 2a reported the use of hearing aids for electro-acoustic stimulation in the ear that received a cochlear implant [20]. The authors reported no statistically significant difference in speech perception in cochlear implant-only vs. hearing aid-assisted states using the NVA open speech recognition test.
PROMs and QOL measures
Seven studies [11, 18, 20, 21, 24, 30, 33] reported PROMs or quality of life (QOL) measures collected via formalized surveys, comprising responses from 75 patients post-implantation: 18 with Usher type 1 [18, 30, 33], 8 with Usher type 2a [20], 22 with Usher type 3 [11, 21], and 27 children with Usher type not specified [24] (Table 2). Survey types included the Gestel-Nijmegen Implant Questionnaire [18], Glasgow Benefit Inventory or Glasgow Children’s Benefit Inventory [20, 21, 24, 30], Nijmegen Cochlear Implant Questionnaire [20, 33], Usher Lifestyle Survey [20, 33], 12-Item Short Form Survey [33], Glasgow Health Status Inventory [21], Health of Equal Terms [13], Hospital Anxiety and Depression Scale [11], and Health Utility Index [24].
All studies using relevant measures reported benefits in hearing-related QOL (HR-QOL) and independence. On general (health and social) QOL surveys such as the Glasgow Benefit Inventory, responses were more varied. General QOL negatively correlated with age of implantation and speech perception among patients with Usher type 1, with two patients reporting no improvement or detriment to their QOL [30]. Compared to non-implanted adult patients with Usher type 1, statistically significant benefits in the HR-QOL domains of sound perception and speech production were noted—the former with greater benefit if implanted in childhood, the latter only among patients implanted in childhood [33]. It must be noted that the sample size in this study was very small. Comparing children implanted aged 1–6 years with and without Usher syndrome at 1-year follow-up, Nair et al. [24] noted that HR-QOL increased to a lesser extent among children with Usher syndrome, a result the authors assigned to the presence of multiple sensory handicaps.
Amongst patients with Usher type 2a and Usher type 3, general QOL was consistently improved following implantation [20, 21] and reported mental health and social trust problems in three patients with Usher type 3 were fewer than reported by twelve Usher type 3 patients without cochlear implants [11]. Amongst patients with Usher type 3, no correlation was identified between Glasgow Benefit Inventory score and patient age, age of implantation, or speech or sound perception threshold [21].
Complications
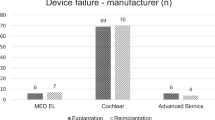
Complications were reported in eight patients, including significant intraoperative bleeding in the youngest child, implanted age 5 months [13], facial nerve stimulation [30], electrode deactivation due to short-circuit[26], implant failure [18, 27, 30, 33], reimplantation [12, 18, 27, 33, 39], and auditory hallucinations in the context of Charles–Bonnet syndrome [14] (Table 1).
Rehabilitation
Fourteen studies reported some form of structured auditory rehabilitation program [8, 12, 16,17,18,19,20, 23,24,25,26,27,28,29], of which four were integrated with an ongoing educational program or schooling [17, 18, 25, 26]. Only one center offered a short (two-week) intensive inpatient auditory rehabilitation course prior to outpatient training as standard [18, 25]. Program lengths varied between one and three years’ post-implantation [12, 18, 23, 24], with reported session frequency weekly [26], fortnightly [23] or 2-monthly [18, 25], and further appointments given according to need [12, 23]. Two pre-lingually deaf patients received pre-implantation auditory-oral therapy [16]. Audioverbal habilitation programs were diverse, comprising speech production, speech reading, auditory training in quiet and group conversation, comprehension and language development, and telephone use [18, 19, 25, 26], with an emphasis on auditory cues in some centers [16] and combined auditory-visual cues in others [18, 25, 26].
