
Abstract
Introduction
Viscosupplementation by repeated intra-articular injections of hyaluronic acid (HA) is used widely in the treatment of symptomatic knee osteoarthritis (OA). The number of injections required can limit the availability of treatment and affect patient compliance. The aim of this study was to assess different dosing regimens of hylan G-F 20, a high molecular-weight cross-linked derivative of HA, in the treatment of pain due to knee OA.
Materials and methods
Pilot, prospective, multi-centre, open-label, randomised trial in 100 patients with unilateral, symptomatic, tibio-femoral OA (Kellgren–Lawrence grade II or III), aged ≥40 years. Patients were randomised to receive varying dosing regimens of hylan G-F 20 (1 × 6 mL, 1 × 4 mL, 2 × 4 mL 2 weeks apart, 3 × 4 mL 1 week apart, or 3 × 2 mL 1 week apart). Adverse events (AE’s) were monitored throughout the study. The primary efficacy endpoint was the change from baseline in the patient-rated knee OA pain assessment (100 mm visual analogue scale (VAS)) at 24 weeks. The secondary efficacy criteria included the WOMAC index, patient and physician global assessments using a 100 mm VAS, and knee OA pain assessment at all other visits. Concomitant use of permitted rescue medications (paracetamol) was also assessed.
Results
The treatment was well tolerated overall. Patients in the 3 × 4 mL group reported the highest percentage of device-related local AE’s (30%) while patients in the 1 × 6 mL and 3 × 2 mL groups reported only 10%. There were no serious device-related AEs.
There was a statistically significant improvement from baseline at week 24 in all efficacy endpoints for all treatment regimens. The 1 × 6 and 3 × 4 and 3 × 2 mL treatment groups showed the greatest mean improvements (−34.9, −32.6 and −36.7 mm respectively) in the patient-rated knee OA pain assessment VAS.
Conclusion
This study suggests that a single 6 mL injection of hylan G-F 20 may be as efficacious, and as well tolerated, as 3 × 2 mL one week apart. A double-blind, controlled trial is needed to confirm these data.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Osteoarthritis (OA) is the most common disease affecting synovial joints and is one of the most prevalent chronic conditions affecting Western populations [11]. The disease is anatomically characterised by cartilage breakdown, osteophyte formation at the margins of the joint and low grade synovial inflammation.
Hyaluronic acid (HA) is the molecule responsible for synovial fluid’s rheological properties, enabling it to act as a lubricant or shock-absorber depending upon the forces exerted upon it [4, 17]. Osteoarthritic synovial fluid is characterised by a decrease in high molecular weight (MW) HA molecules and a reduction in the concentration of HA. These changes reduce the elastoviscosity of the synovial fluid and compromise its ability to protect the joint.
Viscosupplementation is a therapeutic concept based on the injection of exogenous HA, or its derivatives, into the osteoarthritic joint, with the aim of decreasing pain and improving function through the restoration of the synovial fluid’s rheological properties [4]. The therapy’s analgesic effect is thought to be achieved by multiple mechanisms including reducing pain-eliciting nerve activity by providing an elastoviscous protective barrier around the nociceptive afferent fibres in the intercellular matrix. Research suggests that the long-term symptomatic effect of the therapy (which exceeds the residence time of the product in the joint) may be due to the restoration of normal, endogenous synthesis of high quality HA [3].
There are now more than 20 commercial viscosupplement formulations available world-wide from different manufacturers. The MW and residence time in the joint varies between products, and their recommended dosing regimens for the treatment of symptomatic knee OA range from 1 to 5 injections at weekly intervals [6]. The number of injections required to achieve efficacy is inversely related to the MW of the preparation used and, consequently, to the residence time in the joint.
Controlled studies versus placebo have shown that hylan G-F 20, a cross-linked high MW derivative of HA, can decrease pain and improve joint mobility in patients suffering from knee OA [1, 10, 13, 18, 20, 26]. After 3 × 2 mL intra-articular injections administered 1 week apart, peak efficacy occurs between 8 and 12 weeks after the administration of the first injection, and can last for up to 12 months [18]. This regimen was established as being clinically superior to a regimen of 2 × 2 mL administered 2 weeks apart [20].
