
Abstract
Skin fibrosis is one central hallmark of the heterogeneous autoimmune disease systemic sclerosis. So far, there are hardly any standardized and effective treatment options. Pathogenic mechanisms underlying fibrosis comprise excessive and uncontrolled myofibroblast differentiation, increased extracellular matrix protein (ECM) synthesis and an intensification of the forces exerted by the cytoskeleton. A deeper understanding of fibroblast transformation could help to prevent or reverse fibrosis by specifically interfering with abnormally regulated signaling pathways. The transcription factor NF-κB has been implicated in the progression of fibrotic processes. However, the cellular processes regulated by NF-κB in fibrosis as well as the NF-κB isoforms preferentially involved are still completely unknown. In an in vitro model of fibrosis, we consistently observed the induction of the c-Rel subunit of NF-κB. Functional abrogation of c-Rel by siRNA resulted in diminished cell contractility of dermal fibroblasts in relaxed, but not in stressed 3D collagen matrices. Furthermore, directed migration was reduced after c-Rel silencing and total N-cadherin expression level was diminished, possibly mediating the observed cellular defects. Therefore, NF-кB c-Rel impacts central cellular adhesion markers and processes which negatively regulate fibrotic progression in SSc pathophysiology.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Fibrosis leads to fundamental pathological tissue remodeling, a central element of which is the accumulation of several proteins of the extracellular matrix (ECM) [1]. While fibrosis can occur in a variety of diseases, systemic sclerosis is a prototypical fibrotic disease that is associated with dysregulated immune functions and vascular changes [2, 3]. Long-standing fibrosis can also give rise to malignant transformation [4].
An important trigger of fibrosis is the transforming growth factor (TGF)-β1. It mediates activation of key pathways such as Smad signaling and induces fibrotic markers as well as extracellular matrix protein secretion [5]. The mechanisms of skin fibrosis involve a marked proliferation of dermal fibroblasts and their differentiation into contractile myofibroblasts [6]. Myofibroblasts mediate fibrosis-associated tissue reformation through cytoskeletal rearrangements and alpha smooth muscle actin (αSMA) integration. Furthermore, myofibroblasts upregulate the production of ECM proteins such as collagen type I and III, the F-actin-binding protein SM22α and the serine protease inhibitor plasminogen activator-inhibitor (PAI)-1 [7, 8]. Physiologically, myofibroblasts contract wounds and they dissolve by apoptosis. However, for reasons still unknown, this apoptosis does not work in fibrosis [9, 10]. Current antifibrotic therapies aim to target myofibroblasts by small molecule inhibitors affecting Smad or Wnt/β-catenin signaling [11].
The transcription factor NF-κB is a major player in immunity, tumorigenesis and other processes. It mediates signals that are for example of fundamental importance for proliferation and apoptosis [12, 13]. It is activated as homo- or heterodimer of five proteins, p50, p65, c-Rel, p52 and RelB. In fibrosis, NF-κB (p65) activity is increased in dermal and lung fibroblasts [14]. The p65 subunit inhibits collagen I expression in dermal fibroblasts [15], and p50 appears to be a genetic risk locus for systemic sclerosis (SSc) [16, 17].
In connection with fibrosis, the c-Rel subunit has become the focus of scientific interest: c-rel−/− mice were protected against bleomycin-induced fibrosis of the skin [18]. Similarly, c-Rel seems to protect against fibrosis in the liver [19] and the heart [20].
Furthermore, c-rel−/− mice showed B- and T-cell defects [21, 22], particularly in regulatory T-cells [23]. Finally, c-Rel also participates in the regulation of the cell cycle and mitosis in keratinocytes and some epithelial tumor cells [24,25,26].
Since no satisfactory treatment for fibrosis is yet available, a deeper understanding of underlying molecular processes could contribute to the development of such a treatment.
In our study, fibrotic stimulation of human dermal fibroblasts specifically induced c-Rel. Silencing of c-Rel by siRNA led to decreased contractility of non-stressed collagen matrices, whereas contractility of stressed matrices remained unaffected. While viability of dermal fibroblasts remained unchanged, their directed migration was significantly reduced when c-Rel was suppressed. Regarding adhesion marker expression, prominent suppression of N-cadherin resulted, typically upregulated in fibrosis and many cancer types. Thus, c-Rel takes part in the homeostasis of dermal fibroblasts and seems to regulate some steps of fibrotic activation.