Success was partially attributed to personal motivation [8], with parents acting as facilitators of change in studies of implantation in childhood [12, 26]. Children unable to attend the centers regularly due to geographical distance or comorbidity were disadvantaged [23, 26]. Increased time and labor requirements in the assessment and rehabilitation of patients with Usher syndrome compared to normally sighted patients were reported by three studies [8, 27, 28], with two studies [8, 28] reporting a need for tactile communication techniques and interpreters. One study [29] reported no difficulties in delivering speech therapy despite the visual disturbance, but the extent of visual impairment was less severe in their population.
Quality Assessment
All included studies were graded at level 4 of the Oxford CEBM Levels of Evidence. Study quality was generally modest with deficiencies largely attributed to retrospective data collection and lack of clarity regarding patient representativeness (Fig. 2). Heterogeneity of reported outcomes precluded meta-analysis.

Risk of bias assessment using Brazzelli tool [7] and OCEBM grading [6]. Key: Y = yes, N = no, U = unclear. 1. Were participants a representative sample selected from a relevant patient population (e.g. randomly selected from those seeking treatment despite age, duration of disease, primary or secondary disease and severity of disease)? 2. Were the inclusion/exclusion criteria of participants clearly described? 3. Were participants entering the study at a similar point in their disease progression (i.e. severity of disease)? 4. Was selection of patients consecutive? 5. Was data collection undertaken prospectively? 6. Were the groups comparable on demographic characteristics and clinical features? 7. Was the intervention (and comparison) clearly defined? 8. Was the intervention undertaken by someone experienced at performing the procedure? (‘Yes’ if the practitioner received training on conducting the procedure before or conducted same kind of procedure before (i.e. no learning curve).) 9. Were the staff, place and facilities where the patients were treated appropriate for performing the procedure (e.g. access to back-up facilities in hospital or special clinic)? 10. Were any of the important outcomes considered (i.e. on clinical effectiveness, cost-effectiveness, or learning curves)? 11. Were objective (valid and reliable) outcome measures used, including satisfaction scale? 12. Was the assessment of main outcomes blind? 13. Was follow-up long enough (≥ 1 year) to detect important effects on outcomes of interest? 14. Was information provided on non-respondents, dropouts? (‘No’ if participants were those whose follow-up records were available (retrospective).) 15. Were the characteristics of withdrawals/dropouts similar to those that completed the study and therefore unlikely to cause bias? ('Yes’ if no withdrawal/dropout; ‘No’ if dropout rate ≥ 30% or differential dropout (e.g. those having most severe disease. 16. Was length of follow-up similar between comparison groups. 17. Were the important prognostic factors identified (e.g. age, duration of disease, disease severity)? (‘Yes’ if two or more than two factors were identified.). 18. Were the analyses adjusted for confounding factors?
Discussion
This review identified post-implantation auditory and QOL outcomes for 186 patients with Usher syndrome, complementing and updating a previous systematic review on cochlear implant outcomes that included literature up to November 2019 [41] and provides an insight into the impact of cochlear implant on patient’s schooling. Our results suggest that good audiometric and quality of life outcomes can be expected in the majority of patients with Usher types 1, 2 and 3. Our review was, however, limited by the quality of available evidence in the literature particularly for patients with Usher types 2 and 3 where only a limited number of studies reported audiometric outcomes.
For patients with Usher type 1, the extent of implant success after implantation in pre-lingually deafened patients was highly variable. Much of this may be accounted for by the effect of patient age at implantation. Several studies of non-syndromic pediatric patients have identified a transition period between the ages of 24–36 months after which implantation results in relatively inferior perceptive and communicative outcomes [3]. Amongst studies of Usher syndrome, Loundon et al. [29] reported better perceptive results in children implanted below the age of nine years. Amongst pre-lingually deaf patients implanted as adolescents or adults, few achieved open-set speech perception [16, 18, 22, 26, 29, 33, 35], consistent with outcomes reported from non-syndromic populations [42]. Auditory outcomes appear similar to those of pre-lingually deaf patients with normal vision [3, 29, 30, 42]. Henricson et al. [40] found that auditory information processing performance in children with Usher type 1 was similar to that of children with normal hearing, except in tests of phonological working memory and lexical skill. In these subtests, they generally performed better than non-Usher cochlear implant users, but poorer than normal hearing and hearing aid-assisted children, likely due to the relatively earlier implantation of children with diagnosed Usher syndrome in their cohort. The improved outcomes with early implantation seen in this review suggests that genetic screening of children who are born profoundly deaf is important to enable children with Usher syndrome to be identified early and allow for effective counselling for patients and their families regarding the treatment options available.