The overall tolerability of the treatment is good with a low incidence of local adverse events (AE’s) [15, 24]. This incidence appears to be slightly elevated, but remains low compared to other therapies, in repeat courses of treatment [19, 23]. In a recent retrospective trial of 1,489 knees treated with hylan G-F 20 [24], local treatment-related and procedure-related AE rates were reported as 4.2% of patients and 2.4% of injections. The most frequently reported AE’s were joint effusion, joint swelling, arthralgia, joint warmth and injection site erythema. Most AE’s were mild to moderate.
Nevertheless, there is an obvious medical justification for decreasing the total number of injections in order to decrease the risk of severe adverse events such as septic arthritis, associated with any intra-articular procedure, and to improve patient compliance and convenience. There is also significant economic benefit to be gained from reducing the number of injections required to complete treatment as physician time, patient time, clinic overheads and medical disposable costs that could all reduced.
It was hypothesised that this objective could be achieved by increasing the volume of hylan G-F 20 injected. Injections of 4 or 6 mL into the knee joint have never been tested in clinical trials so there are no published data available regarding the risk of local AE’s such as pain, swelling and effusion, or regarding their potential frequency and/or intensity.
The primary objective of this pilot study was therefore to assess the safety and efficacy profiles of new dosing regimens of hylan G-F 20 in patients with knee OA, using higher single dose volumes (4 and 6 mL) and reducing the number of injections (1 or 2) and to compare these results to the dosing regimen currently approved (3 × 2 mL) [21]. The secondary objective was to assess the safety and efficacy profiles of a second cycle of these new dosing regimens. The possible relationship between clinical efficacy and total volume injected (6 mL currently, up to 12 mL in this study) was also examined.
Materials and methods
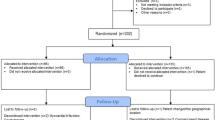
The study was a prospective, multi-centre, randomised, open, five-arm trial conducted in France and Germany. The protocol and patient informed consent form were reviewed and approved by the appropriate independent ethics committee (IEC) and complied with the requirements of the international conference on harmonisation (ICH). Patient’s written, informed consent was required before enrolment in the study. Investigators complied with the Declaration of Helsinki, “Guidance for Good Clinical Practice” and the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use when developing the patient informed consent documents.
Patients
The main inclusion criteria were: Male or female patient aged 40 years or older with an active lifestyle, consulting for OA pain in one knee and scoring ≥50 and ≤80 mm on a 100 mm OA pain visual analogue scale (VAS) where 0 mm = no pain and 100 mm = worst possible pain; tibio-femoral OA (ACR criteria) [2], Kellgren–Lawrence grade II or III [14] diagnosed by standard X-rays taken within 3 months prior to enrolment; no surgical intervention planned in the study knee in the next 6 months. If taking analgesics (except permitted doses of paracetamol ≤3 g/day for rescue analgesia), NSAIDs or cyclooxygenase-2 inhibitors, patient were required to comply with a washout period of 1–3 weeks depending on the half-life of the medication.
The main exclusion criteria were: Patients with bilateral symptomatic knee OA or predominantly patello-femoral involvement of the study knee; knee OA flare with obvious tense effusion, diagnosed by clinical examination, at the study knee; clinical symptoms of meniscal instability or significant valgus/varus that required corrective osteotomy; significant ligamentous instability; any prior viscosupplementation therapy or history of sepsis in the study knee; systemic or intra-articular injection of corticosteroids in any joint within 3 months of enrolment; chondrocalcinosis and microcrystals-mediated arthritis, concomitant inflammatory or other rheumatologic, neurological or cardiovascular diseases which could affect the evaluation of knee pain.