Materials and methods
Antibodies
Primary antibodies directed against the following antigens were used: p50 (Abcam, Cambridge, USA), p65 (Cell Signaling Technology, Danvers, USA), c-Rel (Cell Signaling Technology), RelB (Santa Cruz Biotechnology, Dallas, USA) and p52 (Cell Signaling Technology), αSMA (Abcam), PAI-1 (Santa Cruz Biotechnology), pSmad3 (Abcam), GAPDH (Cell Signaling Technology), focal adhesion kinase, FAK (Abcam), SM22α (ThermoFisher Scientific, Waltham, USA), talin (Abcam), vinculin (SigmaAldrich, St. Louis, USA), actin (Merck Millipore, Burlington, USA), N-cadherin (Abcam) and Calnexin (ENZO Life Sciences, Belgium).
Cell culture and stimulation
BJ dermal fibroblasts (ATCC® CRL-2522™) were cultured in EMEM (SigmaAldrich) growth media supplemented with 10% FCS (Biochrom, Berlin, Germany) and 1% l-glutamine (Lonza, Morristown, USA) at 37 °C with 5% CO2 and were frequently tested for mycoplasma contamination. Cells were stimulated after adherence with rh-TGF-β1 (10 ng/ml, ThermoFisher Scientific).
siRNA transfection
Adherent fibroblasts were transfected using Promofectin siRNA (Promocell, Heidelberg, Germany) with c-Rel siRNA I (SI00045570), c-Rel siRNA II (SI03070599) and AllStars Negative Control siRNA (Qiagen, Hilden, Germany) at 40 nM for 72 h. Untreated and transfection reagent-treated controls were performed to exclude nonspecific effects.
Collagen gel contraction assay
Collagen gels populated with 4 × 104 transfected fibroblasts per well were prepared using a final concentration of 1 mg/ml collagen I (ThermoFisher Scientific) in growth medium and polymerized using 1 M NaOH (Merck, Billerica, USA). Polymerized gels where covered with growth medium and either detached directly (relaxed) or adhered for 24 h including TGF-β stimulation (stressed) followed by final photographic documentation using a LAS4000 device (Fujifilm, Tokyo, Japan). Quantification of collagen gel size was determined using ImageJ Vers. 1.52 (NIH, USA).
Cell migration assay
On each side of the migration insert, 3.5 × 104 transfected cells were seeded on collagen I-coated culture inserts (Ibidi, Gräfelfing, Germany) in medium with low serum concentration (2.5% FCS). Migration into the cell-free gap was documented for indicated time points using an Axiovert200 microscope and the Axiovision software (Zeiss, Jena, Germany). Quantification of the cell-free gap was performed using ImageJ Vers. 1.52 (NIH, USA).
G/F actin fractionation
The G/F actin ratio was determined using the G actin/F actin kit as recommended by the manufacturer (Cytoskeleton Inc., Denver, USA) and analyzed by western blotting. Quantification was performed using ImageJ vers. 1.52 (NIH, USA).
Immunofluorescence
In each well, 6 × 103 transfected cells were cultured in 8 well chamber slides (ThermoFisher Scientific) followed by fixation with 4% paraformaldehyde. After blocking/permeabilization with 5% FCS/0.25% Triton-X/PBS, primary antibodies were incubated followed by anti-mouse Alexa488 (Cell Signaling Technology) and anti-rabbit Alexa 555 (ThermoFisher Scientific) labeled secondary antibodies. Finally, cells were embedded in Fluorescent Mounting Medium (Dako, Santa Clara, USA) supplemented with 1 µg/ml DAPI (SigmaAldrich). Images were acquired using AxioImager M1 and the Axiovision Software Rel. 4.7.1 (Zeiss).
