
Abstract
The genetic variant rs72824905-G (minor allele) in the PLCG2 gene was previously associated with a reduced Alzheimer’s disease risk (AD). The role of PLCG2 in immune system signaling suggests it may also protect against other neurodegenerative diseases and possibly associates with longevity. We studied the effect of the rs72824905-G on seven neurodegenerative diseases and longevity, using 53,627 patients, 3,516 long-lived individuals and 149,290 study-matched controls. We replicated the association of rs72824905-G with reduced AD risk and we found an association with reduced risk of dementia with Lewy bodies (DLB) and frontotemporal dementia (FTD). We did not find evidence for an effect on Parkinson’s disease (PD), amyotrophic lateral sclerosis (ALS) and multiple sclerosis (MS) risks, despite adequate sample sizes. Conversely, the rs72824905-G allele was associated with increased likelihood of longevity. By-proxy analyses in the UK Biobank supported the associations with both dementia and longevity. Concluding, rs72824905-G has a protective effect against multiple neurodegenerative diseases indicating shared aspects of disease etiology. Our findings merit studying the PLCγ2 pathway as drug-target.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The protein product of the phospholipase Cγ2 (PLCG2) gene is involved in the transmembrane transduction of immune signals [30, 42, 45] that determine the fate and function of various immune cell types, both in the periphery and the brain [42, 45]. It is known that gain-of-function mutations in the PLCG2 gene cause autoimmune disorders [40, 46, 58, 59] and resistance to treatment of chronic lymphocytic leukemia [56].
In 2017, a genome-wide association (GWA) study of Alzheimer’s disease (AD) showed that the rare nonsynonymous variant in the PLCG2 gene (rs72824905-G; p.Pro522Arg; NC_000016.9:g.81942028C > G) reduced AD risk (OR = 0.68, p = 5.4 × 10−10) [47]. In both mouse and human brain tissues, PLCG2 has been shown to be overexpressed > 6-log fold in microglia compared to other brain cells [12]. Further, PLCG2 has higher expression levels in pathologically affected brain regions of AD patients, which seems to be driven by microgliosis [7]. Since microglia are the brain’s immune cells, these findings suggest an important role for PLCG2 in the neural immune response. Next to PLCG2, GWA studies of AD identified additional immune- and microglia-related genes that associate with AD, e.g. the triggering receptor expressed on myeloid cells 2 (TREM2) gene and pathway analysis based on these same GWA studies indicate that the immune system plays a key role in the development of AD [47]. Likewise, human genetic studies imply the immune system plays a role in other neurodegenerative diseases such as frontotemporal dementia (FTD) [3], Parkinson’s disease (PD) [13], and multiple sclerosis (MS) [17, 20, 41]. We reasoned that next to AD, PLCG2-related immune signaling may be involved in the etiology of these other neurodegenerative diseases. This led us to question whether the rs72824905-G variant in PLCG2 is also associated with a reduced risk of other neurodegenerative diseases.
Here, we tested whether rs72824905-G protects against other neurodegenerative diseases. We first tested whether rs72824905-G associates with reduced risk of AD, FTD, dementia with Lewy bodies (DLB), progressive supranuclear palsy (PSP), PD, amyotrophic lateral sclerosis (ALS) and MS. Since a reduced risk of neurodegenerative diseases could lead to an increased likelihood to survive to old age, we tested whether rs72824905-G associated with longevity.
Materials and methods
Study populations and genotyping
We present a short description of 16 cohorts, often including multiple sites or studies, which contributed to this manuscript in Suppl. Table 1, Online Resource. Studies were approved by corresponding ethics committees and informed consent was obtained for all participants (Suppl. Table 1, Online Resource). Study characteristics (age, percentage female, apolipoprotein E (APOE) status and age) are described in Suppl. Table 2, Online Resource. In most cohorts, the average age of the controls was lower than that of cases (Suppl. Fig 3, Online Resource). We determined rs72824905-G genotypes (NC_000016.9:g.81942028C > G, p.Pro522Arg) using direct genotyping with a variety of genotyping arrays or TaqMan genotyping. If direct genotyping was not available, we used imputation to 1000 Genomes phase I version 3 [15] or the Haplotype Reference Consortium (HRC) reference panels [37]. Details on genotyping or imputation by study can be found in Suppl. Table 3, Online Resource. We studied participants from European descent.
Study populations of AD, FTD, DLB and PSP patients
We compared rs72824905-G genotypes in a total of 4,985 AD patients and 9,238 controls from eight cohorts. All samples were independent from Sims et al. [47], but include the samples from Conway et al. [7]. We compared in total 2,437 FTD patients with 10,647 controls from four studies and two consortia. Further, we studied 1446 DLB patients with 5509 controls from five cohorts and 882 PSP patients with 3187 controls from five cohorts. Details on sample size by cohort and which cohort contributed to which analysis can be found in Suppl. Table 2, Online Resource.
Study populations of ALS, PD and MS patients
To study the association of rs72824905-G with ALS, PD and MS, we obtained summary statistics from existing GWAS meta-analyses, see Suppl. Table 1, 2, 3, Online Resource, for study descriptions. We present results of a combined total of 28,448 PD patients that were compared with 108,438 controls: data from 27,595 PD patients and 106,951 controls from the International Parkinson Disease Genomics Consortium (IPDGC) [39] were combined with data from 853 PD patients and 1,487 controls from the Mayo Clinic. Furthermore, we studied 10,953 ALS patients and 20,673 controls, which represents the subset of the data presented by van Rheenen et al. [54], for which rs72824905-G was imputed with sufficient quality (imputation quality > 0.3). Last, we studied 4476 MS patients and 5714 controls which were previously described by Dankowski et al. [8].
Study populations of longevity
We investigated the association of rs72824905-G with longevity in five different cohorts; in total, we compared 3516 individuals who reached at least 90 years with 9677 control individuals who died before age 90 years or were last screened before 90 years (Suppl. Table 1–3, Online Resource). The data from Tesi et al. [51] were included in this study. A subset of 1136 Dutch long-lived individuals for whom follow-up data until death were available [22] was included. In this subset, we compared the survival of carriers of rs72824905-G with non-carriers.
Studies of dementia and longevity by-proxy in the UK Biobank
The UK Biobank is a study of genetic and health of a half million people from the United Kingdom [49]. Information from parents or first-degree relatives can be used as a proxy-phenotype for the participants [34]. In this study, we used maternal and paternal history of Alzheimer’s/dementia as proxy for dementia [34, 36] and the reported age of the parents (at completing the survey or death) as proxy phenotype for longevity [44]. In the UK Biobank, the rs72824905-G variant was imputed using the available genotyping arrays and the HRC-reference panel as previously described [25]. The maternal and paternal by-proxy phenotypes were analyzed separate using the genotypes of the participants and the results were meta-analyzed.