Methods
Patients meeting the inclusion and exclusion criteria were randomised to 1 of 5 groups:
- Group 1:
-
: 1 intra-articular injection of 6 mL hylan G-F 20
- Group 2:
-
: 1 intra-articular injection of 4 mL hylan G-F 20
- Group 3:
-
: 2 intra-articular injections of 4 mL hylan G-F 20 administered 2 weeks apart
- Group 4:
-
: 3 intra-articular injections of 4 mL hylan G-F 20 administered 1 week apart
- Group 5:
-
: 3 intra-articular injections of 2 mL hylan G-F 20 administered 1 week apart
Intra-articular injections were performed under strict aseptic technique by a trained physician using a lateral, medial mid patellar or antero-medial injection route (according to the injector’s preference) after aspiration of any synovial fluid.
Patients were followed-up 7 days after each injection then at 3, 8, 16 and 24 weeks after the first injection. Safety and efficacy were assessed at each patient visit. At week 24, patients who scored ≥50 and ≤80 mm on the pain VAS, or patients who experienced a worsening of pain (>15 mm on VAS) compared to week 16, were eligible to receive a second cycle of treatment (Extension Study). Patients undergoing a second cycle of treatment received the same dosing regimen as that dictated by their original randomisation.
Safety assessment
Target knee and systemic AE’s were monitored throughout the study. Additionally, patients assessed overall safety using a four point side-effect rating scale.
Efficacy assessment
The primary efficacy endpoint was the patient-completed study knee OA pain score (VAS) 24 weeks after the first injection, compared to baseline.
The secondary endpoints were the patient-completed study knee OA pain score (VAS) at all other time points; improvement in pain, stiffness and functional impairment as measured by the Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC™) [5], and patient and physician global assessment of OA in the target knee using a 100 mm VAS at all visits. Concomitant use of permitted rescue medication (paracetamol ≤3 g/day) was also recorded.
Statistical methods
The safety analysis was performed on all patients who underwent a first injection. Treatment-emergent AE’s were summarised by the number and percentage of patients for each MedDRA Preferred Term for each treatment group. Post-hoc analyses were performed using Fisher’s Exact Test to compared Incidence of Target Knee Treatment-Emergent AEs between Groups.
The primary efficacy hypothesis was evaluated by the change from baseline to the week 24 evaluation in the patient’s assessment of target knee OA pain during the previous 48 h on VAS. This endpoint was analysed for each treatment group in the ITT population (all patients who underwent the first injection and had at least one post-baseline efficacy assessment) using a paired t test. No statistical analysis was performed between treatment groups.
The secondary endpoints (patient-completed knee OA pain score on VAS at all other time points; total WOMAC score; WOMAC A (pain), B (stiffness), and C (physical function) sub-scores; patient global assessment (VAS) and physician global assessment (VAS)) were analysed similarly. The number and percentage of patients using paracetamol was tabulated for each treatment group at each visit. Paracetamol use was summarised by: number of patients, mean, median, standard deviation and range at each visit for each treatment group.
Results
The ITT population consisted of 100 patients (56 female and 44 male) with a mean age of 61.1 years (range 41–86 years). These and other demographic parameters were similarly distributed between the five treatment groups (Table 1). Patient disposition, re-treatment rates and reasons for discontinuation are shown in Table 2.
Safety
The treatment was well tolerated. There were no serious or severe, device-related AE’s in any of the studied dosing regimens, nor were any new safety concerns identified following initial or repeat treatment. Group 4 (3 × 4 mL) had the highest percentage of patients reporting device-related local AE’s (30%) while Group 1 (1 × 6 mL) and Group 5 (3 × 2 mL) had only 10% (Table 3). These device-related local AE’s consisted mostly of mild or moderate post-injection pain (n = 12 patients) with local inflammation (described as synovitis by some investigators, n = 3) or effusion (n = 1). Post-hoc analyses using Fisher’s Exact Test which compared incidence of target knee treatment-emergent AEs between groups 1–4 compared with Group 5 did not show any statistically significant differences between groups.