RNA isolation, cDNA synthesis and qRT-PCR
RNA was isolated using InnuPrep RNA kit (Analytik Jena, Jena, Germany) and cDNA was generated using SuperScript IV cDNA Synthesis Kit (ThermoFisher Scientific). For qPCR, Eva Green Dye (Solis Biodyne, Tartu, Estonia) was used and respective primer pairs; p50 (fw: 5′-CACTTAGCAATCATCCACCTT-3′, rev: 5′-AGCCCTCAGCAAATCCT-3′), p65 (Qiagen; QT02324308), c-Rel (Qiagen; QT00052472), p52 (fw: 5′-GGGGCATCAAACCTGAAGATTTCT-3′, rev: 5′-TCCGGAACACAATGGCATACTGT-3′), RelB (Qiagen QT00038640), CTGF (fw: 5′-CTCGCGGCTTACCGACTG-3′, rev: 5′-GGCTCTGCTTCTCTAGCCTG-3′), PAI-1 (SERPINE-1) (fw: 5′-CTCTCTCTGCCCTCACCAAC-3′, rev: 5′-GTGGAGAGGCTCTTGGTCTG-3′) αSMA (fw: 5′-CGTGGGTGACGAAGCACAG-3′, rev: 5′-GGTGGGATGCTCTTCAGGG-3′), collagen IAI (fw: 5′-GCTCCTGCTCCTCTTAGCG-3′, rev 5′-CCGTTCTGTACGCAGGTGAT-3′), RNA-polymerase IIA (fw: 5′-GGAGATTGAGTCCAAGTTCA-3′, rev: 5′-GCAGACACACCAGCATAGT-3′), integrin αv (fw: 5′-CACTTCGGCGATGGCTTTTC-3′, rev: 5′-GTAGCAGGAGTCCCGAGAGA-3′), integrin α2 (fw: 5′-GTGGCTTTCCTGAGAACCGA-3′, rev: 5′-GATCAAGCCGAGGCTCATGT-3′), integrin β1 (fw: 5′-ACGCCGCGCGGAAAAGATGA-3′, rev: 5′-GCACCACCCACAATTTGGCCC-3′), Data analysis was performed using QuantStudio5 and QuantStudio Design and Analysis Software (ThermoFisher Scientific).
Protein lysates and Western blotting
Cells were resuspended in RIPA lysis and extraction buffer (ThermoFisher Scientific), incubated for 30 min at 4 °C and centrifuged at 14.000g. Western blots were performed as described previously [26].
MTT assay
For viability testing, 5 × 103 cells were seeded and finally measured using the Non-Radioactive Cell Proliferation Assay (Promega, Mannheim, Germany) and determined 72 h after transfection measuring absorbance at 570 nm and 630 nm using Appliskan (ThermoFisher Scientific).
Statistical analysis
For MTT, migration and collagen contractility assay, one way analysis of variance (ANOVA) was performed followed by post tukey test using Graph Pad PRISM vers. 7 (GraphPad Software, Inc.). For qRT PCR, relative expression software tool (REST) Software (Qiagen, 2009) was used. Differences were considered statistically significant when P ≤ 0.05.
Results
Fibrotic stimulation with TGF-β1 induces myofibroblastic traits in vitro
In a previously described in vitro model of fibrosis [27, 28], human BJ fibroblasts with extended life span were stimulated with TGF-β1 (10 ng/ml) and their expression of different fibrosis markers was investigated and validated (Fig. 1).

Myofibroblast-related changes are induced by TGF-β stimulation. a qPCR of fibrotic markers (PAI-1, Collagen IAI and αSMA) after TGF-β stimulation for 0 h, 24 h, 48 h and 72 h, respectively. Error bars = SEM, * indicates p ≤ 0.05, n = 3–4 independent experiments. b Protein expression of fibrotic markers after 0 h, 24 h, 48 h and 72 h of TGF-β stimulation. One of 3–4 representative western blots is shown, GAPDH served as loading control. c Immunofluorescence staining of filamentous actin binding protein SM22α showing morphologic changes after 0 h, 24 h, 48 h and 72 h of TGF-β stimulation, respectively. Scale bar = 50 µm, n = 3
As detected by quantitative RT-PCR, transcription of plasminogen activator inhibitor-1 (PAI-1) was significantly increased (3.87-fold after 24 h, 3.22-fold after 48 h, 2.9-fold after 72 h). Likewise, collagen IAI (3.49-, 2.64- and 2.88-fold, respectively) and alpha-smooth muscle actin (αSMA; 2.75-, 1.28- and 2.44-fold, respectively) were induced, albeit the increase of αSMA did not reach statistical significance (Fig. 1a).
