
Abstract
Peptide drugs are increasingly used to treat a variety of diseases ranging from cancer, and infections to cardiovascular diseases. However, peptides can suffer from low stability in the bloodstream. Entrapment of peptides into nano-sized carriers of various types has widely been explored, but all of them have spherical shapes. Nanocellulose can in contrast serve as a non-spherical nanoparticle with a high aspect ratio. After the isolation of nanocellulose by TEMPO-mediated oxidation, the material needs to be modified with polymers to generate nanoparticles with high water-solubility that can also favourably interact with peptide drugs. We have here chosen insulin as the model drug, which can strongly interact with cationic polymers. As it is known that cationic polymer may retain charged drugs too tightly, we have selected poly(2-(dimethylamino)ethyl acrylate) PDMAEA as a degradable polymer that undergoes self-hydrolysis to poly(acrylic acid) in water. This polymer was compared to poly(N-(3-(dimethylamino)propyl) acrylamide) PDMAPAA, which is a stable cationic polymer. The cationic polymer was co-grafted with poly(2-hydroxyethyl acrylate) PHEA as a water-soluble neutral polymer using the three-component Passerini reaction. A combination of fluorescence and UV-Vis techniques were used to quantify the amount of polymer that was conjugated to the surface. The polymer-coated nanocellulose was labelled with the fluorescent cyanine dye Cy5 while insulin was labelled with Cy3 creating a FRET system that allows monitoring of the interaction between insulin and polymer in cell growth media. We observed that despite the self-hydrolysis of PDMAEA into a negatively charged polymer, the negatively charged insulin was not released in buffer solution according to the FRET studies. Only the addition of serum-supplemented cell growth media led to insulin release. The limited release was explained with the fact that insulin, as well as other peptides, have a mixture of negative and positive charges, with the pH value and the isoelectric point determining the balance between both. Negative-charged polymers can therefore still interact favourably with negatively charged peptides by interacting with cationic amino acids.
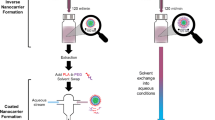
Graphical Abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
There are currently around 80 peptide drugs on the market, and several more have reached clinical trials. It is expected that this field will further increase in the coming years [1]. Notable peptide drugs are peptide hormones such as somatostatin, the natural immunosuppressor cyclosporine, and, the oldest peptide drug of them all, insulin [1]. Insulin, which was first used 100 years ago, is widely used for the treatment of diabetes mellitus as it regulates the absorption of glucose [2]. Despite their success, peptide drugs are faced with several challenges including short circulation time, low hydrolytic stability, and sometimes limited cellular uptake. Strategies to enhance the delivery of peptides are widely explored, and approaches include the use of a drug delivery system, chemical modification of the peptide, co-administration of absorbance enhancers, and the direct using for example microneedles or electroporation [3]. The advantage of a drug delivery system based on nanoparticles is that it can significantly change the pharmacokinetics of the drug without altering the structure. Moreover, it can protect the drug from hydrolysis as the shell can repel enzymes such as proteases and peptidases. A variety of drug delivery carriers are available for peptides, ranging from polymer-peptide conjugates, solid lipid nanoparticles, liposomes to polyelectrolyte complexes [4]. Polyelectrolyte complexes, often also called polyion complex [5] or complex coacervates [6], which are prepared from two macromolecules of opposite charge, have the advantage that the formulation can be prepared by simply mixing the charged peptide drug and the charged drug carrier under mild conditions. Complexation is usually complete as the driving force to bind with the opposite charge usually overcomes entropic factors and the good water-solubility of the peptide. Polyion complexes are widely used to deliver proteins and peptides [5, 7, 8], although there are some challenges in obtaining stable complexes. While binding between polymers and large proteins usually leads to relatively stable complexes, that may only disassemble in the presence of competing proteins [9], very small peptides have often not a sufficient amount of charges to ensure that the electrostatic bond stays intact when the ionic strength increases [10]. More importantly, although proteins and peptides have a certain net charge at a given pH value and isoelectric point, there the peptide or protein surface is a combination of both charges and hydrophobic areas. This means that although a cationic polymer should bind to a protein with an overall negative surface charge, there might still be positively charged pockets on the surface that might repel the protein. An example here is albumin which is overall negatively charged at pH 7. As it carries positive charges around the fatty acid binding pocket, polymers with negative charges bind relatively well [11].
One of the most widely used peptide drugs is insulin, which contains 51 amino acids and consists of a dipeptide structure of two peptide chains connected by a disulphide bridge. As it has an isoelectric point IEP at pH 5.5, the peptide has a negative net charge at pH7 [12]. A large array of different nanoparticles have been explored to enhance the delivery of insulin [13,14,15], which include many approaches where insulin is retained in the carrier using cationic [16, 17] or zwitterionic polymers [18]. It is notable that the field is dominated by spherical nanoparticles, while other shapes are absent. It is however proposed that non-spherical nanoparticles may have superior properties when employed as drug delivery carriers [19]. The elongated shape has a high surface area and therefore high surface functionality that will alter the interface to the biological environment. The high aspect ratio is also known to promote an extended circulation time in the blood stream, may result in better penetration into the tumour, but also facilitate cellular uptake [20].
We therefore propose the immobilization of the cationic polymer onto cellulose nanofibers (CNFs), which are a readily available source of non-spherical nanoparticles suitable for drug delivery [21]. The type of cationic polymer will influence binding strength to the peptide, but will also influence toxicity [22]. Amongst them, poly(2-dimethylaminoethyl acrylate) (PDMAEA) is an attractive candidate due to its non-toxicity and capability to release therapeutics by self-catalysed degradation in aqueous media [23]. According to the study undertaken by Truong et al., the ester group located on the side chain of polymer degrades to poly(acrylic acid) (PAA) and 2-dimethylaminoethanol (DMAE) in water. The degradation rate can be varied depending on the molecular weight of the polymer and the pH of the solution [24,25,26,27]. The polymers and the released side products after degradation were confirmed to be non-toxic [23]. In contrast, poly(N-[3-(dimethylamino)propyl] acrylamide (PDMAPAA) is non-degradable, and the electrostatically bound drug is only released slowly when the electrostatic bond is interrupted upon competition with other proteins present inside the cells [28,29,30]. Both polymers are based on acrylate and acrylamide systems as it was shown recently that polymethacrylates retain peptides too tightly [31].
In this work, we grafted two cationic polymers, PDMAEA and PDMAPAA, onto CNFs using the three component Passerini reaction [32]. In order to achieve high colloidal stability, the cationic polymers were co-grafted with the neutral and water-soluble poly(2-hydroxyethyl acrylate) (PHEA) (Scheme 1). The aim of this work was to evaluate if the polymer-grafted CNF can efficiently bind and release insulin, which we used here as model peptide.