Effects on speech perception and quality of life were more consistent among post-lingually deaf patients with Usher syndrome, with all patients achieving open-set speech recognition. Auditory outcomes are similar to those reported by other studies of post-lingually deafened adults [43]. Among post-lingually deafened adults, QOL benefit of implantation for patients with Usher syndrome was found to be comparable to that of non-Usher patients without visual deficits in two of the studies included in this review [21, 44]. Rehabilitation practices were generally reported poorly, if at all. Janeschik et al. [10] found that in the first three years after implantation, children with Usher syndrome had worse post-implant hearing outcomes compared to other children with hereditary hearing loss but at 48 months after implantation, results between the two groups were comparable. The team attributed this relatively slow improvement in auditory-oral ability to severe progressive visual loss experienced by patients with Usher syndrome. However, the number of Usher syndrome patients in the study were small and rehabilitation efforts were confounded by underlying cognitive deficits and bilingual education in the Usher syndrome group. In a separate study, Hinderink et al. [18] identified a plateau in auditory abilities amongst one adolescent and adults with Usher type 1 beyond 12 months post-implant, consistent with studies of non-syndromic pre-lingually deaf adolescents and adults [45, 46].
None of the studies included in this review identified any correlation between the genotype and post-implantation outcomes. Reported outcome data were sufficiently heterogenous as to preclude meta-analysis of auditory outcomes and quality of life with respect to age at implantation or visual impairment.
Eight studies [12, 17,18,19, 22, 36,37,38] reported educational placement. As recent changes in public policy favor the integration of children with disabilities into mainstream schools where patients are more likely to achieve language development comparable to that of normal-hearing peers [47], the extent of language development reported may be an underestimation of contemporary outcomes in children with Usher syndrome and cochlear implants. Whilst several studies report of individual cases that have successfully enrolled at mainstream schools, no studies report both pre- and post- implantation educational placement for the same patient cohort. This highlights a gap in the published literature of whether cochlear implantation not only improves audiometric outcomes but whether this translates into improvements in educational outcomes. The seven studies identified in this review that reported on QOL measures would suggest improvements in educational outcomes to be present as improvements in QOL post-implantation were found in the majority of cases. There was, however, data that suggested that general QOL negatively correlated with age of implantation and speech perception among patients with Usher type 1 [30]. As cochlear implant costs are estimated to be over $1 million US dollars over a lifetime [48], further research into quantifying educational outcomes post-cochlear implantation in this population may be useful for policy makers, clinicians and patients/parents in making an informed decision regarding cochlear implantation.
The modest quality of included studies is a limitation of this review that reflects the low prevalence of Usher syndrome in the general population and varied reporting practices. Though not possible to quantify, we suspect that some reporting bias in favor of extreme results will be present, particularly among case studies and smaller case series. Future studies would benefit from the use of standardized international reporting measures for auditory outcomes and quality of life, and systematic reporting of educational placement, communication preferences, and educational attainment. Studies should also report data regarding non-implanted patients with Usher syndrome assessed at their center to better determine the representativeness of the implanted cohort. Multi-center collaboration may identify larger cohorts of patients with Usher syndrome, from which more confident estimations of effect may be inferred.
Conclusion
Where reported, cochlear implantation was found to be beneficial in improving sound detection, speech perception, speech intelligibility, and quality of life in the majority of patients with Usher syndrome. As clinical practice has evolved to emphasize early, bilateral implantation and access to oral education it is likely that the outcomes reported in this systematic review underestimate the potential benefits of cochlear implantation among young children with Usher syndrome. Clearly, early identification and implantation is even more important in the prelingually deaf Usher group, as they are likely to proceed to visual loss in time, with resulting multisensory deficits if cochlear implant outcomes are poor due to late implantation.