On the protein level, PAI-1 showed the most pronounced and earliest TGF-β associated induction, while αSMA was induced 48 h and 72 h after TGF-β stimulation. Furthermore, pSmad3 expression analysis served as positive control for TGF-β signaling activation and showed clear induction at all time points of stimulation (Fig. 1b).
Finally, we detected fibrosis-associated changes of the cytoskeleton in this model. As demonstrated by staining of the F-actin binding protein SM22α, contractile actin filaments increased markedly following exposure of the cultures to TGF-β (Fig. 1c). Thus, our in vitro fibrosis model reflected key fibrotic and myofibroblast-associated changes.
NF-κB c-Rel is induced by fibrotic stimulation
When expression and activity of NF-κB proteins were analyzed after incubation of BJ fibroblasts with TGF-β1, only c-Rel mRNA consistently showed elevation (by 48%, 45% and 63% after 24 h, 48 h and 72 h, respectively; Fig. 2a). Correspondingly, the c-Rel protein was also more strongly expressed after TGF stimulation with a maximum after 48 h, while p65, p50, p52 and RelB remained unchanged (Fig. 2b). An analysis of subcellular distribution showed that c-Rel was localized both in unstimulated fibroblasts and after TGF-β stimulation mainly in the cytoplasm and considerably less in the nucleus (Fig. 2c).

NF-κB expression and activity in fibrotically activated dermal fibroblasts. a qPCR of c-Rel, p50, p65, RelB and p52 after TGF-β stimulation for 0 h, 24 h, 48 h and 72 h, respectively. Error bars = SEM, n = 3–4, *p ≤ 0.05. b Western blotting of c-Rel, p50 and p65, p52 and RelB expression after 0 h, 24 h, 48 h and 72 h TGF-β stimulation. GAPDH served as loading control. One representative experiment of four is shown. c Subcellular distribution of c-Rel after TGF-β stimulation for 0 h, 24 h, 48 h and 72 h, respectively. Nuclear (N) and cytoplasmic (CP) lysates are depicted, Lamin A/C and GAPDH served as nuclear and cytoplasmic loading control, respectively. One of four independent experiments is depicted
c-Rel is involved in the contraction of collagen matrices by dermal fibroblasts
The data collected so far suggested that c-Rel modulates fibrosis processes. To test this hypothesis further, its activity was suppressed in fibroblasts by transfection with two siRNA constructs directed against c-Rel. Both unstimulated (homeostatic) and fibrotic conditions showed efficient c-Rel downregulation 72 h after treatment and fibroblast viability was not significantly altered after c-Rel suppression with either of the two constructs (Fig. 3a, b). Of note, this downregulation of c-Rel did not lead to compensatory changes of the major dimerization partners, p50 and p65 (supplementary Fig. 1a).

c-Rel siRNA suppression affects cell contractility. a Western blotting of ctrl siRNA and c-Rel siRNA (c-Rel I and c-Rel II) transfected samples, either unstimulated (left) or stimulated with TGF-β (right) 72 h after transfection showing efficient c-Rel knockdown. GAPDH served as loading control. b MTT viability assay of ctrl, c-Rel I and c-Rel II siRNA transfected cells. The mean absorbance values of three independent experiments are shown (error bars = SEM). c Schematic illustration of 3D collagen contraction assay using dermal fibroblasts in a “relaxed” model (left side) and in a “stressed” model by induction of mechanical stress plus TGF-β stimulation (right side). d, e Collagen contractility assay of ctrl siRNA, c-Rel I and c-Rel II siRNA transfected dermal fibroblasts in “relaxed” (d) and “stressed” lattices (e) 72 h after siRNA transfection. One representative experiment is depicted while below, quantification of cell-collagen-matrix area is depicted. Relative mean values of 3–4 experiments are shown, error bars = SEM, * indicates p ≤ 0.05. f qPCR of c-Rel, PAI-1, and collagen IAI of control and c-Rel siRNA transfected dermal fibroblasts 72 h after transfection. Relative mean values of three independent experiments are shown, RNA-polymerase IIA served as reference gene, error bars = SEM, * indicates p ≤ 0.05
Cytoskeletal remodeling and the exertion of traction forces are essential characteristics of myofibroblastic transdifferentiation. 3D contraction assays using cell-collagen I-matrices were used to mimick these features. Non-stressed conditions were generated by gently releasing the collagen gels (without TGF supplementation) from the vessel wall directly after polymerization (hereinafter referred to as “relaxed” conditions; Fig. 3c left side and Fig. 3d [29]). To simulate fibrotic conditions, the collagen gels were polymerized supplemented with TGF-β and not detached from the vessel wall until 24 h after polymerisation (hereinafter referred to as "stressed" conditions; Fig. 3c right side and Fig. 3e [29]). Following c-Rel suppression, relaxed fibroblast-collagen I-matrices were significantly and reproducibly larger than control matrices (50.6% with c-Rel I siRNA, p ≤ 0.05; 37.7% with c-Rel II siRNA, p ≤ 0.05; Fig. 3b). In contrast, stressed matrices showed minor differences in contractility after c-Rel suppression (Fig. 3e). Thus, c-Rel seemed to significantly affect fibroblast contractility in relaxed but not in stressed collagen matrices. After siRNA-induced significant suppression of c-Rel, the transcription of fibrotic marker PAI-1 was only moderately induced while collagen 1A1 was slightly reduced and no significant expression changes occurred following TGF-β stimulation (Fig. 3f). Protein expression of pSmad3, PAI-1 and αSMA was not significantly altered (supplementary Fig. 1b).