Overall Scheme for the design of cellulose nanoparticles for efficient delivery and release of peptide drugs. Cy5 stands for cyanine 5 dye
Experimental procedure
Materials
N-(3-Dimethylaminopropyl)-N’-ethylcarbodiimide hydrochloride (EDC.HCl, Sigma-Aldrich), anhydrous dichloromethane (DCM, Sigma-Aldrich, ≥ 99.8%), 1-amino-3,3-diethoxypropane (Sigma-Aldrich, 97%), 4-(dimethylamino)pyridine (DMAP, Sigma-Aldrich, ≥ 99%), magnesium sulfate (MgSO4, Sigma-Aldrich, ≥ 99.5%), acetone (Sigma-Aldrich, ≥ 99.5%), hydrochloric acid (HCl, Sigma-Aldrich, 32%), hexane (Sigma-Aldrich, 95%), ethyl acetate (Sigma-Aldrich), 2-hydroxyethyl acrylate (HEA, Sigma-Aldrich), N,N-dimethylformamide (DMF, Sigma-Aldrich), methanol, 2-(dimethylamino)ethyl acrylate (DMAEA, Sigma-Aldrich), N-(3-(dimethylamino)propyl)acrylamide (Sigma-Aldrich, 97%), diethyl ether, 2,3,3‐trimethyl‐3H‐indole (Sigma-Aldrich, 98%), 6-bromohexanoic acid (Sigma-Aldrich, 97%), potassium iodide (Ajax Finechem Pty Ltd), acetonitrile (Chem-Supply Pty. Ltd.), malonaldehyde bis(phenylimine) monohydrochloride (Sigma-Aldrich, 97%), 1,2,3,3-tetramethyl-3H-indolium iodide (Sigma-Aldrich, 98%), acetic anhydride, pyridine (Chem-Supply Pty. Ltd.), chloroform (Sigma-Aldrich, ≥ 99.5%), deuterated chloroform (CDCl3, Sigma-Aldrich, 100%), deuterium oxide (D2O, Sigma-Aldrich), deuterated dimethyl sulfoxide (DMSO-d6, Sigma-Aldrich, 100%), N,N′-dicyclohexylcarbodiimide (DCC, Sigma-Aldrich, 99%), anhydrous N,N-dimethylformamide (DMF, Sigma-Aldrich, 99.8%), and cyclohexyl isocyanide (Sigma-Aldrich, 98%) were used without purification. Azobisisobutyronitrile (AIBN) was purified by recrystallization in methanol. Cellulose nanomaterials (CNFs) with acid functionalities were fibrillated from microcrystalline cellulose (MCCs, Sigma-Aldrich) according to the procedure reported previously [32]. The synthesis of the aldehyde-terminated RAFT agent, N-(2-formylethyl)-3-(benzylthiocarbonothioylthio)- propionamide, was described earlier [33, 34], but is also included in the supporting info.
Synthesis
Synthesis of aldehyde-terminated polymers
Synthesis of poly(2-hydroxyethyl acrylate) PHEA37
The hydrophilic monomer, 2-hydroxyethyl acrylate, was polymerized via RAFT polymerization using synthesized aldehyde-terminated RAFT agent. In a glass vial, 2-hydroxyethyl acrylate (HEA) (2.00 g, 17.22 mmol), RAFT-aldehyde (68.78 mg, 210.04 µmol), and the initiator AIBN (2.75 mg, 16.80 µmol) were dissolved in N,N-dimethylformamide (DMF). The molar ratio of [HEA]/[RAFT]/[AIBN] was 82:1:0.08, and a monomer concentration was 2 M. The flask containing the mixture was sealed with rubber septa, and the solution was degassed with nitrogen purging for 45 min. The polymerization was performed at 65 °C for 2 h to achieve 46% monomer conversion. Then, the polymerization was ceased by introducing air to the mixture and placing the vial in an ice bath for 15 min. The resulting polymer was purified via dialysis against methanol (MeOH) for 2 days and then dried under vacuum. The pure polymer was characterized via 1H-NMR and SEC. Mn (SEC) = 15000 g mol-1 and Ɖ = 1.21.
Synthesis of poly(2-(dimethylamino)ethyl acrylate) PDMAEA12
The polymerization procedure of PDMAEA12 was similar to the synthesis of PHEA37. Firstly, 2-(dimethylamino) ethyl acrylate monomer/DMAEA (3.00 g, 20.95 mmol), RAFT-aldehyde (83.93 mg, 0.21 mmol), and AIBN initiator (3.43 mg, 20.91 μmol) were dissolved in DMF (7.27 mL) in a round bottom flask. The concentration of monomer was 2 M, and the molar ratio of [DMAEA]/[RAFT]/[AIBN] was 100:1:0.1. The solution was degassed with nitrogen purging for 45 min and then placed into a preheated oil bath at 65 °C. The polymerization was stopped after 4 h by introducing air to the solution and placing the vial in an ice bath for 15 min. The resulting polymer was purified by precipitation four times into hexane, and the obtained pure polymer was dried under vacuum. The monomer conversion and molecular weight of the pure polymer were analysed via 1H-NMR and SEC. Mn (SEC) = 2740 g mol-1 and Ɖ = 1.27.
Synthesis of poly(N-(3-(Dimethylamino)propyl) acrylamide) PDMAPAA16
N-(3-(dimethylamino)propyl) acrylamide (234 mg, 1.5 mmol) was dissolved in DMF (0.1 mL) in a 4-mL glass vial, followed by the addition of acetic acid (0.15 mL). Secondly, the mixture of RAFT-aldehyde (12.3 mg, 37.5 μmol) and AIBN (0.62 mg, 3.8 μmol) in DMF (1.15 mL) was added to the solution. The monomer concentration was 1 M, and the molar ratio of [DMAPAA]/[RAFT]/[AIBN] was 40:1:0.1. The mixture was then degassed with nitrogen purging for 1 h at room temperature, followed by placing in a preheated oil bath at 70 °C. After 3 h, the polymerization was stopped by cooling the solution in an ice bath and introducing air. The obtained solution was then submitted to precipitation in a mixture of hexane/diethyl ether (30 mL, 2:1), and the polymer was collected by centrifugation. For further purification, the crude polymer was dissolved in 1 mL of propanol and precipitated two more times. The final polymer as a yellow product was then dried under vacuum.
Fluorescent labelling of PHEA37 with cyanine-5
The preparation of fluorescently labelled PHEA37 was carried out by using synthesized cyanine 5 acid under mild reaction condition. Cyanine dye (3 mg, 4.59 μmol) and N,N′-dicyclohexylcarbodiimide (DCC) (11 mg, 53.40 μmol) were dissolved in anhydrous DMF (1.5 mL) and stirred at room temperature for 30 min. After that, PHEA37 (700 mg, 0.15 mmol) and 4-(dimethylamino)pyridine (DMAP) (7 mg, 57.30 μmol) dissolved in anhydrous DMF (1 mL) were added to the solution and stirred overnight. The obtained mixture was then dialysed against methanol (1 kDa MWCO) to remove unreacted fluorescent dye, followed by removing the solvent and drying under vacuum.