We compared rs72824905-G genotypes of 32,262 participants whose mother was reported to have dementia with the genotypes of 346,999 participants whose mothers did not have dementia. Likewise, we compared 16,968 participants whose father had dementia with 358,468 whose fathers did not have dementia.
For the analysis of longevity-by-proxy, we chose the age of 90 years as a cut-off for the minimum age reached by the parents. By principle, phenotype by-proxy analyses suffer from dilution effect [34, 36]; therefore, a more extreme parental age cut-off of 95 years was also studied. In this analysis, we compared 35,256 UK Biobank participants who had a mother who reached at least 90 years (7790 mothers reached the age of 95 years) with 342,810 participants whose mother did not reach 90 years of age. Likewise, we compared 17,558 UK Biobank participants with a father who reached at least 90 years (3,043 fathers reached the age of 95 years) with 353,100 participants whose father did not reach 90 years of age.
Statistical analysis
R (version 3.5.1) was used for all analysis [50]. Logistic regression models were fitted within studies to assess the association of rs72824905-G with AD, FTD, DLB, and PSP patients, and long-lived individuals, compared to controls. For each study, we calculated the odds ratio’s (OR) and 95% confidence intervals (CI). We accounted for population substructure by adjusting for principal components or by comparing cases and controls from the same study or country of origin. We meta-analyzed the effect estimates (log(OR)) from the studies using inverse-variance fixed-effect meta-analyses (R-package ‘rmeta’ v3.0). The fraction of variance that is due to heterogeneity was estimated by the I2 statistic [21]. We visualized survival of rs72824905-G carriers compared to non-carriers using Kaplan–Meier curves. Differences in survival were tested using a Cox proportional hazards model correcting for (age at inclusion, sex and relatedness).
For MS, the results originate from a single study, which used ancestry principal components (PCs) to adjust for population stratification [8]. The statistical methods of the GWAS meta-analyses of ALS and PD were previously described [39, 54]. In short, individual cohorts calculated logistic regression models and then summary statistics of cohorts were combined using inverse-variance fixed-effect meta-analyses. PCs were used to adjust for population stratification. Analysis in the UK Biobank were performed using logistic regression models adjusted for genotyping array and the first 12 PCs. Effect estimates of the paternal and maternal analysis were combined using inverse-variance fixed-effect meta-analysis (R-package ‘rmeta’ v3.0). We reported two-sided p values and considered p values < 0.05 as significant; p values are not corrected for multiple testing.
Power analysis
For all diseases studied, we performed power analysis using the online tool Genetic Association Study (GAS) power Calculator implementing the methods described in Skol et al. [48]. We calculated power of our analysis to attain a p value of 0.05 and used the total number of cases and controls from our analysis. We assumed an additive model, a minor allele frequency of 0.009 and a disease frequency of 0.01 for all diseases (higher disease frequency assumption would lead to higher power estimates). We report the power for an OR between 1 and 2. This corresponds to protective OR (the inverse OR = 1/OR) between 0.50 and 1.
Results
An overview of study sample, contributing studies, corrections applied by study and counts of carriers split by case–control status is shown in Table 1.
Association with brain diseases
We replicated the association of rs72824905-G in PLCG2 with a reduced AD risk (OR = 0.57, p = 6.0 × 10−4, I2 = 0%). In addition, we found that rs72824905-G associated with a reduced risk of both DLB (OR = 0.54, p = 0.045, I2 = 0%) and FTD (OR = 0.61, p = 0.011, I2 = 0%). In contrast, we found no evidence that rs72824905-G is associated with PSP (OR = 1.46, p = 0.19, I2 = 0%), ALS (OR = 1.07, p = 0.52, I2 = 0%), PD (OR = 1.18, p = 0.10, I2 = 0) and MS (OR = 0.99, p = 0.95). The association of rs72824905-G with these seven brain diseases is shown in Fig. 1. In Suppl. Figs. 2–7, Online Resource, we show the association estimates for each study separately in the meta-analyses for AD, DLB, FTD, PSP, ALS and PD (the MS study consisted of a single study).

Association results of rs72824905-G with seven brain diseases and longevity. *P values < 0.05. Numbers (N) of cases (patients or long-lived individuals) and controls studied. The figure shows the odds-ratio (box) of the rs72824905-G with the 95% confidence intervals (whiskers)
Association with longevity
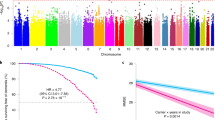
In line with a reduced risk of neurodegenerative diseases, we found that rs72824905-G associated with a 1.49 (95% CI 1.12–1.98) increased likelihood (p = 6.3 × 10−3, I2 = 0%) to reach the age of 90 years. Although no heterogeneity was observed between studies, it is of interest that a cohort of centenarians who were selected based on being 100 years old and cognitively healthy (description of ‘100-plus Study’ in Suppl. Table 1, Online Resource) was most enriched with rs72824905-G (OR = 2.36, 95% CI 1.34–4.15, p = 2.8 × 10−3) (Suppl. Fig 8, Online Resource). Next, we tested whether carrying the rs72824905-G variant was associated with longer survival after the age of 90 years in 1,136 Dutch long-lived individuals of which 96.3% were followed until death [median age at inclusion 93.2, IQR 91.6–95.0 years, 63% female; mean survival after inclusion was 3.3 years; inter quartile range (IQR) 1.4–5.8 years]. We found that 28 carriers survived a median of 4.7 years (IQR = 1.9–7.4) while 1108 non-carriers survived a median of 3.3 years (IQR = 1.4–5.8) (Suppl. Fig 9, Online Resource). However, the difference was not significant (HR 0.75, 95% CI 0.51–1.09, p = 0.078), likely due to the low number of rs72824905-G carriers in the analysis, as a consequence of variant rareness (MAF ~ 1%).
Association with by-proxy dementia and longevity
In line with the protection against AD, the by-proxy analysis showed that PLCG2 variant carriers had a reduced risk of having a parent with dementia, OR = 0.88 (0.81–0.95, p = 1.9 × 10−3) (Fig. 2). Next, we tested the association of rs72824905-G with longevity-by-proxy. Carriers of the rs72824905-G variant did not have an increased likelihood of having a parent who reached 90 years of age compared to non-carriers (OR = 1.05, p = 0.24). However, carriers did have an increased likelihood of having a parent who reached 95 years (OR = 1.19, p = 2.1 × 10−2). The threshold of 95 years was chosen as too few parents reached the age of 100 years.

Association results of rs72824905-G with dementia by-proxy and longevity by-proxy analysis in the UK Biobank. *P values < 0.05. The figure shows the odds-ratio (box) of the rs72824905-G with the 95% confidence intervals (whiskers)
Power analysis
Power analysis (Suppl. Fig 10, Online Resource) showed that the PD, MS and ALS analysis had adequate statistical power (power > 0.8) to detect a protective association (p = 0.05) with an OR ~ 0.68 (the OR for AD reported in Sims et al. [47].). The PSP analysis had the lowest statistical power (0.32 at the expected OR = 0.67).