Impaired motility in c-Rel suppressed dermal fibroblasts
In addition to contractility, which we had demonstrated to be influenced by c-Rel, altered motility and migration ability of fibroblasts also play an important role in wound healing and fibrosis [30]. In the following experiments, the migration of normal (control transfected) fibroblasts was therefore compared in artificial "wounds" (standardized scratch assays) with that of fibroblasts whose c-Rel was suppressed by siRNA for periods of up to 24 h.
After 6 h, c-Rel suppression reduced directed cell migration by 47.9% for siRNA I and 56.9% for siRNA II compared to control transfected fibroblasts, the latter difference being already statistically significant at this early stage (p < 0.05). After 10 h the difference between control cells and the two c-Rel suppressed populations was also consistently detectable (46.6% and 55.0%, respectively; with p < 0.05 and p < 0.01). The delayed migration of c-Rel-suppressed fibroblasts was still visible after 24 h, with the values of the c-Rel-suppressed populations slowly approaching the already completely closed "wound" of the control cells (p < 0.05; Fig. 4a).

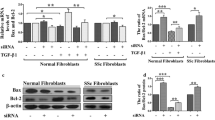
Silencing of c-Rel impairs dermal fibroblast migration and N-cadherin expression. a Migration assay of control siRNA (ctrl), c-Rel I siRNA and c-Rel II siRNA transfected fibroblasts. Representative images are shown 0 h, 10 h and 24 h after assay start, quantitative evaluation shows mean values of three independent experiments compared to assay start. Error bars = SEM. *p ≤ 0.05, **p ≤ 0.01. b Western blotting of N-cadherin and Talin of control siRNA, c-Rel siRNA I and II 72 h after transfection. One of three experiments is shown, GAPDH and Calnexin served as loading control. c qPCR of integrin αV (ITGAV), β1 (ITGB1), α2 (ITGA2) of control and c-Rel siRNA transfected fibroblasts 72 h after transfection. The mean values of three-four independent experiments are shown (error bars = SEM). d G and F actin distribution of control siRNA, c-Rel siRNA I and II transfected cells. The mean pixel intensities of three independent experiments are depicted (error bars = SEM) and one representative western blot experiment is shown below. e SM22α and vinculin immunofluorescence of control siRNA, c-Rel I siRNA and c-Rel II siRNA transfected fibroblasts. Single representative cells of one of three independent experiments processed identically are shown. Nuclei were counterstained with DAPI in merged illustrations, size bar = 25 µm
To understand the molecular mechanisms, we analyzed expression of adhesion marker N-cadherin and focal adhesion protein talin by western blot analysis and determined strong downmodulation of N-cadherin following c-Rel suppression (Fig. 4b).
However, the expression of the classical adhesion receptor integrin subunits β1 (ITGB1), α2 (ITGA2) and αV (ITGAV) was not significantly altered by c-Rel suppression (Fig. 4c). The significant impairment of N-cadherin expression following c-Rel suppression was also not accompanied by a change in the relative proportions of globular (G) and filamentous (F) actin, as demonstrated by fractional analysis (Fig. 4d). Finally, immunofluorescence staining of vinculin and SM22α showed that also the formation of focal adhesions was not affected by c-Rel suppression (Fig. 4e).