Hydrolysis kinetics of PDMAEA12 in PBS at pH 7.4
The hydrolysis of PDMAEA12 in phosphate buffer saline (PBS) at pH 7.4 was performed at room temperature and at 37 °C. Firstly, 7 mg of PDMAEA12 was dissolved in the mixture of D2O and 2 × concentrated PBS (1:1 v/v) at the concentration of 2.35 mg mL−1. After that, two NMR tubes were filled with 650 μL of solution and incubated at room temperature and 37 °C. The 1H-NMR measurements (400 MHz) were performed at different time intervals: 15 min, 20 h, and 40 h. The percentage of hydrolysis was calculated according to following equation based on the integrated intensity of signals at δ = 3.62 ppm and 3.81 ppm. (I = intensity of the peak):
Co-grafting PHEA35-Cy5 and PDMAEA12
The covalent tethering of homopolymers onto CNFs was performed via a three-component Passerini reaction under mild conditions. The dried CNFs (10 mg, 16.5 μmol of -COOH groups) was dispersed in DMF (0.3 mL) in a glass vial and sonicated using the bench sonicator for 15–30 min. On the other hand, PHEA37-Cy5 (20 mg, 4.32 μL) dissolved in DMF (0.2 mL) was added to three vials containing 35 mg, 25 mg, and 10.7 mg of PDMAEA12, respectively, followed by the addition of mixture to the CNFs dispersion. Finally, the cyclohexyl isocyanide (17.56 mg, 0.16 mmol, 20 μL) was slowly added to the mixture. After overnight stirring at room temperature, the polymer modified CNFs were collected by precipitating in water (1.5 mL) and centrifugation at 15000 rpm for 10 min. The purification was repeated six times until almost clear supernatant was achieved. After freeze drying the collected modified cellulose nanomaterials, the samples were submitted for characterizations using UV-Vis, FT-IR, DLS, and 1H-NMR. Table 1 lists the initial ratios of CNFs and homopolymers added to perform the surface modification reaction.
Co-grafting PHEA37-Cy5 and PDMAPAA16
For comparing the release profiles of nanoparticles tethered with different cationic polymers, a CNFs sample grafted with PHEA37-Cy5 and PDMAPAA16 was also prepared. The employed experimental procedure was the same as above, and the initial molar ratio of -COOH from CNFs/PHEA37-Cy5/PDMAPAA16 was 1:0.36:1.3. The exact mass and moles of components added are described in Table 2.
Calculation of grafting efficiency
The determination of grafting efficiency was performed using two characterizations techniques: fluorescence and UV-Vis spectroscopy. Firstly, the calibration curves for both polymer chains PHEA37-Cy5 and PDMAEA12 or PDMAPAA16 were prepared by dissolving the polymers at various concentrations in in milli-Q water.
Calibration curves of PHEA37-Cy5
Fluorescent analysis
The standard solutions were obtained by serial dilution of 0.125 mg mL-1 to 0.0312, 0.0156, 0.0078, 0.0039, and 0.0020 mg mL−1. The excitation and emission wavelengths were 648 nm and 657 nm.
UV-Vis analysis
The standard solutions were obtained by serial dilution of 0.2496 mg mL-1 to 0.1248, 0.0624, 0.0312, 0.0156, and 0.0078 mg mL-1. The absorption peak representing C = S in RAFT group was observed at λmax = 313 nm.
Calibration curve of PDMAEA12
UV-Vis analysis
The standard solutions were obtained by serial dilution of 0.4992 mg mL-1 to 0.2496, 0.1248, 0.0624, 0.0312, 0.0156, and 0.0078 mg mL-1. The absorption peak representing C = S in RAFT group was observed at λmax = 313 nm.
Calibration curve of PDMAPAA16
UV-Vis analysis
A range of standard solutions were prepared at desired concentrations in mg mL-1: 0.033, 0.052, 0.081, 0.126, 0.175, 0.216, 0.267, 0.329, 0.405, 0.500, and 0.640. The absorption peak representing C = S in RAFT group was observed at λmax = 307 nm.
Procedure for the Calculation of grafting efficiency
The polymer-grafted CNFs were dissolved in milli-Q water at a concentration of 1 mg mL−1. The sample was first analysed using fluorescent spectroscopy for the determination of the amount of grafted PHEA37-Cy5 by taking advantage of the conjugated cyanine 5 fluorescent dye which has the emission at λmax = 657 nm (PDMAEA12 and PDMAPAA16 were not fluorescent). The obtained mass of PHEA37-Cy5 from fluorescent spectrophotometer was used to calculate the possible UV-Vis absorption intensity using a standard curve. Finally, the total amount of grafted polymers on the surface of cellulose nanomaterials was determined by UV-Vis absorption intensity, from which the calculated intensity of PHEA37-Cy5 was subtracted. The remaining intensity was equal to the intensity of either PDMAEA12 or PDMAPAA16, which was converted to mass using its calibration curve.
Hydrolysis kinetics of (PHEA37-Cy5-PDMAEA12)-g-CNFs in PBS at pH 7.4
The hydrolysis of synthesized polymer-grafted CNFs in phosphate buffer saline (PBS) at pH 7.4 was performed at room temperature and at 37 °C. 1 mg of nanoparticles was dissolved in a mixture of D2O and 2 × concentrated PBS (1:1 v/v) at the concentration of 0.5 mg mL-1. After that, two NMR tubes were filled with 650 μL of solution, sonicated for 15 min to obtain fully dispersed nanoparticles, and incubated at room temperature and 37 °C. The 1H-NMR measurements (400 MHz) were performed at different time intervals: 15 min, 20 h, and 40 h. The percentage of hydrolysis was determined by comparing the integrated intensity of signals at δ = 3.62 ppm and 3.83 ppm. The calculation was carried out by using the following equation (I = intensity of the peak):
Fluorescent labelling of insulin with cyanine-3 (Cy-3)
A stock solution of fluorescent dye was prepared by dissolving Cy-3 carrying NHS ester (1 mg, 1.59 μmol) in 100 μL of DMSO. Afterwards, the labelling was carried out according to the following procedure: Cy3-NHS (0.22 mg, 3.49 × 10-7 mol), insulin (2.00 mg, 3.49 × 10-7 mol) were mixed in 28 μL of milli-Q water in the presence of 0.2 M sodium bicarbonate buffer (50 μL). The reaction was then left in a water shaker overnight, followed by purification by dialysis against milli-Q water for 1 day using 3500 MWCO membrane. Finally, the red-coloured sample was collected from the dialysis bag and subsequently freeze-dried. The resulting 300 μg of dried sample was dissolved in 1.5 mL of milli-Q water and kept in the fridge for further experiments. The emission spectrum of insulin-Cy3 (800 ng in 1 mL of pH 7.4 PBS) was then recorded at an excitation of 520 nm, which is the wavelength employed for FRET study.
Monitoring insulin loading via FRET analysis
The precise composition of the nanoparticles used is summarised in Table 3. 250 μg of NPs-Cy5 was dispersed in 1 mL of phosphate buffer saline (PBS, pH 7.4), and the emission spectrum was recorded at 520 nm. Then, insulin-Cy3 prepared in stock solution was added serially: 100 ng to a total of 1200 ng, to observe increasing binding behaviour on nanoparticles. To be noted here is that after each addition of insulin to NPs, the dispersion was stirred for 5 min at room temperature to allow the protein attachment on NPs. The experimental conditions including the instrumental voltage have been kept the same for all analyses. The FRET ratio was calculated according to the equation below. Here, Cy-3 serves as the donor, while Cy-5 molecule behaves as an acceptor.