Discussion
A recent study showed the protective effect against AD risk of the p.Pro552Arg nonsynonymous amino acid change in PLCG2 (rs72824905-G) [47]. We replicated this protective effect in independent AD patients and controls. Additionally, we found that the variant also protected against FTD and DLB, but not against ALS, PD and MS. The analysis of PSP was inconclusive because of insufficient power. We also found that rs72824905-G associated with increased likelihood of longevity, which is according to expectations, since overall dementia is the leading cause of death at older age [1]. Indeed, the strongest effect of PLCG2 variant was observed in cognitively healthy centenarians, individuals where an absence of dementia and extreme longevity is combined. Our findings were supported by analyses of by-proxy phenotypes for dementia and longevity in the UK Biobank. Taken together, the association of the rs72824905-G variant with a decreased risk of multiple dementia types and the increased risk of longevity warrants thorough investigation of the molecular mechanisms underlying this protective effect.
Thus far, the common APOE ɛ4 allele and the rare TREM2.R47H variant are strong genetic risk factors shared across AD, FTD and DLB (not TREM2 [55]). [2, 11, 18, 27, 47] The HLA-locus and the microtubule-associated protein tau (MAPT) loci (not individual variants) also have (suggestive) effects on the risks of AD, FTD and DLB [3, 11, 18, 43]. The APOE gene has been implied in a multitude of pathways [52], TREM2 and HLA are involved in immunity [2], and MAPT encodes the tau protein. These shared genetic risk factors indicate a partial overlap in AD, DLB and FTD etiology. It is of interest that, like the PLCG2 variant, APOE and the HLA-DR locus were also associated with longevity [4, 10, 28, 38]. A possible explanation is that APOE, PLCG2 and HLA are involved in the processing of accumulated aging-associated proteins [9]. In line with this hypothesis, rs72824905-G is associated with reduced pTau181 in the CSF of memory clinic patients with pathologic Aβ1-42 CSF levels (L. Kleineidam et al. submitted). It is well known that having a dementia-associated neurodegenerative disease is associated with shorter life-span [57]. Conversely, resilience to diseases is associated with a longer life-span [24]. It is likely that the association of rs72824905-G with longevity is due to the protection against dementia-associated neurodegenerative diseases. However, with the available data we cannot exclude that rs72824905-G has an independent effect of rs72824905-G on the risk of longevity and/or the risk of maintaining cognitive health. In line with this observation, we anecdote one cognitively healthy centenarian who is homozygous for the APOE ɛ4 risk allele, but also carried the rs72824905-G allele. On MRI scan and amyloid scan (PiB-PET), this person has some global atrophy and only amyloid-β positivity in the precuneus and in the frontal lobes (Fig. 3). At the age of 90 years, the dementia risk for homozygous carriers of the APOE ɛ4 genotype is approximately 80% [53] and virtually all are amyloid positive by age 90 [26]. The literature reports only a handful of centenarians who are homozygous for the APOE ɛ4 allele [14, 16, 23]. It is unknown if these individuals were cognitively healthy. This case shows that cognitively healthy aging in the presence of the APOE ɛ4ɛ4 genotype is possible, likely due to the protective effect of other genetic variants, such as the rs72824905-G variant in PLCG2 [26, 53].

MRI scan and PiB-PET scan, of a 102-year-old centenarian carrying the homozygote APOE ɛ4 genotype as well as the rs72824905-G allele in PLCG2. MRI scan (Titan 3T MR scanner) shows some hippocampal atrophy (MTA grade 2), some global cortical atrophy (GCA-scale grade 1), but pronounced posterior cortical atrophy (grade 2), moderate white matter hyperintensities (Fazekas grade 2), no lacunar infarcts or microbleeds. PET-PiB (scan after admission of 396 MBq C-11 PIB, 20-min image starting 90 min after administration): Abnormal retention in the posterior cingulate/precuneus and frontal lobes. Neuropsychological testing around time of scanning showed average performance on global cognitive functioning/MMSE, memory, attention, working memory, fluency and visuo-spatial tests compared to the cohort of cognitively healthy centenarians. The result of the clock drawing test is shown. The patient was asked to draw a clock and put the time at 10 before 11
The mechanism that explains the protective effect of rs72824905-G variant in the PLCG2 gene is currently unclear. We find that the associations of the rs72824905-G variant with disease risk differ between diseases that have overlapping pathological features. For example, we observe that carrying the rs72824905-G variant is protective against DLB, but not against PD, while a common characteristic for both diseases is the presence of α-synuclein-positive Lewy bodies. The same holds for pathologies associated with the FTD-ALS and PSP spectrum of diseases (e.g. TDP-43, FUS inclusions as well as aggregations of tau). The observation that our results do not point to a single pathological condition does not preclude that PLCG2 is involved in a single biological process. In fact, determining the involvement of the PLCG2-related pathway might be an asset in pathological classifications of diseases, e.g. differentiating between DLB and PD. Thus far, only one publication investigated the functional effect of the rs72824905-G variant in in vitro experiments [35]. The authors suggest that in the mouse and human brain, PLCɣ2 is expressed in microglia [12]. They show that PLCɣ2 mRNA co-localized with microglia-specific markers in healthy brain tissue and is expressed in microglia near amyloid-β plaques in an APP mouse model [35]. Furthermore, functional characterization of PLCɣ2 with the p.Pro552Arg amino acid substitution suggested only a slight increase in activity compared to wild-type PLCɣ2 [35]. While additional functional experiments will be needed to confirm these findings, these experiments suggest that the functional changes induced by the PLCɣ2 p.Pro552Arg genetic variant may be subtle and, therefore, difficult to pinpoint. This is according to expectations, as major changes to the immune system will most likely be harmful. Indeed, known germline mutations in PLCG2 cause the immune disorders PLAID (PLCG2-associated antibody deficiency and immune dysregulation) and APLAID (autoinflammatory PLAID) [40, 46, 59] while somatic variants in PLCɣ2 are associated with resistance to treatment of leukemia [56] (reviewed in Koss et al. [32].). The mutations that cause PLAID and APLAID contribute to a strong hyperactivation of PLCɣ2 upon activation. In the case of APLAID (caused by a p.Ser707Tyr substitution), the auto-inflammation has been suggested to be partially driven by PLCɣ2-dependent activation of the pyrin (PYD)-domain-containing protein 3 (NLRP3) inflammasome [5]. The potential of PLCG2 to activate the inflammasome is further supported by in vitro experiments [31]. The NLRP3 inflammasome is a crucial signaling node in microglia that ultimately controls the maturation of pro-inflammatory interleukin (IL)-1β and IL-18 [19] and has been linked to a multitude of neurodegenerative diseases [60]. Although functional studies will need to elucidate the effects of the rs72824905-G on PLCɣ2 function, we speculate that subtle changes in the NLRP3 inflammasome activation may explain its protective effect.