Data availability
Not applicable.
References
Tsang SH, Aycinena ARP, Sharma T (2018) Ciliopathy: Usher syndrome. Adv Exp Med Biol 1085:167–170. https://doi.org/10.1007/978-3-319-95046-4_32
Lentz J, Keats BJB (1993) Usher syndrome type I. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A (eds) GeneReviews®. University of Washington, Seattle, WA
Forli F et al (2011) Systematic review of the literature on the clinical effectiveness of the cochlear implant procedure in paediatric patients. Acta Otorhinolaryngol Ital 31(5):281–298
Toms M, Pagarkar W, Moosajee M (2020) Usher syndrome: clinical features, molecular genetics and advancing therapeutics. Ther Adv Ophthalmol. https://doi.org/10.1177/2515841420952194
Thierry B et al (2015) Cochlear implantation and vestibular function in children. Int J Pediatr Otorhinolaryngol 79(2):101–104. https://doi.org/10.1016/j.ijporl.2014.11.002
OCEBM Levels of Evidence Working Group et al (2011) The Oxford levels of evidence 2. Oxford Centre for Evidence-Based Medicine. https://www.cebm.ox.ac.uk/resources/levels-of-evidence/ocebm-levels-of-evidence. Accessed May 2020.
Brazzelli M et al (2015) Collagenase clostridium histolyticum for the treatment of Dupuytren’s contracture: systematic review and economic evaluation. Health Technol Assess 19(90):1–202. https://doi.org/10.3310/hta19900
Derinsu U, Ciprut A (2002) Cochlear implantation in a patient with Usher’s syndrome. Marmara Med J 15(4):258–261
Withers SJ, Gibson WP, Greenberg SL, Bray M (2011) Comparison of outcomes in a case of bilateral cochlear implantation using devices manufactured by two different implant companies (Cochlear Corporation and Med-El). Cochlear Implants Int 12(2):124–126. https://doi.org/10.1179/146701010X12711475887315
Janeschik S, Teschendorf M, Bagus H, Arweiler-Harbeck D (2013) Influence of etiologic factors on speech perception of cochlear-implanted children. Cochlear Implants Int 14(4):190–199. https://doi.org/10.1179/1754762812Y.0000000017
Wahlqvist M, Moller C, Moller K, Danermark B (2016) Implications of deaf blindness: the physical and mental health and social trust of persons with Usher Syndrome Type 3. J Vis Impair Blind 110(4):245–256
Alzhrani F, Alhussini R, Hudeib R, Alkaff T, Islam T, Alsanosi A (2018) The outcome of cochlear implantation among children with genetic syndromes. Eur Arch Otorhinolaryngol 275(2):365–369. https://doi.org/10.1007/s00405-017-4832-0
Alsanosi AA (2015) Simultaneous bilateral cochlear implantation in a five-month-old child with Usher syndrome. J Laryngol Otol 129(9):919–922. https://doi.org/10.1017/S0022215115001760
Serrador-García M, Santos-Bueso E, Sáenz-Francés F, Díaz-Valle D, Martínez-De-La-Casa-Borrelia JM, García-Feijóo J (2012) Charles Bonnet plus syndrome: apropos of a case. Eur J Ophthalmol 22(5):836–839. https://doi.org/10.5301/ejo.5000130
Ruiz AP, Garcia Gomez JM (2013) Labyrinthitis ossificans in a cochlear implant patient with Usher syndrome. Otol Neurotol 34(3):e10–e11. https://doi.org/10.1097/MAO.0b013e3182814d15
Liu XZ et al (2008) Cochlear implantation in individuals with Usher type 1 syndrome. Int J Pediatr Otorhinolaryngol 72(6):841–847. https://doi.org/10.1016/j.ijporl.2008.02.013
Jenison VW, Fears BT, Lusk RP (1995) Cochlear implantation of two siblings with Usher syndrome—case studies. Semin Hear 16(3):240–245. https://doi.org/10.1055/s-0028-1083721