Twenty-four patients (24%) were re-treated in the extension study (Table 2). No safety concerns were raised by re-treatment with the same injection schedules. Four patients that were re-treated experienced five target knee AE’s (Table 3). No patients experienced AE’s in Group 1, while one patient reported an AE in each of Groups 2–5. None of the target knee, treatment-emergent AE’s was serious. One patient in Group 4 (3 × 4 mL) discontinued from the study due to synovitis with effusion at the target knee. One case of synovitis in Group 5 (3 × 2 mL) was severe.
Efficacy
Treatment with hylan G-F 20 resulted in a statistically significant improvement from baseline to week 24 in all endpoints for all treatment regimens (Fig. 1). The largest changes were observed in Group 5 (3 × 2 mL) with a mean change [SD] from baseline at week 24 in the patient-completed knee OA pain VAS score of −36.7 mm [26.9]. Groups 1 (1 × 6 mL) and 4 (3 × 4 mL) consistently showed similar mean improvement (respectively −34.9 mm [16.4] and −32.6 mm [25.3]). Smaller changes (−24.0 mm [22.9] and −24.3 mm [28.3]) were found in Group 3 (2 × 4 mL) and Group 2 (1 × 4 mL).

Mean target knee OA pain scores (VAS) at each visit for each treatment group
Table 4 summarises the rankings of mean response to treatment for all primary and secondary endpoints by treatment group. When the results of all assessment criteria were added together, Group 4 (3 × 4 mL) was ranked first. Group 1 (1 × 6 mL) was ranked second and was either first or second in 5 of the 6 endpoints.
Twenty-four patients (those who had a VAS pain score ≥50 and ≤80 mm at week 24, or those who had a 15 mm increase in pain between weeks 16 and 24) were re-treated in the extension study. The group with the highest number of re-treated patients (n = 7) was Group 3 (2 × 4 mL). Group 1 (1 × 6 mL) had the lowest number of patients qualifying for repeat treatment (Table 2).
Discussion
The principal objective of this pilot study was to examine the safety of alternate dosing regimens of hylan G-F 20 to that currently approved for the treatment of symptomatic knee OA (3 × 2 mL). Though this study was not powered to demonstrate statistical difference on efficacy analysis, trends in efficacy data were examined using patient-completed questionnaires and physician assessments to provide insight into possible alternate dosage regimens with higher volumes and lower numbers of injections. Another objective was to examine the safety of giving a second course of these alternate dosing regimens.
The data presented here suggest that alternative protocols could be proposed to replace the current 3 × 2 mL regimen of viscosupplementation with hylan G-F 20, offering similar clinical efficacy without a greater percentage of adverse events. In patients with hip [7, 8, 22], shoulder [12] and ankle [25] OA, experience with a single intra-articular injection of 2 mL hylan G-F 20 has demonstrated significant immediate, and sustained, symptomatic effect for up to 6 months.
Among the hypotheses explaining these efficacy effects is the suggestion that these joints have a smaller volume than the knee and consequently a single injection of 2 mL hylan G-F 20 would exhibit a greater ‘fill’ ratio. Increasing the ‘fill’ in the knee by injecting a single dose of a higher volume (6 mL as opposed to 2 mL currently) could therefore be likely to provide improved immediate and sustained efficacy.
Given the known safety profile of hylan G-F 20 in its current usage [9, 16] and the volume of the knee space, it was thought that the injection of the proposed volumes should not pose major safety concerns. This has been confirmed by the present data which show no more target knee AE’s in the 1 × 6 mL group than in the 3 × 2 mL group. The percentage of device- and procedure-related AE’s (all mild or moderate in intensity) in these two groups was similar to that previously published in clinical trials using a 3 × 2 mL dosing regimen. In contrast, the 3 × 4 mL group experienced about three times more local AE’s than expected [15, 24]. Nevertheless none of these AE’s was serious and no severe, acute, inflammatory reactions were reported.