Monitoring insulin release via FRET analysis
(PHEA37-Cy5-PDMAEA12)-g-CNFs and (PHEA37-Cy5-PDMAPAA16)-g-CNFs, (500 μg) were dispersed in 1mL PBS (pH 7.2 or pH5). After sonicating the dispersed nanoparticles using a bench sonicator, 2 μg of insulin (4 μL, 0.5 μg μL-1 stock solution) was added. Mass ratio between nanoparticles and insulin was recorded as 1: 0.004. Then, the FRET ratio changes were continuously monitored at different time intervals, t = 0, 5 min, 10 min, 20 min, 30 min, 1 h, 2 h, 4 h, 8 h, and 20 h, using fluorescent spectroscopy. To be noted here is that the mixture was stirred for 5 min before each measurement, and the experiment condition was kept the same throughout the analysis. The same procedure was employed to study the insulin release in cell growth medium (DMEM).
Results and discussions
Synthesis of polymer-coated nanocellulose
Nanocellulose including CNF has been widely used to deliver drugs [21], including for the delivery of doxorubicin [35,36,37], siRNA [38], and DNA [39, 40]. Although it is possible to absorb drugs directly to the surface of cellulose nanomaterials, grafting with polymers can enhance colloidal stability, prevent protein absorption, and enhance the drug loading capacity [21]. We have recently developed a technique to graft polymers onto TEMPO-oxidized CNFs via Passerini reaction using the carboxylic acid groups on the surface of TEMPO-oxidized CNF, aldehyde-terminated polymer, and an isocyanide. The advantage of this technique is the high efficiency in aqueous solution under ambient temperature [32]. In the first step, the synthesis of an RAFT agent that can generate aldehyde-terminated polymers is necessary. Although aldehyde-based RAFT agents were described earlier [32, 41], the ester functionality makes the final polymer prone to hydrolysis. Therefore, a RAFT agent with a hydrolytically more stable amide functionality was employed. Evidence of the successful synthesis of the chain transfer agent was gathered from 1H- and 13C-NMR (ESI, Figs. S1 and S2) (Scheme 2).

Synthesis of polymers by RAFT polymerization
The aldehyde-functionalized chain transfer agent was subsequently employed in the synthesis of PHEA as the water-soluble building block of the final grafted CNF and PDMAEA (pKa = 8.41) or PDMAPAA (pKa = 10.35) as the binding site for the negatively charged insulin [42]. As PDMAPAA might bind relatively strongly, there is a possibility that insulin may not be released. This problem was alleviated by the hydrolytic nature of the PDMAEA, which converts the cationic polymers into negatively charged carboxylic acid, leading to the release of insulin in time-, temperature-, and pH-dependent manner.
The homopolymerizations were performed via RAFT polymerization in the presence of an aldehyde-functionalized RAFT agent and AIBN as an initiator. The aim was to generate a PHEA polymer length that was around 2–3 times longer than that of the cationic polymers in order to create a relatively neutral surface that can shield the charges. The polymerization conditions listed in Table 1 were used after a range of preliminary experiments helped to identify the most suitable conditions to obtain the desired polymer length. It was found that the polymerization of DMAPAA is more controlled when small amounts of acid are added, which protonates the amino group and lowers the likelihood of RAFT agent cleavage. The monomer conversion was determined by comparing the integrations of resonance signals corresponding to the methylene protons from the polymer chain and residual monomer. Then, the crude products were purified by different techniques: PHEA by dialysis against methanol for 2 days, PDMAEA by repeated precipitation in hexane, and PDMAPAA by precipitation in a mixture of hexane/diethyl ether. The purity of homopolymers and the existence of aldehyde moiety, which was crucial for future surface grafting reactions, were also confirmed by 1H-NMR (ESI, Figs. S3 and S4 and Fig. 1a). The 1H-NMR spectra were recorded in CDCl3 but also in DMSO-d6 as this allowed the comparison of the phenyl group of the R-group of the RAFT agent with the aldehyde group that belongs to the Z-group (insert in Fig. 1a). The intensity of both signals is in relatively good agreement although the aldehyde functionality is slightly less than 1. While this could point to some loss of aldehyde functionality in the process, it also needs to be considered that aldehyde can exchange with water in wet solvents, thus the signal is reduced in intensity. The molecular weight and molecular weight distribution of the homopolymers were determined by SEC in DMF as eluent (Table 4, ESI, Fig. 1b). The resulting polymers show symmetrical unimodal molecular weight distribution without a shoulder and tailing peak, indicative of the absence of significant side reactions.

a 1H-NMR spectrum of aldehyde-functionalized PDMAEA12 in CDCl3with the aldehyde signal magnified; the box shows the phenyl and aldehyde functionality recorded using d6-DMSO. b SEC traces of synthesized homopolymers: PDMAPAA16 (in red), PDMAEA12 (in black), and PHEA37 (in blue). Samples were dissolved in DMF
Prior to the attachment of the polymers onto CNF, PHEA was labelled with cyanine 5 dye [43], which was synthesised via a two-step process as described in the experimental part and characterized by 1H-NMR and UV-Vis analysis (ESI, Figs. S7 and S8) [43, 44]. The fluorophore was conjugated to the side chains of PHEA37 via DCC/DMAP coupling reaction in anhydrous DMF resulting in the attachment of ~ one Cy5 dye per polymer chain (ESI, Fig. S9).
The surface grafting reaction of the polymers onto CNF was previously performed in aqueous media; however, PDMAEA can be degraded slowly to poly(acrylic acid) (PAA) and DMAE via a self-catalysed hydrolysis process in water as illustrated in Scheme 1 [23, 45]. It was therefore deemed necessary to study the degradation rate of PDMAEA in aqueous media (mixture of D2O and 2 × concentrated PBS (1: 1 v/v)) at pH 7.4. The samples were incubated at room temperature and 37 °C at various time intervals up to 40 h. Using 1H-NMR, shown in Fig. 2, the rate of degradation can be elucidated (ESI, Fig. S8), which revealed that after 20 h, around 27% (ambient temperature) and 43% (37 °C) have been degraded. This suggests that after the binding of negatively charged drugs, the drug can be released within a short time in biological media due to the charge conversion. However, this study also shows that aqueous solutions are not suitable for the Passerini reaction, and therefore DMF was employed as solvent.