Hinderink JB, Brokx JP, Mens LH, van den Broek P (1994) Results from four cochlear implant patients with Usher’s syndrome. Ann Otol Rhinol Laryngol 103(4 Pt 1):285–293. https://doi.org/10.1177/000348949410300405
Imtiaz F et al (2012) USH1G with unique retinal findings caused by a novel truncating mutation identified by genome-wide linkage analysis. Mol Vis 18:1885–1894
Hartel BP et al (2017) Cochlear implantation in patients with Usher Syndrome Type IIa increases performance and quality of life. Otol Neurotol 38(6):e120–e127. https://doi.org/10.1097/MAO.0000000000001441
Pietola L et al (2012) Speech recognition and communication outcomes with cochlear implantation in Usher syndrome type 3. Otol Neurotol 33(1):38–41. https://doi.org/10.1097/MAO.0b013e31823dbc56
Hoshino ACH, Echegoyen A, Goffi-Gomez MVS, Tsuji RK, Bento RF (2017) Outcomes of late implantation in Usher syndrome patients. Int Arch Otorhinolaryngol 21(2):140–143. https://doi.org/10.1055/s-0036-1583306
Broomfield SJ, Bruce IA, Henderson L, Ramsden RT, Green KMJ (2013) Cochlear implantation in children with syndromic deafness. Int J Pediatr Otorhinolaryngol 77(8):1312–1316. https://doi.org/10.1016/j.ijporl.2013.05.022
Nair G, Dham R, Sekhar A, Kumar RS, Kameswaran M (2020) Cochlear implantation in children with Usher’s syndrome: a South Asian experience. Indian J Otolaryngol Head Neck Surg 72(1):140–144. https://doi.org/10.1007/s12070-019-01759-y
Vermeulen L, Vandijk J, Hinderink H, Vandenbroek P (1994) Some results of cochlear implants in 4 persons with Ushers syndrome type-I. In: Low vision: research and new developments in rehabilitation, vol 11, pp 419–422
Dawson PW et al (1992) Cochlear implants in children, adolescents, and prelinguistically deafened adults: speech perception. J Speech Hear Res 35(2):401–417. https://doi.org/10.1044/jshr.3502.401
El-Kashlan HK, Boerst A, Telian SA (2001) Multichannel cochlear implantation in visually impaired patients. Otol Neurotol 22(1):53–56. https://doi.org/10.1097/00129492-200101000-00010
Saeed SR, Ramsden RT, Axon PR (1998) Cochlear implantation in the deaf-blind. Am J Otol 19(6):774–777
Loundon N et al (2003) Usher syndrome and cochlear implantation. Otol Neurotol 24(2):216–221. https://doi.org/10.1097/00129492-200303000-00015
Pennings RJE, Damen GWJA, Snik AFM, Hoefsloot L, Cremers CWRJ, Mylanus EAM (2006) Audiologic performance and benefit of cochlear implantation in Usher syndrome type I. Laryngoscope 116(5):717–722. https://doi.org/10.1097/01.mlg.0000205167.08415.9e
Shiomi Y, Naito Y, Hirano S, Fujiki N, Honjo I (1997) Cortical activity of a patient with Usher’s syndrome using a cochlear implant. Am J Otolaryngol 18(6):412–414. https://doi.org/10.1016/s0196-0709(97)90063-6
Mesnildrey Q, Venail F, Carlyon RP, Macherey O (2020) Polarity sensitivity as a potential correlate of neural degeneration in Cochlear implant users. JARO J Assoc Res Otolaryngol 21(1):89–104. https://doi.org/10.1007/s10162-020-00742-7
Damen GWJA, Pennings RJE, Snik AFM, Mylanus EAM (2006) Quality of life and cochlear implantation in Usher syndrome type I. Laryngoscope 116(5):723–728. https://doi.org/10.1097/01.mlg.0000205128.86063.17
Gifford RH, Revit LJ (2010) Speech perception for adult cochlear implant recipients in a realistic background noise: effectiveness of preprocessing strategies and external options for improving speech recognition in noise. J Am Acad Audiol 21(7):441–488. https://doi.org/10.3766/jaaa.21.7.3