Strengths and weaknesses
The most important strength of our study is that we investigated the effect of the rs72824905-G variant in seven neurological diseases in more than 53,000 patients and almost 150,000 controls. The AD cases and controls studied here were all independent from the AD patients and controls in which the protective effect of rs72824905-G was first identified [47], but includes the samples used in Conway et al. [7] and Tesi et al. [51]. This report offers a robust replication of the protection against AD. Some may argue that the protective effect observed in FTD and DLB cases is driven by misclassified AD cases. However, the effect size of rs72824905-G in these cases is very similar to the protective effect in AD, which makes it unlikely that the effect can be ascribed purely to misclassified AD. Moreover, the age of the controls was mostly younger than that of cases, making the protection from dementia not a longevity effect.
The large numbers under study were necessary because rs72824905-G has a minor allele frequency (MAF) ~ 1% in European ancestry populations, which makes it a relatively rare genetic variant. Therefore, we ensured that our samples provided adequate statistical power to observe a similar protective effect of rs72824905-G against other neurodegenerative diseases. Despite the large sample sizes, we found no evidence for this effect in our sample of PD, ALS and MS, which makes it unlikely that larger meta-analyses will observe an association between rs72824905-G and these three diseases. An association of rs72824905-G with an increased risk of PSP has been reported previously [7]. In our analysis, which includes additional PSP cases, we were not able to replicate this finding. Larger studies are needed to determine the association with PSP. Including as large as possible samples came with the consequence that we were not able to correct for population stratification using ancestry principal components in all studies. Therefore, we used PCs were possible and further matching cases and controls by study or country of origin. Finally, we indicate that the identified effects need to be replicated in other ethnicities in which rs72824905-G occurs. We note that in some ethnicities rs72824905-G plays no role as the frequency is very low in African (MAF = 0.0012) and African-American populations (MAF = 0.0004), and is not observed in East Asian [7, 33].
Conclusions
Our study shows that the rs72824905-G allele in PLCG2 associates with a decreased risk for AD, FTD, DLB and concurrently with an increased chance of longevity. The protective effect of the rs72824905-G allele was not observed in ALS, PD and MS cases, which suggests that PLCG2-associated processes overlap in the etiology of AD, FTD and DLB, but not in the etiologies of ALS, PD and MS (PSP too little power). Explaining the protective effect of the PLCγ2 protein on brain immune function may contribute to the design of successful therapeutic intervention strategies applicable to those at risk for neurodegenerative diseases.
Change history
18 January 2020
The IPDGC (The International Parkinson Disease Genomics Consortium) and EADB (Alzheimer Disease European DNA biobank) are listed correctly as an author to the article, however, they were incorrectly listed more than once.
References
Alzheimer’s A (2016) 2016 Alzheimer’s disease facts and figures. Alzheimers Dement 12:459–509
Borroni B, Ferrari F, Galimberti D, Nacmias B, Barone C, Bagnoli S et al (2014) Heterozygous TREM2 mutations in frontotemporal dementia. Neurobiol Aging 35(934):e937-910. https://doi.org/10.1016/j.neurobiolaging.2013.09.017
Broce I, Karch CM, Wen N, Fan CC, Wang YP, Tan CH et al (2018) Immune-related genetic enrichment in frontotemporal dementia: an analysis of genome-wide association studies. Plos Med. https://doi.org/10.1371/journal.pmed.1002487
Broer L, Buchman AS, Deelen J, Evans DS, Faul JD, Lunetta KL et al (2015) GWAS of longevity in CHARGE consortium confirms APOE and FOXO3 candidacy. J Gerontol A Biol Sci Med Sci 70:110–118. https://doi.org/10.1093/gerona/glu166
Chae JJ, Park YH, Park C, Hwang IY, Hoffmann P, Kehrl JH et al (2015) Connecting two pathways through Ca2+ signaling: NLRP3 inflammasome activation induced by a hypermorphic PLCG2 mutation. Arthritis Rheumatol 67:563–567. https://doi.org/10.1002/art.38961
Chen JA, Wang Q, Davis-Turak J, Li Y, Karydas AM, Hsu SC et al (2015) A multiancestral genome-wide exome array study of Alzheimer disease, frontotemporal dementia, and progressive supranuclear palsy. JAMA Neurol 72:414–422. https://doi.org/10.1001/jamaneurol.2014.4040
Conway OJ, Carrasquillo MM, Wang X, Bredenberg JM, Reddy JS, Strickland SL et al (2018) ABI3 and PLCG2 missense variants as risk factors for neurodegenerative diseases in Caucasians and African Americans. Mol Neurodegener 13:53. https://doi.org/10.1186/s13024-018-0289-x
Dankowski T, Buck D, Andlauer TF, Antony G, Bayas A, Bechmann L et al (2015) Successful replication of GWAS hits for multiple sclerosis in 10,000 germans using the exome array. Genet Epidemiol 39:601–608. https://doi.org/10.1002/gepi.21933
Deane R, Sagare A, Hamm K, Parisi M, Lane S, Finn MB et al (2008) apoE isoform-specific disruption of amyloid beta peptide clearance from mouse brain. J Clin Invest 118:4002–4013. https://doi.org/10.1172/JCI36663
Deelen J, Beekman M, Uh HW, Broer L, Ayers KL, Tan Q et al (2014) Genome-wide association meta-analysis of human longevity identifies a novel locus conferring survival beyond 90 years of age. Hum Mol Genet 23:4420–4432. https://doi.org/10.1093/hmg/ddu139
Ferrari R, Wang Y, Vandrovcova J, Guelfi S, Witeolar A, Karch CM et al (2017) Genetic architecture of sporadic frontotemporal dementia and overlap with Alzheimer’s and Parkinson’s diseases. J Neurol Neurosurg Psychiatry 88:152–164. https://doi.org/10.1136/jnnp-2016-314411
Friedman BA, Srinivasan K, Ayalon G, Meilandt WJ, Lin H, Huntley MA et al (2018) Diverse brain myeloid expression profiles reveal distinct microglial activation states and aspects of Alzheimer’s disease not evident in mouse models. Cell Rep 22:832–847. https://doi.org/10.1016/j.celrep.2017.12.066
Gagliano SA, Pouget JG, Hardy J, Knight J, Barnes MR, Ryten M et al (2016) Genomics implicates adaptive and innate immunity in Alzheimer’s and Parkinson’s diseases. Ann Clin Transl Neur 3:924–933. https://doi.org/10.1002/acn3.369