Together, our findings showed that c-Rel suppression interferes with N-cadherin expression potentially disturbing cell contraction and migration, while only marginally, if at all, touching on cytoskeleton or focal adhesion characteristics.
Discussion
Fibrotic diseases such as systemic sclerosis (SSc) comprise complex pathological tissue alterations, which, in addition to misregulated immune functions and vascular changes, include organ fibrosis in particular [31]. In SSc patients, fibrosis usually starts in the skin and can proceed to organs such as lung, kidney and heart [32]. Therefore, a better understanding of signaling mechanisms in skin-derived fibroblasts may help to develop new therapeutic targets preventing uncontrolled fibrotic proceeding of SSc. While the pathogenesis of fibrosis is still largely enigmatic, the c-Rel subunit of NF-κB appears to be associated with fibrotic changes in various organs such as the skin, liver and heart as suggested by c-Rel-deficient mice [18,19,20]. However, a direct association of c-Rel with fibrosis-relevant mesenchymal cells has scarcely been investigated so far.
In this situation, we showed induction of c-Rel in fibroblasts in an in vitro fibrosis model both on the transcriptional and the translational level. No other NF-κB subunit was similarly induced in fibroblasts by TGF-β. This is somewhat surprising, since NF-κB as a central transcription factor is involved in numerous regulatory processes [33] and TGF-β can exert rather pleiotropic effects [34]. However, in some respects c-Rel is unusual among NF-кB proteins as it possesses a broader nuclear recognition site and, consecutively, has a larger spectrum of target genes compared to other NF-κB subunits [35]. Moreover, it is thought to be associated with a more pronounced capability to suppress genes in a proinflammatory surrounding [36].
Following suppression of c-Rel, N-cadherin expression was reduced, possibly mediating diminished contractility and migration of human dermal fibroblasts. Supporting our findings, N-cadherin was recently shown to be involved in injury-triggered migration (“swarming”) and contraction of fascia fibroblasts important for scar formation [37].
Subsequent analyses need to reveal whether N-cadherin serves as a direct target of c-Rel and whether also typical intracellular target molecules such as α- and β-catenin are affected. Although N-cadherin and actin cytoskeleton dynamics are intricately intertwined, no changes appeared neither in G/F actin content nor in F-actin signal intensity. Possibly, associated cytoskeletal mechanisms include rather actomyosin-based dynamics [38]. Moreover, c-rel−/− mice revealed decreased bladder smooth muscle contraction depending on CPI-17 (protein kinase C-potentiated inhibitory protein of 17 kDa), a protein affecting myosin light chain phosphorylation [39]. This suggests an alternative impact of c-Rel on myosin dynamics. However, muscle and non-muscle cells such as dermal fibroblasts may be regulated differently.
Myofibroblasts resemble smooth muscle cells in their expression and cytoskeletal inclusion of αSMA [32]. However, in contrast to smooth muscle cells the former are permanently contractile presumably also through activation of the Rho/ROCK/myosin light chain phosphatase pathway [40]. Thus, our results described here, together with the outlined molecular details, could be a good starting point for further mechanistic studies on the contractility of myofibroblasts in fibrotic diseases.
Another result, which at first glance seems somewhat curious, was that c-Rel suppression apparently reduced the contractility of fibroblasts in "relaxed", but not in "stressed" gels. On closer examination, however, there appear to be important differences between these two conditions: Former analyses revealed that fibroblasts in “relaxed” matrices have low proliferative activity and a generally pro-inflammatory phenotype, whereas fibroblasts in “stressed” matrices show more myofibroblast traits such as αSMA expression (which was not affected by c-Rel suppression in our experiments) as well as higher contractility [29]. A global influence on the entire fibrotic “machinery” does not seem to occur, because in our models we could not find a general influence of c-Rel suppression on fibrotic markers. Therefore it seems conceivable that c-Rel performs its main function in a “pre-fibrotic” state of fibroblasts, which the "relaxed" matrices may simulate in vitro. If this hypothesis can be confirmed in vivo in future studies, a therapeutic modulation of c-Rel could influence early inflammatory phases of fibrosis and thus have a preventive effect. In any case, we could show that NF-кB c-Rel is an interesting candidate influencing an essential adhesion marker and thus cellular processes in skin fibroblasts with potential consequences for the pathophysiology of fibrosis and/or fibrotic diseases.