Left: 1H-NMR spectra of a PDMAEA12 in D2O and PBS (2 × concentrated, pH 7.4) incubated at room temperature and 37 °C. The spectra were recorded at different time intervals; right) (PHEA37-Cy5-PDMAEA12)-g-CNFs in D2O and PBS (2 × concentrated, pH 7) incubated at room temperature and 37 °C at different time intervals (15 min, 20 h, and 40 h)
As the reaction needed to be in a water-free system, freeze-dried CNFs were used and redispersed in a small volume of DMF. Initially, three grafted samples were prepared by keeping the molar ratio of carboxyl groups on CNFs (determined by pH titration) and PHEA37-Cy5 constant at 1:0.25, but varying the molar ratio of PDMAEA12 (1.04, 0.74, and 0.31 respectively). The mixture was then stirred overnight with cyclohexyl isocyanide at ambient temperature. The obtained polymer-grafted CNFs were purified four times by precipitation in water (the precipitation did not cause any degradation of the polymer as this step was completed quickly). It should be emphasized here that this is not a true precipitation as all reactants are water-soluble. At high enough concentrations, the product forms gel-like particles. While this alone is not sufficient to separate the product from free polymer, extensive centrifugation at 15,000 rpm helps to isolate the product. The colourless supernatant was taken as a sign of the complete removal of unreacted polymer chains since PHEA has been labelled with Cy5, which renders the polymer solution blue (ESI, Fig. S9, picture insert). The amount of grafted PHEA can be calculated using the fluorescent intensity of the conjugated Cy5 (ESI, Fig. S7). The overall amount of grafted polymer can be obtained using UV–Vis spectroscopy, which quantifies the amount of RAFT groups that are still preserved after the Passerini reaction [32]. The percentage of grafting efficiency, calculated by using the calibration curves shown in ESI, Fig. S10, and the molar ratio of homopolymers grafted onto CNFs are listed in Table 2. The grafting efficiency in organic solvent is lower compared to aqueous solutions, not only due to the generally lower yields of these reactions in organic solvent but also due to the generally poor re-dispersibility of CNFs in organic solvents. Therefore, it is crucial to always sonicate the CNFs dispersion by using a bench sonicator before starting the reaction. Table 5 lists the relationship between the molar ratio of PDMAEA12 to PHEA37-Cy5 in the feed prior to the reaction and in the final grafted sample. While the grafting efficiency of PHEA remains constant at around 7 ± 1%, the percentage of conjugated PDMAEA varies depending on the amount of cationic polymer in the feed. Surprisingly, independent of the amount of cationic polymer in the feed, the final amount grafted on the polymers is constant, and, in all cases, a molar ratio of PHEA and PDMAEA12 of roughly 1:2 was obtained. Despite the comparable compositions, the zeta potential varies suggesting slight differences in structure. It is not clear what drives this effect, but it might be caused by electrostatic interactions of CNF and PDMAEA12 that pre-assemble the cationic polymer onto the negatively charged CNF before the Passerini reaction. At this point, it is not possible to conclude that all polymers on the surface are truly connected by Passerini reaction as the newly formed functional group is not visible in 1H NMR. Although we washed the final product thoroughly, we cannot exclude that some polymer remained that is only electrostatically bound.
Successful surface grafting was further confirmed by FT-IR spectroscopy, which revealed a new peak in the spectrum corresponding to the amide II band at 1573 cm-1 of PDMAEA in (PHEA37-Cy5-PDMAEA12)-g-CNFs (Fig. 3). Moreover, the TEM analysis confirmed that the structural integrity of the nanocellulose is maintained. The dark outline indicated the presence of polymers.

Characterisation of polymer coated CNFs using a ATR-FT-IR spectroscopy, b TEM analysis (scale bar 1 µm), and c dispersibility test of (PHEA37-Cy5-PDMAEA12)-g-CNFs in aqueous condition at different pH levels. The concentration of nanoparticles was 250 μg mL-1
The conditions used to obtain Sample 2 in Table 5 were now chosen to graft the non-degradable cationic polymer PDMAPAA16. The final molar ratio of components on grafted nanoparticles was obtained as 1:0.01:0.02 for COOH on CNFs, PHEA37-Cy5, and PDMAPAA16, with the grafting efficiency of 7.6% and 6.2% respectively for PHEA37-Cy5 and PDMAPAA16. From FT-IR and the spectroscopic analysis, it can be concluded that both polymers have been successfully grafted. Per 15–20 COOH groups on the surface of CNF (determined by titration) [32], one polymer chain was grafted, which means that a large amount of potentially negatively charged carboxylate functionalities are remaining. Two-thirds of the polymers were cationic polymers, while around one-third were PHEA.
Prior to the analysis of the polymer modified CNF in buffer and cell growth media, the hydrolytic stability of (PHEA37-Cy5-PDMAEA12)-g-CNFs was again studied to identify the rate of hydrolysis. The emergence of 2-dimethylaminoethanol is visible over the 40 h time window reflecting hydrolysis rates similar to that of the free PDMAEA (Fig. 2). The amount of hydrolysis after 2 h was found to be negligible, and therefore further stability testing in cell growth media was carried out within that time frame. So far, only the potential hydrolysis of the PDMAEA polymers has been discussed, but it should also be noted that the polymers are connected to CNF by a relatively labile trithiocarbonate functionality. In alkaline conditions, these groups can be readily cleaved, resulting in the liberation of the polymer from CNF. Although we did not observe any colour changes in water that might indicate such an event, we cannot exclude that cleavage might occur at extended storage or in biological media.
The dispersibility of these polymers is strongly dependent on the pH value, which affects the degree of protonation of the cationic polymers PDMAEA and PDMAPAA and the residual carboxy groups of CNF in aqueous media. The degree of ionization will therefore affect the solubility of the polymer-coated CNF as shown in Fig. 3. More important is however the stability of these nanoparticles in biological media. In biological fluids (cell cultures or body fluids), the extracellular proteins can immediately associate with synthetic materials such as nanoparticles, creating the so-called protein corona, which can be sufficiently long-lived to alter the aggregation state and surface properties of nanoparticles [46, 47]. This in turn critically affects the interactions of the nanoparticles with the cells, altering biological responses [48]. Several studies have been conducted to investigate the influence of nanoparticle properties such as size, shape, and surface structure on protein adsorption, which affects cellular uptake and cytotoxicity [49]. As variations in surface properties of nanoparticles (e.g. size and surface charge) may have a significant impact on protein adsorption, we herein investigated the behaviour of synthesized CNFs in terms of their hydrodynamic size in serum-containing cell culture medium. The changes in the hydrodynamic size were then recorded by DLS after exposed to cell culture medium for various time points up to 2 h showing a slight increase in hydrodynamic diameter (ESI, Fig. S11).