Garatachea N, Marin PJ, Santos-Lozano A, Sanchis-Gomar F, Emanuele E, Lucia A (2015) The ApoE gene is related with exceptional longevity: a systematic review and meta-analysis. Rejuvenation Res 18:3–13. https://doi.org/10.1089/rej.2014.1605
Genomes Project C, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, et al (2015) A global reference for human genetic variation. Nature 526:68–74. https://doi.org/10.1038/nature15393
Gerdes LU, Jeune B, Ranberg KA, Nybo H, Vaupel JW (2000) Estimation of apolipoprotein E genotype-specific relative mortality risks from the distribution of genotypes in centenarians and middle-aged men: apolipoprotein E gene is a “frailty gene,” not a “longevity gene”. Genet Epidemiol 19:202–210. https://doi.org/10.1002/1098-2272(200010)19:3%3c202:AID-GEPI2%3e3.0.CO;2-Q
Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH (2010) Mechanisms underlying inflammation in neurodegeneration. Cell 140:918–934. https://doi.org/10.1016/j.cell.2010.02.016
Guerreiro R, Ross OA, Kun-Rodrigues C, Hernandez DG, Orme T, Eicher JD et al (2018) Investigating the genetic architecture of dementia with Lewy bodies: a two-stage genome-wide association study. Lancet Neurol 17:64–74. https://doi.org/10.1016/S1474-4422(17)30400-3
He Y, Hara H, Nunez G (2016) Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem Sci 41:1012–1021. https://doi.org/10.1016/j.tibs.2016.09.002
Hemmer B, Kerschensteiner M, Korn T (2015) Role of the innate and adaptive immune responses in the course of multiple sclerosis. Lancet Neurol 14:406–419. https://doi.org/10.1016/S1474-4422(14)70305-9
Higgins JP, Thompson SG (2002) Quantifying heterogeneity in a meta-analysis. Stat Med 21:1539–1558. https://doi.org/10.1002/sim.1186
Holstege H, Beker N, Dijkstra T, Pieterse K, Wemmenhove E, Schouten K et al (2018) The 100-plus Study of cognitively healthy centenarians: rationale, design and cohort description. Eur J Epidemiol. https://doi.org/10.1007/s10654-018-0451-3
Ishioka YL, Gondo Y, Fuku N, Inagaki H, Masui Y, Takayama M (2016) Effects of the APOE epsilon4 allele and education on cognitive function in Japanese centenarians. Age (Dordr) 38:495–503. https://doi.org/10.1007/s11357-016-9944-8
Ismail K, Nussbaum L, Sebastiani P, Andersen S, Perls T, Barzilai N et al (2016) Compression of morbidity is observed across cohorts with exceptional longevity. J Am Geriatr Soc 64:1583–1591. https://doi.org/10.1111/jgs.14222
Jansen IE (2017) Genetic meta-analysis identifies 9 novel loci and functional pathways for Alzheimers disease risk. Biorxiv 1:258533
Jansen WJ, Ossenkoppele R, Knol DL, Tijms BM, Scheltens P, Verhey FRJ et al (2015) Prevalence of cerebral amyloid pathology in persons without dementia a meta-analysis. J Am Med Assoc 313:1924–1938. https://doi.org/10.1001/jama.2015.4668
Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J et al (2013) Variant of TREM2 associated with the risk of Alzheimer’s disease. N Engl J Med 368:107–116. https://doi.org/10.1056/NEJMoa1211103
Joshi PK, Pirastu N, Kentistou KA, Fischer K, Hofer E, Schraut KE et al (2017) Genome-wide meta-analysis associates HLA-DQA1/DRB1 and LPA and lifestyle factors with human longevity. Nat Commun 8:910. https://doi.org/10.1038/s41467-017-00934-5
Keogh MJ, Wei W, Wilson I, Coxhead J, Ryan S, Rollinson S et al (2017) Genetic compendium of 1511 human brains available through the UK Medical Research Council Brain Banks Network Resource. Genome Res 27:165–173. https://doi.org/10.1101/gr.210609.116
Kim D, Jun KS, Lee SB, Kang NG, Min DS, Kim YH et al (1997) Phospholipase C isozymes selectively couple to specific neurotransmitter receptors. Nature 389:290–293. https://doi.org/10.1038/38508
Kleineidam L, Chouraki V, Próchnicki T, van der Lee S, Madrid-Márquez L, Wagner-Thelen H et al (2018) PLCG2 protective variant p.P522R modulates Tau pathology and disease progression in patients with mild cognitive impairment. Lancet. https://ssrn.com/abstract=3307649
Koss H, Bunney TD, Behjati S, Katan M (2014) Dysfunction of phospholipase Cgamma in immune disorders and cancer. Trends Biochem Sci 39:603–611. https://doi.org/10.1016/j.tibs.2014.09.004
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T et al (2016) Analysis of protein-coding genetic variation in 60,706 humans. Nature 536:285–291. https://doi.org/10.1038/nature19057
Liu JZ, Erlich Y, Pickrell JK (2017) Case-control association mapping by proxy using family history of disease. Nat Genet 49:325–331. https://doi.org/10.1038/ng.3766
Magno L, Lessard CB, Martins M, Lang V, Cruz P, Asi Y et al (2019) Alzheimer’s disease phospholipase C-gamma-2 (PLCG2) protective variant is a functional hypermorph. Alzheimers Res Ther 11:16. https://doi.org/10.1186/s13195-019-0469-0
Marioni RE, Harris SE, Zhang Q, McRae AF, Hagenaars SP, Hill WD et al (2018) GWAS on family history of Alzheimer’s disease. Transl Psychiatry 8:99. https://doi.org/10.1038/s41398-018-0150-6
McCarthy S, Das S, Kretzschmar W, Delaneau O, Wood AR, Teumer A et al (2016) A reference panel of 64,976 haplotypes for genotype imputation. Nat Genet 48:1279–1283. https://doi.org/10.1038/ng.3643
McDaid AF, Joshi PK, Porcu E, Komljenovic A, Li H, Sorrentino V et al (2017) Bayesian association scan reveals loci associated with human lifespan and linked biomarkers. Nat Commun. https://doi.org/10.1038/ncomms15842
Nalls MA (2018) Parkinson’s disease genetics: identifying novel risk loci, providing causal insights and improving estimates of heritable risk. BioRxiv. https://doi.org/10.1101/388165
Ombrello MJ, Remmers EF, Sun G, Freeman AF, Datta S, Torabi-Parizi P et al (2012) Cold urticaria, immunodeficiency, and autoimmunity related to PLCG2 deletions. N Engl J Med 366:330–338. https://doi.org/10.1056/NEJMoa1102140
Patsopoulos NA (2018) Genetics of multiple sclerosis: an overview and new directions. Cold Spring Harb Perspect Med 8:45. https://doi.org/10.1101/cshperspect.a028951
Patterson RL, van Rossum DB, Ford DL, Hurt KJ, Bae SS, Suh PG et al (2002) Phospholipase C-gamma is required for agonist-induced Ca2+ entry. Cell 111:529–541
Peuralinna T, Myllykangas L, Oinas M, Nalls MA, Keage HA, Isoviita VM et al (2015) Genome-wide association study of neocortical Lewy-related pathology. Ann Clin Transl Neurol 2:920–931. https://doi.org/10.1002/acn3.231
Pilling LC, Kuo CL, Sicinski K, Tamosauskaite J, Kuchel GA, Harries LW et al (2017) Human longevity: 25 genetic loci associated in 389,166 UK biobank participants. Aging (Albany NY) 9:2504–2520. https://doi.org/10.18632/aging.101334
Rhee SG (2001) Regulation of phosphoinositide-specific phospholipase C. Annu Rev Biochem 70:281–312. https://doi.org/10.1146/annurev.biochem.70.1.281