Jatana KR, Thomas D, Weber L, Mets MB, Silverman JB, Young NM (2013) Usher syndrome: characteristics and outcomes of pediatric cochlear implant recipients. Otol Neurotol 34(3):484–489. https://doi.org/10.1097/MAO.0b013e3182877ef2
Chute PM, Nevins ME (1995) Cochlear implants in people who are deaf-blind. J Vis Impair Blind 89(3):297–301
Waltzman SB, Scalchunes V, Cohen NL (2000) Performance of multiply handicapped children using cochlear implants. Am J Otol 21(3):329–335. https://doi.org/10.1016/s0196-0709(00)80040-x
Vincent C et al (2012) Bilateral cochlear implantation in children: localization and hearing in noise benefits. Int J Pediatr Otorhinolaryngol 76(6):858–864. https://doi.org/10.1016/j.ijporl.2012.02.059
Lyutenski S, Zellhuber N, Helbig R, James P, Bloching M (2021) Cochlear reimplantation from mid-scala to lateral wall electrode array: Surgical and hearing outcome. Clin Case Rep 9(6):e04210. https://doi.org/10.1002/ccr3.4210
Henricson C, Wass M, Lidestam B, Möller C, Lyxell B (2012) Cognitive skills in children with Usher syndrome type 1 and cochlear implants. Int J Pediatr Otorhinolaryngol 76(10):1449–1457. https://doi.org/10.1016/j.ijporl.2012.06.020
Davies C et al (2021) The outcomes of cochlear implantation in Usher syndrome: a systematic review. J Clin Med. https://doi.org/10.3390/jcm10132915
Teoh SW, Pisoni DB, Miyamoto RT (2004) Cochlear implantation in adults with prelingual deafness. Part I. Clinical results. Laryngoscope 114(9):1536–1540. https://doi.org/10.1097/00005537-200409000-00006
Bond M et al (2009) The effectiveness and cost-effectiveness of cochlear implants for severe to profound deafness in children and adults: a systematic review and economic model. Health Technol Assess 13(44):1–330. https://doi.org/10.3310/hta13440
UK Cochlear Implant Study Group (2004) Criteria of candidacy for unilateral cochlear implantation in postlingually deafened adults I: theory and measures of effectiveness. Ear Hear 25(4):310–335. https://doi.org/10.1097/01.aud.0000134549.48718.53
Manrique M, Cervera-Paz FJ, Huarte A, Perez N, Molina M, García-Tapia R (1999) Cerebral auditory plasticity and cochlear implants. Int J Pediatr Otorhinolaryngol 49(Suppl 1):S193–S197. https://doi.org/10.1016/s0165-5876(99)00159-7
Snik AFM, Makhdoum MJA, Vermeulen AM, Brokx JPL, van den Broeka P (1997) The relation between age at the time of cochlear implantation and long-term speech perception abilities in congenitally deaf subjects. Int J Pediatr Otorhinolaryngol 41(2):121–131. https://doi.org/10.1016/S0165-5876(97)00058-X
Langereis M, Vermeulen A (2015) School performance and wellbeing of children with CI in different communicative-educational environments. Int J Pediatr Otorhinolaryngol 79(6):834–839. https://doi.org/10.1016/j.ijporl.2015.03.014
Cheng AK, Rubin HR, Powe NR, Mellon NK, Francis HW, Niparko JK (2000) Cost-utility analysis of the cochlear implant in children. JAMA 284(7):850–856. https://doi.org/10.1001/jama.284.7.850
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cornwall, H.L., Lam, C.M., Chaudhry, D. et al. Outcomes of cochlear implantation in Usher syndrome: a systematic review. Eur Arch Otorhinolaryngol 281, 1115–1129 (2024). https://doi.org/10.1007/s00405-023-08304-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00405-023-08304-2