Analysis of insulin loading and release using FRET analysis
The two drug carriers were subsequently loaded with insulin by mixing the drug carrier and insulin in buffer solution. Loading is driven by the electrostatic interaction between the cationic polymer and the negative charged insulin. Loading into nanoparticles via electrostatic interaction is usually complete, and no free peptide or protein is in solution unless the ratio between cationic and anionic charges is unfavourable. In our case, it also needs to be considered that the surface of CNF has still an excess of unreacted carboxylate groups that might repel insulin if not compensated by an oversupply of cationic charges. Analysis of the interaction and release of the peptide in water or buffer solution is straightforward, but more interesting is to learn how these polyion complex nanoparticle behave in biological media, where competing proteins are abundant. Biological environments can be simulated by serum containing cell growth media, which contains not only vitamins and amino acids, but also a large variety of proteins. Hence, the release was monitored using fluorescently labelled peptide and drug carrier, respectively. The fluorescence pair chosen here displays Förster resonance energy transfer (FRET), where the energy is transferred from the donor molecule (D) to the nearby acceptor (A) [50]. As FRET strongly depends on D-A spacing, it is widely used as a nano-sized ruler to measure the distance of the drugs from the nanoparticles. To perform this study, insulin was labelled with Cy3, which serves as a FRET donor while PHEA-Cy5 on CNFs as an acceptor. In this experiment, the excitation wavelength was set at 520nm, at which Cy3 expressed emission, but Cy5 did not (ESI, Fig. S12). First, the FRET formation upon insulin association on nanoparticles was attempted using (PHEA37-Cy5-PDMAEA12)-g-CNFs in pH7.4 PBS. As expected, the Cy5 emission signal appeared and then increased upon serial addition of insulin-Cy3 indicating the close interaction of insulin with the nanoparticles (Fig. 4). The FRET ratio was calculated by comparing the emission intensity of Cy5 at 670 nm with that of Cy3 at 574 nm after exciting at 520 nm as shown in the equation below:

FRET experiment on insulin-Cy3 binding on (PHEA37-Cy5-PDMAEA12)-g-CNFs at pH7.4 PBS. NPs concentration = 250 μg mL−1, λmax(excitation) = 520 nm. a Emission spectra of nanoparticles with no insulin added initially and after serial addition of insulin-Cy3. Picture insert shows the dispersion of nanoparticles after addition of 100 ng of insulin-Cy3. b The diagram presenting the calculated FRET ratio vs mass ratio between insulin-Cy3 and NPs-Cy5. NPs, nanoparticles
The FRET ratio quickly increased to its maximum value of around 0.2 after adding only 0.1 wt% of insulin compared to the drug carrier, and the value remained constant when more insulin was added, indicative that all added insulin was loaded onto the drug carrier until a loading capacity of 0.5 wt% was reached (Fig. 4). We did not investigate the maximum loading capacity as our main concern was to understand how the type of cationic polymer will affect the release of insulin in biological media. PDMAEA and PDMAPAA differ by the degradable ester and the non-degradable amide linkages. One of these polymers, PDMAEMA, will undergo charge conversion resulting in the formation of negative charged polymers that in theory should push out the peptide. The release of insulin was therefore monitored via FRET analysis by firstly adding 2 μg of insulin-Cy3 to 500 μg of nanoparticles resulting in a loading content of 0.4 wt%, which is very low for an actual drug delivery experiment, but suitable for the FRET experiment devoid of any interferences.
The release of insulin from the loaded nanoparticles was subsequently monitored over 20 h using FRET at pH7 and pH5 in buffer solution (Fig. 5) as well as in serum supplemented cell growth media (Fig. 6). It is expected that with the release of insulin, the FRET ratio declines, indicative of a location of insulin outside the drug carrier. The samples were freshly prepared by mixing insulin and drug carrier, followed by monitoring the FRET ratio after 5 min. Over the next hour, the FRET ratio started to increase in intensity. This suggests that although initial binding is fast, it takes around 1 h to optimise the interaction between insulin and the cationic polymer. Rearrangements result in denser structures and therefore closer proximity of Cy3 and Cy5, thus higher FRET ratios. This is similar in all systems, independent of the type of polymer, the pH value, and the type of solution. After around 1 h, the different systems start behaving differently.

Insulin release study from two different polymers: a from (PHEA37-Cy5-PDMAEA12)-g-CNFs and b from (PHEA37-Cy5-PDMAPAA16)-g-CNFs. The experiments were performed in pH7.2 and pH5 PBS. Mass ratio between nanoparticles-Cy5 and insulin-Cy3 = 4 × 10-3. t = 0 is the measurement straight after the addition of insulin-Cy3

Insulin release study from two different polymers: a from (PHEA37-Cy5-PDMAEA12)-g-CNFs and b from (PHEA37-Cy5-PDMAPAA16)-g-CNFs. The experiments were performed in cell growth media (DMEM). Mass ratio between nanoparticles-Cy5 and insulin-Cy3 = 4 × 10-3. t = 0 is the measurement straight after addition of insulin-Cy3
PDMAEA/water
In case of the degradable (PHEA37-Cy5-PDMAEA12)-g-CNFs, the positive charges are converted slowly into negatively charged poly(acrylic acid), which is supposed to repel the peptide. This was demonstrated to be very successful in the case of the delivery of siRNA [51], but it appears here that there is a limited release of insulin. It must be considered that peptides, in contrast to siRNA, consist of negative and positive charges, with a balance that is a function of the surrounding pH value [52]. The negative charged polyacrylate can therefore interact with positive charged areas of the peptide (Scheme 3). Moreover, the high local concentration of carboxylates is likely to result in further protonation of insulin, approaching the IEP of insulin of 5.5. Lowering the pH value to 5.0 magnifies this process as insulin is now positive charged. As the pH value is now below the pKa value of nanocellulose and poly(acrylic acid), the amount of negative charges is significantly reduced. However, the positive charge of insulin at pH 5 compensates for this. Insulin is now bound stronger; the FRET ratio increases.
PDMAPAA/water
In (PHEA37-Cy5-PDMAPAA16)-g-CNFs, the polymer is well below its pKa value, and it is therefore positively charged at pH 7 and pH 5 (Scheme 3). At pH 7, a slow decline in the FRET ratio is observed indicative of some minor release of insulin, while at pH5, the polymer is strongly retained. This is not immediately conceivable as now insulin is also positively charged. Most likely, some interactions with other groups such as hydrophobic interactions or H-bonding hold the insulin in place [53].

Proposed mechanism of the insulin release considering degree of ionisation of polymer and insulin at different pH value, degradation of PDMAEA side chains, and competing protein available in serum-supplemented cell growth media
It can therefore be concluded that both polymers, at neutral and acidic pH, can retain insulin in water and prevent release. The self-hydrolysable PDMAEA did not have any advantages and cannot accelerate the release in buffer solution. The difference in release becomes apparent when the insulin-loaded polymer-coated CNFs are immersed in cell growth media, where an abundance of negatively charged proteins immediately interact with the nanoparticles (ESI, Figs. S11, S13 and S14). These proteins can either replace the insulin (which would lead to release) or form a thick protein corona around the nanoparticles that act as a barrier for insulin release. It is well known that positively charged polymers can quickly adsorb protein. However, in the case of (PHEA37-Cy5-PDMAEA12)-g-CNFs, the emergence of PAA after self-hydrolysis might lower the amount of protein adsorbed. Insulin is then free to interact with the cell growth media; therefore, the FRET ratio is reduced, indicative of insulin release (Scheme 3). In summary, it appears that there is no difference between these two polymers in water although PDMAEA undergoes charge conversion. However, there is a difference when competing peptides and proteins are present as the permanently positive charged polymer PDMAPAA binds differently compared to the degrading PDMAEA. This work highlights the complexity of binding between polymer and peptides as it is not only governed by electrostatic interactions, but also other forces such as the presence of opposite charges [52], hydrophobicity [31], and the length of the spacer between charge and polymer backbone [54], among other influences. However, FRET allowed us here to gain quickly insight into the events [50].