Schade A, Walliser C, Wist M, Haas J, Vatter P, Kraus JM et al (2016) Cool-temperature-mediated activation of phospholipase C-gamma2 in the human hereditary disease PLAID. Cell Signal 28:1237–1251. https://doi.org/10.1016/j.cellsig.2016.05.010
Sims R, van der Lee SJ, Naj AC, Bellenguez C, Badarinarayan N, Jakobsdottir J et al (2017) Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer’s disease. Nat Genet 49:1373–1384. https://doi.org/10.1038/ng.3916
Skol AD, Scott LJ, Abecasis GR, Boehnke M (2006) Joint analysis is more efficient than replication-based analysis for two-stage genome-wide association studies. Nat Genet 38:209–213. https://doi.org/10.1038/ng1706
Sudlow C, Gallacher J, Allen N, Beral V, Burton P, Danesh J et al (2015) UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. Plos Med 12:e1001779. https://doi.org/10.1371/journal.pmed.1001779
Team RC (2018) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna
Tesi N, van der Lee SJ, Hulsman M, Jansen IE, Stringa N, van Schoor N et al (2018) Centenarian controls increase variant effect sizes by an average twofold in an extreme case-extreme control analysis of Alzheimer’s disease. Eur J Hum Genet. https://doi.org/10.1038/s41431-018-0273-5
Tudorache IF, Trusca VG, Gafencu AV (2017) Apolipoprotein E—a multifunctional protein with implications in various pathologies as a result of its structural features. Comput Struct Biotechnol J 15:359–365. https://doi.org/10.1016/j.csbj.2017.05.003
van der Lee SJ, Wolters FJ, Ikram MK, Hofman A, Ikram MA, Amin N et al (2018) The effect of APOE and other common genetic variants on the onset of Alzheimer’s disease and dementia: a community-based cohort study. Lancet Neurol 17:434–444. https://doi.org/10.1016/S1474-4422(18)30053-X
van Rheenen W, Shatunov A, Dekker AM, McLaughlin RL, Diekstra FP, Pulit SL et al (2016) Genome-wide association analyses identify new risk variants and the genetic architecture of amyotrophic lateral sclerosis. Nat Genet 48:1043. https://doi.org/10.1038/ng.3622
Walton RL, Soto-Ortolaza AI, Murray ME, Lorenzo-Betancor O, Ogaki K, Heckman MG et al (2016) TREM2 p.R47H substitution is not associated with dementia with Lewy bodies. Neurol Genet 2:85. https://doi.org/10.1212/nxg.0000000000000085
Woyach JA, Furman RR, Liu TM, Ozer HG, Zapatka M, Ruppert AS et al (2014) Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. N Engl J Med 370:2286–2294. https://doi.org/10.1056/NEJMoa1400029
Xie J, Brayne C, Matthews FE, Medical Research Council Cognitive F, Ageing Study c (2008) Survival times in people with dementia: analysis from population based cohort study with 14 year follow-up. BMJ 336:258–262. https://doi.org/10.1136/bmj.39433.616678.25
Yu P, Constien R, Dear N, Katan M, Hanke P, Bunney TD et al (2005) Autoimmunity and inflammation due to a gain-of-function mutation in phospholipase C gamma 2 that specifically increases external Ca2+ entry. Immunity 22:451–465. https://doi.org/10.1016/j.immuni.2005.01.016
Zhou Q, Lee GS, Brady J, Datta S, Katan M, Sheikh A et al (2012) A hypermorphic missense mutation in PLCG2, encoding phospholipase Cgamma2, causes a dominantly inherited autoinflammatory disease with immunodeficiency. Am J Hum Genet 91:713–720. https://doi.org/10.1016/j.ajhg.2012.08.006
Zhou K, Shi L, Wang Y, Chen S, Zhang J (2016) Recent Advances of the NLRP3 Inflammasome in Central Nervous System Disorders. J Immunol Res 2016:9238290. https://doi.org/10.1155/2016/9238290
Acknowledgements
The following studies and consortia have contributed to this manuscript. Amsterdam dementia Cohort (ADC): Research of the Alzheimer center Amsterdam is part of the neurodegeneration research program of Amsterdam Neuroscience. The Alzheimer Center Amsterdam is supported by Stichting Alzheimer Nederland and Stichting VUmc fonds. The clinical database structure was developed with funding from Stichting Dioraphte. Genotyping of the Dutch case-control samples was performed in the context of EADB (European Alzheimer DNA biobank) funded by the JPco-fuND FP-829-029 (ZonMW projectnumber 733051061). 100-Plus study: We are grateful for the collaborative efforts of all participating centenarians and their family members and/or relations. This work was supported by Stichting Alzheimer Nederland (WE09.2014-03), Stichting Diorapthe, horstingstuit foundation, Memorabel (ZonMW projectnumber 733050814) and Stichting VUmc Fonds. Genotyping of the 100-Plus Study was performed in the context of EADB (European Alzheimer DNA biobank) funded by the JPco-fuND FP-829-029 (ZonMW projectnumber 733051061). German Study on Ageing, Cognition and Dementia in Primary Care Patients (AgeCoDe): This study/publication is part of the German Research Network on Dementia (KND), the German Research Network on Degenerative Dementia (KNDD; German Study on Ageing, Cognition and Dementia in Primary Care Patients; AgeCoDe), and the Health Service Research Initiative (Study on Needs, health service use, costs and health-related quality of life in a large sample of oldest-old primary care patients (85+; AgeQualiDe)) and was funded by the German Federal Ministry of Education and Research (grants KND: 01GI0102, 01GI0420, 01GI0422, 01GI0423, 01GI0429, 01GI0431, 01GI0433, 01GI0434; grants KNDD: 01GI0710, 01GI0711, 01GI0712, 01GI0713, 01GI0714, 01GI0715, 01GI0716; grants Health Service Research Initiative: 01GY1322A, 01GY1322B, 01GY1322C, 01GY1322D, 01GY1322E, 01GY1322F, 01GY1322G). Alfredo Ramirez was partly supported by the ADAPTED consortium: Alzheimer’s disease Apolipoprotein Pathology for Treatment Elucidation and Development, which has received funding from the Innovative Medicines Initiative 2 Joint Undertaking under grant agreement No 115975. Brain compendium: This work was funded by the UK Medical Research Council (13044). P.F.C. is a Wellcome Trust principal Fellow (212219/Z/18/Z) and a UK NIHR Senior Investigator, who receives support from the Medical Research Council Mitochondrial Biology Unit (MC_UU_00015/9), and the National Institute for Health Research (NIHR) Biomedical Research Centre based at Cambridge University Hospitals NHS Foundation Trust and the University of Cambridge. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR, or the Department of Health.Clinical AD, Sweden: We would like to thank UCL Genomics for performing the genotyping analyses. Danish data: The studies behind the Danish long-lived cases received funding from The National Program for Research Infrastructure 2007 (grant no. 09-063256), the Danish Agency for Science Technology and Innovation, the Velux Foundation, the US National Institute of Health (P01 AG08761), the Danish Agency for Science, Technology and Innovation/The Danish Council for Independent Research (grant no. 11-107308), the European Union’s Seventh Framework Programme (FP7/2007-2011) under grant agreement no. 259679, the INTERREG 4 A programme Syddanmark-Schleswig-K.E.R.N. (by EU funds from the European Regional Development Fund), the CERA Foundation (Lyon), the AXA Research Fund, Paris, and The Health Foundation (Helsefonden), Copenhagen, Denmark. The GOYA study was conducted as part of the activities of the Danish Obesity Research Centre (DanORC, www.danorc.dk) and The MRC centre for Causal Analyses in Translational Epidemiology (MRC CAiTE). The genotyping for GOYA was funded by the Wellcome Trust (WT 084762). GOYA is a nested study within The Danish National Birth Cohort which was established with major funding from the Danish National Research Foundation. Additional support for this cohort has been obtained from the Pharmacy Foundation, the Egmont Foundation, The March of Dimes Birth Defects Foundation, the Augustinus Foundation, and the Health Foundation. Fundació ACE (FACE): We would like to thank patients and controls who participated in this project. We are indebted to Trinitat Port-Carbó and her family for their support of Fundació ACE research programs. Fundació ACE collaborates with the Centro de Investigación Biomédica en Red sobre Enfermedades Neurodegenerativas (CIBERNED, Spain) and is one of the participating centers of the Dementia Genetics Spanish Consortium (DEGESCO). Agustín Ruiz has received support from the EU/EFPIA Innovative Medicines Initiative Joint Undertaking ADAPTED Grant No. 115975 and by grants PI13/02434 and PI16/01861. Acción Estratégica en Salud, integrated in the Spanish National R + D + I Plan and financed by ISCIII (Instituto de Salud Carlos III)-Subdirección General de Evaluación and the Fondo Europeo de Desarrollo Regional (FEDER- “Una manera de Hacer Europa”), by Fundación bancaria “La Caixa” and Grifols SA (GR@ACE project). Genetics of Healthy Ageing Study (GEHA – NL): The work described in this paper was funded mainly by the EU GEHA Project contract no. LSHM-CT-2004-503-270. Gothenburg Birth Cohort (GBC) Studies: We would like to thank UCL Genomics for performing the genotyping analyses. The studies were supported by The Stena Foundation, The Swedish Research Council (2015-02830, 2013-8717), The Swedish Research Council for Health, Working Life and Wellfare (2013-1202, 2005-0762, 2008-1210, 2013-2300, 2013-2496, 2013-0475), The Brain Foundation, Sahlgrenska University Hospital (ALF), The Alzheimer’s Association (IIRG-03-6168), The Alzheimer’s Association Zenith Award (ZEN-01-3151), Eivind och Elsa K:son Sylvans Stiftelse, The Swedish Alzheimer Foundation. International FTD-Genomics Consortium (IFGC): International FTD-Genomics Consortium (IFGC): The authors thank the IFGC for providing relevant data to support the analyses presented in this manuscript. Further acknowledgments for IFGC (https://ifgcsite.wordpress.com/), e.g. full members list and affiliations, are found in the online supplementary files. IPDGC (The International Parkinson Disease Genomics Consortium): We also would like to thank all members of the International Parkinson Disease Genomics Consortium (IPDGC). See for a complete overview of members, acknowledgements and funding http://pdgenetics.org/partners. Kompetenznetz Multiple Sklerose (KKNMS): This work was supported by the German Ministry for Education and Research (BMBF) as part of the “German Competence Network Multiple Sclerosis” (KKNMS) (grant nos. 01GI0916 and 01GI0917) and the Munich Cluster for Systems Neurology (SyNergy). TA was supported by the BMBF through the Integrated Network IntegraMent, under the auspices of the e:Med Programme (01ZX1614J). BH was supported by the EU Horizon 2020 project MultipleMS.Longitudinal Aging Study Amsterdam (LASA) is largely supported by a grant from the Netherlands Ministry of Health, Welfare and Sports, Directorate of Long-Term Care. The authors are grateful to all LASA participants, the fieldwork team and all researchers for their ongoing commitment to the study. Leiden Longevity Study: This study was supported by a grant from the Innovation-Oriented Research Program on Genomics (SenterNovem IGE05007), the Centre for Medical Systems Biology, and the Netherlands Consortium for Healthy Ageing (Grant 050-060-810), all in the framework of the Netherlands Genomics Initiative/Netherlands Organization for Scientific Research (NWO) and by Unilever Colworth.Maria Carolina Dalmasso: Georg Forster Research Award (Alexander von Humboldt Foundation). Mayo Clinic AD, DLB, PD, PSP: We thank the patients and their families for their participation, without whom these studies would not have been possible. Funding for this work was supported by National Institute on Aging [RF AG051504 to NET.; U01 AG046139 to NET]; and National Institute of Neurological Disorders and Stroke [R01 NS080820 to NET; P50 NS072187]. The Mayo Clinic is a Lewy Body Dementia Association (LBDA) Research Center of Excellence, American Parkinson Disease Association (APDA) Information and Referral Center and Center for Advanced Research, NINDS Tau Center without Walls (U54-NS100693) and is supported by Mayo Clinic AD and related dementias genetics program, The Little Family Foundation, the Mangurian Foundation for Lewy body research and NINDS R01 NS078086 (to OAR). The PD program at the Mayo Clinic Florida is also supported by the Mayo Clinic Center for Regenerative Medicine, Mayo Clinic Center for Individualized Medicine, Mayo Clinic Neuroscience Focused Research Team (Cecilia and Dan Carmichael Family Foundation, and the James C. and Sarah K. Kennedy Fund for Neurodegenerative Disease Research at Mayo Clinic in Florida), the gift from Carl Edward Bolch, Jr., and Susan Bass Bolch, and The Sol Goldman Charitable Trust. Samples included in this study are from the brain bank at Mayo Clinic in Jacksonville which is supported by CurePSP|Society for Progressive Supranuclear Palsy and the Tau Consortium. NDRU cohort: We would like to thank the NIH Neuro Brain Bank for contributing tissue samples; this study was supported in part by grants from the National Institutes of Health: U19-AG03365, P50 NS38377, and P50-AG005146. Tissue