Availability of data and material
Not applicable.
Code availability
Not applicable.
References
Wynn TA, Ramalingam TA (2012) Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med 18(7):1028–1040
Sticherling M (2019) Systemic sclerosis—the dermatological perspective. J German Soc Dermatol JDDG 17(7):716–728
Ciechomska M, van Laar J, O’Reilly S (2015) Current frontiers in systemic sclerosis pathogenesis. Exp Dermatol 24(6):401–406
Heck J, Olk J, Kneitz H, Hamm H, Goebeler M (2020) Long-standing morphea and the risk of squamous cell carcinoma of the skin. J German Soc Dermatol JDDG 18(7):669–673
Kim KK, Sheppard D, Chapman HA (2018) TGF-beta1 signaling and tissue fibrosis. Cold Spring Harbor Persp Biol 10(4):a022293
Leask A, Holmes A, Abraham DJ (2002) Connective tissue growth factor: a new and important player in the pathogenesis of fibrosis. Curr Rheumatol Rep 4(2):136–142
Ghosh AK, Vaughan DE (2012) PAI-1 in tissue fibrosis. J Cell Physiol 227(2):493–507
Davis J, Molkentin JD (2014) Myofibroblasts: trust your heart and let fate decide. J Mol Cell Cardiol 70:9–18
Hinz B (2007) Formation and function of the myofibroblast during tissue repair. J Invest Dermatol 127(3):526–537
Darby IA, Zakuan N, Billet F, Desmouliere A (2016) The myofibroblast, a key cell in normal and pathological tissue repair. Cell Mol Life Sci 73(6):1145–1157
Yazdani S, Bansal R, Prakash J (2017) Drug targeting to myofibroblasts: implications for fibrosis and cancer. Adv Drug Deliv Rev 121:101–116
Karin M, Lin A (2002) NF-kappaB at the crossroads of life and death. Nat Immunol 3(3):221–227
Karin M (2006) NF-kappaB and cancer: mechanisms and targets. Mol Carcinog 45(6):355–361
Mia MM, Bank RA (2015) The IkappaB kinase inhibitor ACHP strongly attenuates TGFbeta1-induced myofibroblast formation and collagen synthesis. J Cell Mol Med 19(12):2780–2792
Beauchef G, Bigot N, Kypriotou M et al (2012) The p65 subunit of NF-kappaB inhibits COL1A1 gene transcription in human dermal and scleroderma fibroblasts through its recruitment on promoter by protein interaction with transcriptional activators (c-Krox, Sp1, and Sp3). J Biol Chem 287(5):3462–3478
Martin JE, Broen JC, Carmona FD et al (2012) Identification of CSK as a systemic sclerosis genetic risk factor through Genome Wide Association Study follow-up. Hum Mol Genet 21(12):2825–2835
Lopez-Isac E, Acosta-Herrera M, Kerick M et al (2019) GWAS for systemic sclerosis identifies multiple risk loci and highlights fibrotic and vasculopathy pathways. Nat Commun 10(1):4955
Fullard N, Moles A, O’Reilly S et al (2013) The c-Rel subunit of NF-kappaB regulates epidermal homeostasis and promotes skin fibrosis in mice. Am J Pathol 182(6):2109–2120
Gieling RG, Elsharkawy AM, Caamano JH et al (2010) The c-Rel subunit of nuclear factor-kappaB regulates murine liver inflammation, wound-healing, and hepatocyte proliferation. Hepatology 51(3):922–931
Gaspar-Pereira S, Fullard N, Townsend PA et al (2012) The NF-kappaB subunit c-Rel stimulates cardiac hypertrophy and fibrosis. Am J Pathol 180(3):929–939
Kontgen F, Grumont RJ, Strasser A et al (1995) Mice lacking the c-rel proto-oncogene exhibit defects in lymphocyte proliferation, humoral immunity, and interleukin-2 expression. Genes Dev 9(16):1965–1977
Grumont RJ, Rourke IJ, O’Reilly LA et al (1998) B lymphocytes differentially use the Rel and nuclear factor kappaB1 (NF-kappaB1) transcription factors to regulate cell cycle progression and apoptosis in quiescent and mitogen-activated cells. J Exp Med 187(5):663–674
Isomura I, Palmer S, Grumont RJ et al (2009) c-Rel is required for the development of thymic Foxp3+ CD4 regulatory T cells. J Exp Med 206(13):3001–3014