In summary, the risk/benefit profile of a single 6 mL injection appears to be good and this regimen could be developed as an alternative to the currently approved 3 × 2 mL regimen for the treatment of symptomatic knee OA. There is indeed a medical need to have effective and safe single injection products since repeated intra-articular injections can be a limiting factor for viscosupplementation treatment. A 6 mL single injection could be particularly useful for patients undergoing concomitant anti-thrombotic therapy (i.e. vitamin K antagonists, aspirin, clopidogrel). In those patients with active and busy lifestyles, or who have travelling challenges due to distance or schedule, a single dose treatment regimen may improve patient compliance. Additionally, a single dose regimen would reduce the risk of procedure-related local AE’s, particularly infectious arthritis and offer medico-economic benefits. Furthermore, not only a single injection allows a major compliance from the patients and reduces risks connected to intra-articular injection, but also has its importance in pharmaco-economics: a minor number of injections shortens medical costs connected to hospital visits, medications, work time of physicians and nurses and patients absenteeism.
These data support the need for a large-scale, prospective clinical trial comparing the safety and efficacy of a single 6 mL intra-articular injection of hylan G-F 20 to placebo, in patients suffering from knee osteoarthritis.
References
Adams ME, Atkinson MH, Lussier AJ, Schulz JI, Siminovitch KA, Wade JP, Zummer M (1995) The role of viscosupplementation with hylan G-F 20 (Synvisc) in the treatment of osteoarthritis of the knee: a Canadian multicenter trial comparing hylan G-F 20 alone, hylan G-F 20 with non-steroidal anti-inflammatory drugs (NSAIDs) and NSAIDs alone. Osteoarthritis Cartilage 3:213–225
Altman R, Alarcon D, Appelrouth D, The American College of Rheumatology subcommittee on criteria for osteoarthritis (1991) The American College of Rheumatology criteria for the classification and reporting of osteoarthritis of the hip. Arthritis Rheum 34:505–511
Bagga H, Burkhardt D, Sambrook P, March L (2006) Long term effects of intra-articular hyaluronan on synovial fluid in osteoarthritis of the knee. J Rheumatol 33:946–950
Balazs EA (1958) Physical chemistry of hyaluronic acid. Fed Proc 17(4):1086–1093
Bellamy N, Buchanan WW, Goldsmith CH, Campbell J, Stitt WJ (1995) Validation of WOMAC. A health status instrument for measuring clinically important patient outcomes to rheumatic drug therapy in patients with osteoarthritis of the hip or knee. J Rheumatol 15:1833–1840
Bellamy N, Campbell J, Robinson V, Gee T, Bourne R, Wells G (2006) Viscosupplementation for the treatment of osteoarthritis of the knee. Cochrane Database Syst Rev 2:CD005321
Brocq O, Tran G, Breuil V, Grisot C, Flory P, Euller-Ziegler L (2002) Hip osteoarthritis: short-term efficacy and safety of viscosupplementation by hylan G-F 20. An open-label study in 22 patients. Joint Bone Spine 69:388–391
Conrozier T, Bertin P, Mathieu P, Charlot J, Bailleul F, Trèves R, Vignon E, Chevalier X (2003) Intra-articular injections of hylan G-F 20 in patients with symptomatic hip osteoarthritis: an open-label, multicentre, pilot study. Clin Exp Rheumatol 21:605–610
Conrozier T, Mathieu P, Schott AM, Laurent I, Hajri T, Crozes P, Grand P, Laurent H, Marchand F, Meignan F, Noel E, Rozand Y, Savoye JF, Vignon E (2003) Factors predicting long-term efficacy of hylan G-F 20 viscosupplementation in knee osteoarthritis. Joint Bone Spine 70:128–133
Dickson DJ, Hosie G (2001) A double blind, placebo-controlled comparison of hylan G-F 20 against diclofenac in knee osteoarthritis. J Clin Res 4:41–52