samples for genotyping were provided by the Johns Hopkins Morris K. Udall Center of Excellence for Parkinson’s Disease Research (NIH P50 NS38377) and the Johns Hopkins Alzheimer’s Disease Research Center. We are grateful for the support of the entire BIOCARD study team at Johns Hopkins University. Additionally, we acknowledge the contributions of the Geriatric Psychiatry Branch (GPB) in the intramural program of NIMH who initiated the BIOCARD study. We would like to thank the NIA Baltimore Longitudinal Study of Aging for contributing tissue samples to the Johns Hopkins Alzheimer’s Disease Research Center. DNA panels from the NINDS Human Genetics Resource Center DNA and Cell Line Repository (http://ccr.coriell.org/ninds) were used in this study, as well as clinical data. We thank the following brain banks for providing brain tissues: Banner Sun Health Research Institute, New York Brain Bank, Newcastle Brain Tissue Resource, Human Brain and Spinal Fluid Resource Center, Netherlands Brain Bank Amsterdam, Mount Sinai Brain Bank, Harvard Brain Bank, Duke University Brain Bank, Virginia Commonwealth University Brain Bank, and the Georgetown University Brain Bank. We also thank the following research centers for providing patient samples: University of Michigan, University College London Institute of Neurology, University of Maryland, University of California – San Diego, and University of Miami. We are grateful to members of the North American Brain Expression Consortium for contributing DNA samples. The authors thank the patients and families who have donated DNA samples and brain tissue for scientific research. The research was supported in part by the Intramural Research Program of the NIH National Institute of Neurological Disorders and Stroke and the National Institute on Aging (project numbers: ZIA-NS003154, Z01-AG000949).” Oviedo: This work was partly supported by Grant from Fondo de Investigaciones Sanitarias-Fondos FEDER EuropeanUnion to Victoria Alvarez PI15/00878. Pascual Sánchez-Juan: Pascual Sánchez-Juan is supported by CIBERNED and Carlos III Institute of Health, Spain (PI08/0139, PI12/02288, and PI16/01652), jointly funded by Fondo Europeo de Desarrollo Regional (FEDER), Unión Europea, “Una manera de hacer Europa”. Project MinE: The ProjectMinE study was supported by the ALS Foundation Netherlands and the MND association (UK) (Project MinE, www.projectmine.com). Risk and modifying factors in Fronto Temporal Dementia (RiMoD-FTD): follows: The SPIN cohort: We are indebted to patients and their families for their participation in the “Sant Pau Initiative on Neurodegeneration cohort”, at the Sant Pau Hospital (Barcelona). This is a multimodal research cohort for biomarker discovery and validation that is partially funded by Generalitat de Catalunya (2017 SGR 547 to JC), as well as from the Institute of Health Carlos III-Subdirección General de Evaluación and the Fondo Europeo de Desarrollo Regional (FEDER- “Una manera de Hacer Europa”) (grants PI11/02526, PI14/01126, and PI17/01019 to JF; PI17/01895 to AL), and the Centro de Investigacion Biomedica en Red Enfermedades Neurodegenerativas programme (Program 1, Alzheimer Disease to AL). We would also like to thank the Fundació Bancària Obra Social La Caixa (DABNI project) to JF and AL; and Fundacion BBVA (to AL), for their support in funding this follow-up study. San Sebastian: We would like to thank patients and control volunteers who participated in this study. We thank Ana Gorostidi for his work at the Biodonostia HRI Genomics Platform. This work was partly supported by CIBERNED. Adolfo López de Munain is supported by Fundación Salud 2000 (PI2013156) and Diputación Foral de Gipuzkoa (Exp. 114/17). UK Biobank analysis: This work was funded by The Netherlands Organization for Scientific Research (NWO VICI 453-14-005). The analyses were carried out on the Genetic Cluster Computer, which is financed by the Netherlands Scientific Organization (NWO: 480-05-003), by the VU University, Amsterdam, The Netherlands, and by the Dutch Brain Foundation, and is hosted by the Dutch National Computing and Networking Services SurfSARA. This research has been conducted using the UK Biobank resource under application number 16406. We are grateful to the numerous participants, researchers, and staff who collected and contributed to the data.
Data availability statement
The authors declare that the data supporting the findings of this study are available within the paper and its online Resource.
Funding
No funding was received specifically for this study. The studies and consortia all have their respective funding sources described in the acknowledgement section.
Author information
Authors and Affiliations
Consortia
Contributions
SvdL and HH had full access to the meta-analysis results and wrote the draft of the manuscript. SvdL, OJC, MMC, LK, EBvdA, NS, JAC, AZ, TFMA, MD-F, JS-S, HZ, MN, CB, JES, JM-F, SM-G, MJK, KB, CL, IdR, WW, BJ, XW, NB, NT, JEC, SLS, AK, MCD, MA, NE-T, SWS and Aru analyzed parts of the data. MJTR provided compute for this study. MCD compiled demographic information of all Spanish cohorts. SvdL, EBvdA, NS, MD-F, JS-S, SM-G, CL, IdR, WW, BJ, NB, NT, NE-T, SWS, Aru, IJ, IH, AL, MW, JF, IS, MAF, OP, MZ, SR-H, II-G, AO, FTB, MH, CM, BI, MS, EM-R, JWI, MNMvB, DA, HW, ERR, BFB, RCP, TJF, JAvG, MJU, LT, WM, OD-I, MBo, MB, EM, ALdM, AP, ZKW, OAR, DWD, NRG-R, DK, RR, AWL, YALP, PS, TG, PFC, BH, JT, FM, EAN, TIAS, PS-J, DP, GC, JC, KC, AR, WMvdF and HH contributed to data collection. JT and OP contributed to neuropathological diagnoses. MW, IS, SR-H, II-G, MS, HW, LT, WM, MBo, MB, ZKW, AWL, PS, TG, PFC, BH, MAH, TIAS, PH, JC, KC, NE-T, SWS, AR, Aru, ES, WMvdF and HH secured funding for their respective studies. MMC, MW, SR-H, MH, JEC, MS, MNMvB, HW, WM, NMvS, PS, TG, PFC, BH, MAH, EAN, PH, NE-T, SWS, AR, ES, WMvdF and HH supervised their respective studies. HH was responsible for the overall study supervision. All authors critically revised the manuscript for important intellectual content and approved the final manuscript.
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
van der Lee, S.J., Conway, O.J., Jansen, I. et al. A nonsynonymous mutation in PLCG2 reduces the risk of Alzheimer’s disease, dementia with Lewy bodies and frontotemporal dementia, and increases the likelihood of longevity. Acta Neuropathol 138, 237–250 (2019). https://doi.org/10.1007/s00401-019-02026-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-019-02026-8