Conclusion
In summary, we reported a design of fluorescent cellulose-based nanomaterials (PHEA37-Cy5-PDMAEA12)-g-CNFs via the three-component Passerini reaction to efficiently bind insulin. This was achieved by mixing a water-soluble neutral polymer, PHEA, with a cationic polymer before grafting. Two polymers were compared, the permanently charged PDMAPAA and the hydrolysable PDMAEA. The idea was to create a system that would initially bind strongly to the negatively charged peptide insulin, but upon charge conversion, release the peptide, driven by electrostatic repulsion. Although it was planned to vary the composition between cationic polymer and neutral polymers grafted to CNF by altering the ratio between both polymers before the Passerini reaction, it was found that the composition of the grafted polymers remains constant independent of the feed, suggesting preassembly of the cationic polymer around the negative charged CNF prior to reaction. FRET analysis was then used to monitor the release of the loaded insulin as the peptide was labelled with Cy3, while PHEA carried a Cy5 tag. In water, despite the obvious PDMAEA hydrolysis to PAA, insulin remains close to the drug carrier as the FRET ratio remains constant. This highlights that the delivery of peptide or protein drugs by electrostatic interaction is not only determined by the peptide net charge, but also by hydrophobic and negative charged pockets on insulin. Release was only achieved when the system was placed in cell growth media as the serum proteins can now compete with insulin for binding. However, only the degradable PDMAEA allowed the release of insulin, most likely because the conversion of positive charges to negative charges altered how the polymer interacts non-specifically with serum proteins.
In conclusion, we showed how FRET can help to gain insight into very complex system. The negatively charged CNF and the positively charged cationic polymer both seemed to compete for various sites on insulin, while CNF and the cationic polymer also bind to each other, potentially resulting in the cationic polymer collapsing on the CNF surface.
Data availability
No datasets were generated or analysed during the current study.
References
Muttenthaler M, King GF, Adams DJ, Alewood PF (2021) Trends in peptide drug discovery. Nat Rev Drug Discov 20:309–325
Mathieu C, Martens P-J, Vangoitsenhoven R (2021) One hundred years of insulin therapy. Nat Rev Endocrinol. 17(12):715–725
Bruno BJ, Miller GD, Lim CS (2013) Basics and recent advances in peptide and protein drug delivery. Ther Deliv 4:1443–1467
Du AW, Stenzel MH (2014) Drug carriers for the delivery of therapeutic peptides. Biomacromol 15:1097–1114
Harada A, Kataoka K (2018) Polyion complex micelle formation from double-hydrophilic block copolymers composed of charged and non-charged segments in aqueous media. Polym J 50:95–100
Sing CE, Perry SL (2020) Recent progress in the science of complex coacervation. Soft Matter 16:2885–2914
Chen F, Stenzel MH (2018) Polyion complex micelles for protein delivery. Aust J Chem 71:768–780
Zhao L, Skwarczynski M, Toth I (2019) Polyelectrolyte-based platforms for the delivery of peptides and proteins. ACS Biomater Sci Eng 5:4937–4950
Chen F, Raveendran R, Cao C, Chapman R, Stenzel MH (2019) Correlation between polymer architecture and polyion complex micelle stability with proteins in spheroid cancer models as seen by light-sheet microscopy. Polym Chem 10:1221–1230
Kim J-H, Kim Y-S, Park K, Kang E, Lee S, Nam HY et al (2008) Self-assembled glycol chitosan nanoparticles for the sustained and prolonged delivery of antiangiogenic small peptide drugs in cancer therapy. Biomaterials 29:1920–1930
Xu YD, Lai RY, Procházková E, Stenzel MH (2021) Saturation transfer difference NMR spectroscopy for the elucidation of supramolecular albumin–polymer interactions. ACS Macro Lett 10:819–824
Wilcox G (2005) Insulin and insulin resistance. Clin Biochem Rev 26:19–39
Mansoor S, Kondiah PPD, Choonara YE, Pillay V (2019) Polymer-based nanoparticle strategies for insulin delivery. Polymers 11:1380
Sharma G, Sharma AR, Nam J-S, Doss GPC, Lee S-S, Chakraborty C (2015) Nanoparticle based insulin delivery system: the next generation efficient therapy for Type 1 diabetes. J Nanobiotech 13:74
Wong CY, Al-Salami H, Dass CR (2017) Potential of insulin nanoparticle formulations for oral delivery and diabetes treatment. J Controlled Release 264:247–275
Li Y, Ji W, Peng H, Zhao R, Zhang T, Lu Z et al (2021) Charge-switchable zwitterionic polycarboxybetaine particle as an intestinal permeation enhancer for efficient oral insulin delivery. Theranostics 11:4452–4466
Wang J, Wang Z, Chen G, Wang Y, Ci T, Li H et al (2021) Injectable biodegradable polymeric complex for glucose-responsive insulin delivery. ACS Nano 15:4294–4304
Chen X, Yang D (2020) Functional zwitterionic biomaterials for administration of insulin. Biomater Sci 8:4906–4919
Zhu X, Vo C, Taylor M, Smith BR (2019) Non-spherical micro- and nanoparticles in nanomedicine. Mater Horiz 6:1094–1121
Jia W, Wang Y, Liu R, Yu X, Gao H (2021) Shape transformable strategies for drug delivery. Adv Funct Mater 31:2009765
Khine YY, Stenzel MH (2020) Surface modified cellulose nanomaterials: a source of non-spherical nanoparticles for drug delivery. Mater Horiz 7:1727–1758
Samal SK, Dash M, Van Vlierberghe S, Kaplan DL, Chiellini E, van Blitterswijk C et al (2012) Cationic polymers and their therapeutic potential. Chem Soc Rev 41:7147–7194
Truong NP, Jia Z, Burges M, McMillan NA, Monteiro MJ (2011) Self-catalyzed degradation of linear cationic poly (2-dimethylaminoethyl acrylate) in water. Biomacromol 12:1876–1882
Zhao W, Fonsny P, FitzGerald P, Warr GG, Perrier S (2013) Unexpected behavior of polydimethylsiloxane/poly(2-(dimethylamino)ethyl acrylate) (charged) amphiphilic block copolymers in aqueous solution. Polym Chem 4:2140–2150
Ros S, Kleinberger RM, Burke NAD, Rossi NAA, Stöver HDH (2018) Charge-shifting polycations with tunable rates of hydrolysis: effect of backbone substituents on poly[2-(dimethylamino)ethyl acrylates]. Macromolecules 51:5752–5761
Ros S, Wang J, Burke NAD, Stöver HDH (2020) A mechanistic study of the hydrolysis of Poly[N, N-(dimethylamino)ethyl acrylates] as charge-shifting polycations. Macromolecules 53:3514–3523
Whitfield R, Anastasaki A, Truong NP, Wilson P, Kempe K, Burns JA et al (2016) Well-defined PDMAEA stars via Cu(0)-mediated reversible deactivation radical polymerization. Macromolecules 49:8914–8924