Felson DT (1990) The epidemiology of knee osteoarthritis: results from the Framingham Osteoarthritis Study. Semin Arthritis Rheum 20(3 (Suppl 1)):42–50
Goupille P, Hagena FW, Laprelle E, Goebel F, Noel E, Hardy P (2007) Prospective study of the safety and efficacy of hylan G-F 20 in symptomatic shoulder osteoarthritis. Poster presentation (P277) at the AAOS annual meeting, San Diego
Kahan A, LLeu PL, Salin L (2003) Prospective randomized study comparing the medicoeconomic benefits of hylan G-F 20 vs. conventional treatment in knee osteoarthritis. Joint Bone Spine 70:276–281
Kellgren JH, Lawrence JS (1957) Radiological assessment of osteoarthrosis. Ann Rheum Dis 16:494–501
Kemper F, Gebhardt U, Meng T, Murray C (2005) Tolerability and short-term effectiveness of hylan G-F 20 in 4,253 patients with osteoarthritis of the knee in clinical practice. Curr Med Res Opin 21:1261–1269
Lussier A, Cividino AA, McFarlane CA, Olszynski WP, Potashner WJ, De Medicis R (1996) Viscosupplementation with hylan for the treatment of osteoarthritis: findings from clinical practice in Canada. J Rheumatol 23:1579–1585
Pelletier JP, Martel-Pelletier J (1993) The pathophysiology of osteoarthritis and the implication of the use of hyaluronan and hylan as therapeutic agents in viscosupplementation. J Rheumatol 20(Suppl 39):19–23
Raynauld J-P, Torrance GW, Band PA, Goldsmith CH, Tugwell P, Walker V, Schultz M, Bellamy N, Canadian Knee OA Study Group (2002) A prospective, randomized, pragmatic, health outcomes trial evaluating the incorporation of hylan G-F 20 into the treatment paradigm for patients with knee osteoarthritis (Part 1 of 2): clinical results. Osteoarthritis Cartilage 10:506–517
Raynauld J-P, Goldsmith CH, Bellamy N, Torrance GW, Polisson R, Belovich D, Pericak D, Tugwell P (2005) Effectiveness and safety of repeat courses of hylan G-F 20 in patients with knee osteoarthritis. Osteoarthritis Cartilage 13:111–119
Scale D, Wobig M, Wolpert W (1994) Viscosupplementation of osteoarthritic knees with hylan: a treatment schedule study. Curr Ther Res 55:220–232
Synvisc instructions for use (2006) Genzyme biosurgery
Tikiz C, Unlu Z, Sener A, Efe M, Tuzun C (2006) Comparison of the efficacy of lower and higher molecular weight viscosupplementation in the treatment of hip osteoarthritis. Clin Rheumatol 24:244–250
Waddell DD, Cefalu CA, Bricker DC (2003) An open-label study of a second course of hylan G-F 20 for the treatment of pain associated with knee osteoarthritis. Curr Med Res Opin 19(6):499–507
Waddell DD, Bricker DC (2006) Clinical experience with the effectiveness and tolerability of hylan G-F 20 in 1,047 patients with osteoarthritis of the knee. J Knee Surg 19:19–27
Witteveen AGH, Giannini S, Guido G, Jerosch J, Lohrer H, van Dijk CN (2006) Prospective study of the safety and efficacy of hylan G-F 20 (Synvisc®) in patients with symptomatic ankle osteoarthritis. Poster presentation at the OARSI annual meeting, Prague
Wobig M, Dickhut A, Maier R, Vetter G (1998) Viscosupplementation with hylan G-F 20: a 26-week controlled trial of efficacy and safety in the osteoarthritic knee. Clin Ther 20:410–423
Acknowledgments
This study was sponsored by Genzyme Europe b.v. This study complied with the current laws of the countries in which it was performed.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Conrozier, T., Jerosch, J., Beks, P. et al. Prospective, multi-centre, randomised evaluation of the safety and efficacy of five dosing regimens of viscosupplementation with hylan G-F 20 in patients with symptomatic tibio-femoral osteoarthritis: a pilot study. Arch Orthop Trauma Surg 129, 417–423 (2009). https://doi.org/10.1007/s00402-008-0601-2
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00402-008-0601-2