Slotta C, Schluter T, Ruiz-Perera LM et al (2017) CRISPR/Cas9-mediated knockout of c-REL in HeLa cells results in profound defects of the cell cycle. PLoS ONE 12(8):e0182373
Priebe MK, Dewert N, Amschler K et al (2019) c-Rel is a cell cycle modulator in human melanoma cells. Exp Dermatol 28(2):121–128
Lorenz VN, Schön MP, Seitz CS (2014) c-Rel downregulation affects cell cycle progression of human keratinocytes. J Invest Dermatol 134(2):415–422
Carthy JM, Meredith AJ, Boroomand S et al (2015) Versican V1 overexpression induces a myofibroblast-like phenotype in cultured fibroblasts. PLoS ONE 10(7):e0133056
Desmouliere A, Geinoz A, Gabbiani F et al (1993) Transforming growth factor-beta 1 induces alpha-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J Cell Biol 122(1):103–111
Kessler D, Dethlefsen S, Haase I et al (2001) Fibroblasts in mechanically stressed collagen lattices assume a “synthetic” phenotype. J Biol Chem 276(39):36575–36585
Kis K, Liu X, Hagood JS (2011) Myofibroblast differentiation and survival in fibrotic disease. Expert Rev Mol Med 13:e27
Eckes B, Moinzadeh P, Sengle G et al (2014) Molecular and cellular basis of scleroderma. J Mol Med 92(9):913–924
van Caam A, Vonk M, van den Hoogen F et al (2018) Unraveling SSc pathophysiology; the myofibroblast. Front Immunol 9:2452
Liu T, Zhang L, Joo D, Sun SC (2017) NF-kappaB signaling in inflammation. Signal Transduct Tar 2:17023
David CJ, Massague J (2018) Contextual determinants of TGFbeta action in development, immunity and cancer. Nat Rev Mol Cell Biol 19(7):419–435
Kunsch C, Ruben SM, Rosen CA (1992) Selection of optimal kappa B/Rel DNA-binding motifs: interaction of both subunits of NF-kappa B with DNA is required for transcriptional activation. Mol Cell Biol 12(10):4412–4421
de Jesus TJ, Ramakrishnan P (2020) NF-kappaB c-Rel dictates the inflammatory threshold by acting as a transcriptional repressor. iScience 23(3):100876
Jiang D, Christ S, Correa-Gallegos D et al (2020) Injury triggers fascia fibroblast collective cell migration to drive scar formation through N-cadherin. Nat Commun 11:5653
Sandbo N, Dulin N (2011) Actin cytoskeleton in myofibroblast differentiation: ultrastructure defining form and driving function. Transl Res 158(4):181–196
Boopathi E, Hypolite JA, Zderic SA et al (2013) GATA-6 and NF-kappaB activate CPI-17 gene transcription and regulate Ca2+ sensitization of smooth muscle contraction. Mol Cell Biol 33(5):1085–1102
Bochaton-Piallat ML, Gabbiani G, Hinz B (2016) The myofibroblast in wound healing and fibrosis: answered and unanswered questions. F1000Research 5:752
Acknowledgements
This work was supported by the “Deutsche Stiftung Sklerodermie/Edith Busch Stiftung”. We thank U. Unkelbach and S. Köchy for excellent technical support.
Funding
Open Access funding enabled and organized by Projekt DEAL. This work was supported by the “Deutsche Stiftung Sklerodermie/Edith Busch Stiftung”.
Author information
Authors and Affiliations
Contributions
MPS and VNL designed the study, interpreted the data and wrote the manuscript. LM, FTK and ALJ performed the laboratory work, LM and FTK and VNL analyzed the data. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflict of interest to declare.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Micus, L.C., Trautschold-Krause, F.S., Jelit, A.L. et al. NF-кB c-Rel modulates pre-fibrotic changes in human fibroblasts. Arch Dermatol Res 314, 943–951 (2022). https://doi.org/10.1007/s00403-021-02310-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00403-021-02310-2