Singhsa P, Diaz-Dussan D, Manuspiya H, Narain R (2018) Well-defined cationic N-[3-(dimethylamino)propyl]methacrylamide hydrochloride-based (Co)polymers for siRNA delivery. Biomacromol 19:209–221
van de Wetering P, Moret EE, Schuurmans-Nieuwenbroek NME, van Steenbergen MJ, Hennink WE (1999) Structure−activity relationships of water-soluble cationic methacrylate/methacrylamide polymers for nonviral gene delivery. Bioconjug Chem 10:589–597
Such GK, Johnston APR (2019) Understanding cell interactions using modular nanoparticle libraries. Aust J Chem 72:595–599
Raveendran R, Chen F, Kent B, Stenzel MH (2020) Estrone-decorated polyion complex micelles for targeted melittin delivery to hormone-responsive breast cancer cells. Biomacromol 21:1222–1233
Khine YY, Ganda S, Stenzel MH (2018) Covalent tethering of temperature responsive pNIPAm onto TEMPO-oxidized cellulose nanofibrils via three-component passerini reaction. ACS Macro Lett 7:412–418
Li Y, Milewska M, Khine YY, Ariotti N, Stenzel MH (2022) Trehalose coated nanocellulose to inhibit the infections by S. aureus. Polym Chem 13:1502–1509
Al-Nakashli R, Raveendran R, Khine YY, Cao C, McKay S, Lu H et al (2023) Drug-loading content influences cellular uptake of polymer-coated nanocellulose. Mol Pharm 20:2017–2028
Wang H, He J, Zhang M, Tam KC, Ni P (2015) A new pathway towards polymer modified cellulose nanocrystals via a “grafting onto” process for drug delivery. Polym Chem 6:4206–4209
Khine YY, Batchelor RR, Lu H, Raveendran R, Stenzel MH (2020) Photo-induced modification of nanocellulose: the design of self-fluorescent drug carriers. Macromol Rapid Commun 41:1900499. https://doi.org/10.1002/marc.201900499
Li N, Zhang H, Xiao Y, Huang Y, Xu M, You D et al (2019) Fabrication of cellulose-nanocrystal-based folate targeted nanomedicine via layer-by-layer assembly with lysosomal pH-controlled drug release into the nucleus. Biomacromol 20:937–948
Ndong Ntoutoume GMA, Grassot V, Brégier F, Chabanais J, Petit J-M, Granet R et al (2017) PEI-cellulose nanocrystal hybrids as efficient siRNA delivery agents—synthesis, physicochemical characterization and in vitro evaluation. Carbohydr Polym 164:258–267
Hu H, Yuan W, Liu F-S, Cheng G, Xu F-J, Ma J (2015) Redox-responsive polycation-functionalized cotton cellulose nanocrystals for effective cancer treatment. ACS Appl Mater Interfaces 7:8942–8951
Pötzinger Y, Rabel M, Ahrem H, Thamm J, Klemm D, Fischer D (2018) Polyelectrolyte layer assembly of bacterial nanocellulose whiskers with plasmid DNA as biocompatible non-viral gene delivery system. Cellulose 25:1939–1960
Kim Y, Pourgholami MH, Morris DL, Stenzel MH (2011) An optimized RGD-decorated micellar drug delivery system for albendazole for the treatment of ovarian cancer: from RAFT polymer synthesis to cellular uptake. Macromol Bioscience 11:219–233
Insua I, Wilkinson A, Fernandez-Trillo F (2016) Polyion complex (PIC) particles: preparation and biomedical applications. Europ Polym J 81:198–215
Kvach MV, Ustinov AV, Stepanova IA, Malakhov AD, Skorobogatyi MV, Shmanai VV et al (2008) A convenient synthesis of cyanine dyes: reagents for the labeling of biomolecules. Eur J Org Chem 2008:2107–2117
Harrison RH, Steele JA, Chapman R, Gormley AJ, Chow LW, Mahat MM et al (2015) Modular and versatile spatial functionalization of tissue engineering scaffolds through fiber-initiated controlled radical polymerization. Adv Funct Mater 25:5748–5757
Sun F, Feng C, Liu H, Huang X (2016) PHEA-g-PDMAEA well-defined graft copolymers: SET-LRP synthesis, self-catalyzed hydrolysis, and quaternization. Polym Chem 7:6973–6979
Del Pino P, Pelaz B, Zhang Q, Maffre P, Nienhaus GU, Parak WJ (2014) Protein corona formation around nanoparticles–from the past to the future. Mater Horiz 1:301–313
Tenzer S, Docter D, Kuharev J, Musyanovych A, Fetz V, Hecht R et al (2013) Rapid formation of plasma protein corona critically affects nanoparticle pathophysiology. Nat Nanotech 8:772
Liu N, Tang M, Ding J (2020) The interaction between nanoparticles-protein corona complex and cells and its toxic effect on cells. Chemosphere 245:125624
Walkey CD, Olsen JB, Guo H, Emili A, Chan WC (2012) Nanoparticle size and surface chemistry determine serum protein adsorption and macrophage uptake. J Am Chem Soc 134:2139–2147
Mustafa AZ, Kent B, Chapman R, Stenzel MH (2022) Fluorescence enables high throughput screening of polyelectrolyte–protein binding affinities. Polym Chem 13:6108–6113
Truong NP, Gu W, Prasadam I, Jia Z, Crawford R, Xiao Y et al (2013) An influenza virus-inspired polymer system for the timed release of siRNA. Nat Commun 4:1902
Zhang S, Saito K, Matsumoto H, Minagawa M, Tanioka A (2008) Characterization of insulin adsorption behavior on amphoteric charged membranes. Polym J 40:837–841
De Luca S, Chen F, Seal P, Stenzel MH, Smith SC (2017) Binding and release between polymeric carrier and protein drug: pH-mediated interplay of Coulomb forces, hydrogen bonding, van der Waals interactions, and entropy. Biomacromol 18:3665–3677
Li K, Chen F, Wang Y, Stenzel MH, Chapman R (2020) Polyion complex micelles for protein delivery benefit from flexible hydrophobic spacers in the binding group. Macromol Rapid Commun 41:e2000208
Acknowledgements
The authors acknowledge the facilities and the scientific and technical assistance of the NMR facility of the Mark Wainwright Analytical Centre. The authors would like to thank the Australian Research Council ARC for funding (FL200100124). YV is grateful to UNSW Sydney for a research training PhD Scholarship.
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions The authors received funding from the Australian Research Council ARC (FL200100124).
Author information
Authors and Affiliations
Contributions
Y.Y.K. wrote the manuscript and carried out polymer grafting and FRET analysis and drafted the manuscript. Y.L. prepared the polymers. W.G. contributed to the analysis of polymer coated nanocellulose. M.H.S. designed study and revised the manuscript.
Corresponding author
Ethics declarations
Ethical approval
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This is dedicated to Prof Stephan Förster to his 60th birthday.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Khine, Y., Li, Y., Ge, W. et al. A non-spherical nanoparticles system for the delivery of peptides using polymer grafted nanocellulose. Colloid Polym Sci (2024). https://doi.org/10.1007/s00396-024-05265-8
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00396-024-05265-8