
Abstract
Four strategies to incorporate a fluorinated monomer (perfluoro octyl acrylate, POA) into a waterborne polymeric dispersion are investigated. Due to the very low water solubility of the POA monomer, three of the strategies use miniemulsion droplets containing the whole POA monomer in the initial charge. The rest of the comonomers of the formulation (methyl methacrylate, MMA, and n-butyl acrylate, BA) are partially incorporated in the initial miniemulsion or fed to the reactor as a preemulsion. In the fourth strategy, a conventional seeded semibatch emulsion polymerization is carried out using cyclodextrin in the seed and feeding the POA/MMA/BA preemulsion to the reactor. Each process strategy led to a distinct particle morphology and hence a particular film morphology. We found that the strategy that produced core–shell particles with the core composed by pure polyPOA yielded the films that showed the best corrosion protection as measured in salt-spray test (1200 h standing without damage).
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Most of the organic coatings used for demanding anticorrosion applications (marine and large infrastructures) are based on solventborne systems. Nevertheless, due to the stringent environmental regulations (VOC’s restriction), the scientific and industrial community are devoting large efforts to the development of waterborne coatings [1]. The use of waterborne polymeric coatings on metal substrates can present the flash rust problem, which can be avoided by the use of a polymerizable phosphate surfactant, as it has been recently reported [2]. Conventional acrylic latexes (methyl methacrylate/butyl acrylate, MMA/BA: 50/50) containing such phosphate surfmer have shown interesting corrosion resistance properties even under harsh conditions (full corrosion protection provided up to 400 h of exposure in the salt spray test) [2]. However, in order to improve this corrosion protection for demanding applications, the barrier properties of the acrylic polymeric film must be improved.
One possibility is to include in the formulation fluorinated (meth)acrylate monomers, FMA [3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26]. These monomers are known because they provide good film-forming and low surface free energy polymers in addition to good chemical, thermal, and photochemical stability properties. It is worth noting that long perfluoro alkyl reagents (C8 and larger) are under scrutiny and strict regulations are forecast for a near future. Nonetheless, they have been extensively investigated to produce materials with superhydrophobic or superoleophobic properties and they have been reported in applications that include self-cleaning coatings, anti-corrosion coatings, anti-icing coatings, surface coatings for textiles, paper, and leather [7, 11, 19, 27,28,29,30,31]. In particular, copolymers with long side chains composed of a perfluoroakyl group (-CnF2n + 1) have received a great deal of attention. The conformational arrangement, distribution, and orientation of perfluoroaklyl side chains and their packing at the air-film interface have been considered a key parameter affecting the performance of the coating [32, 33]. A large number of FMAs with different fluorine content and side chain lengths have been employed to produce majorly coatings (also adhesives) by emulsion polymerization or related techniques [3, 4, 8,9,10,11,12, 17,18,19, 34]. It is worth noting that despite the low (or negligible) water solubility of most of the FMA monomers employed, in the majority of the cases, seeded semibatch emulsion polymerization processes (i.e., two stage process towards the production of a core/shell like morphology) with the fluorinated monomer added in the second stage (aiming at incorporating the FMA in the shell and minimizing its percentage in the formulation) have been reported. Some works have identified the challenge of incorporating such hydrophobic monomers in emulsion polymerization (limited transport rate in the aqueous phase and poor colloidal stability yielding low conversions and large amounts of coagulum) and have used solvents to increase the hydrophobicity of the aqueous phase [4]. Also, fluorinated (reactive) surfactants have been used to increase the affinity with the FMA monomer and facilitate its incorporation [9, 25]. Miniemulsion polymerization is a well-known technique to incorporate hydrophobic monomers or materials into polymer particles and has also been used to incorporate FMAs [11,12,13, 35]. Other alternatives to incorporate FMAs in emulsion polymerization include modification of the FMAs by introduction of hydroxyl groups in the structure to make FMAs more hydrophilic [6], or the use of cyclodextrins as phase transfer catalyst that facilitate the transport of the FMAs throughout the aqueous phase, enhancing monomer conversion and reducing colloidal stability issues observed in the final latexes [24, 36]. The amounts of FMAs targeted in these works were broad with most of them in the range 10–30 wt%.
It is worth to mention that, apart from the copolymer composition, molar mass distribution, branching and crosslinking distribution, and particle size distribution, the properties of a waterborne coating are strongly linked to the morphology of the film, which in turn depends on the morphology of the particles from which it has been cast. Achieving core–shell morphology latexes with FMAs incorporated in the shell phase has been the target of most of the works discussed above. Noteworthy is that most of the works assumed that the two-stage semibatch emulsion polymerization processes used, with addition of the FMAs in the second stage, led to core–shell morphology. However, this was not convincingly demonstrated in many reports. In addition, the analysis of the morphology of the films cast from the “core–shell” latexes is scarce [3, 6, 11, 24]. Most of the works analyzed the contact angle of the films, and the composition of the air-film (and film-substrate) using XPS, but as far as we know, none of the works addressed the relationship of different latex particle morphologies with the obtained film morphologies. The effect of the film morphology on the anticorrosion performance of the films is also missing.
In this work, we explored the increase of the hydrophobicity of waterborne coatings containing phosphate moieties by incorporating a fluorinated monomer, perfluorooctyl acrylate (POA), in the formulation. For this purpose, several process strategies have been implemented to efficiently incorporate the FMAs into the polymer particles. The proposed strategies used a combination of miniemulsion polymerization and different monomer feeding profiles, as well as cyclodextrin to facilitate the diffusion of the hydrophobic fluorinated monomer through the water phase to the polymer particle in conventional semibatch emulsion polymerization. The particle morphologies achieved in each process strategy were analyzed and correlated with the final film morphologies and the anticorrosion performance of the films (by electrochemical impedance spectroscopy (EIS) and salt spray chamber).
Experimental
Materials
For latex synthesis, we used methyl methacrylate (MMA), n-butyl acrylate (BA) (Quimidroga), and 1H,1H,2H,2H-perfluorooctyl acrylate (POA, Interchim) without further purification. Dodecyl diphenyl oxide disulfonate (Dowfax 2A1 45%, Dow Chemical Company) was used as an anionic emulsifier. Sipomer® PAM200, a phosphate ester of polypropylene glycol monomethacrylate (Solvay), was used as a surfmer. Azobisisobutyronitrile (AIBN, Fluka) radical initiator was used as received. Sodium bicarbonate (NaHCO3, Sigma-Aldrich) was used as a buffer to reduce the electrostatic interaction among droplets and to control the viscosity of the miniemulsion. Cyclodextrin (CD, Cavasol W7, Wacker) was kindly provided by Wacker and used as received. Deionized water (MiliQ quality) was used as continuous phase in all reactions performed.
Synthesis of POA containing latexes
Four process strategies were followed to incorporate the perfluorooctyl acrylate (POA) into latexes with 50% solids content, with an overall composition of POA/MMA/BA 30/40/30 (w/w/w; 10/57/33 mol basis), including Sipomer PAM200 phosphated surfmer in the formulation. The strategies are summarized in Fig. 1. PMB1, PMB2, and PMB3 latexes were synthesized by seeded semibatch emulsion copolymerization in which all POA contents (and variable amounts of MMA and BA) were initially incorporated in miniemulsion droplets. The miniemulsions were prepared as follows: the aqueous phase was composed by Dowfax 2A1 surfactant (DOW) and deionized water in a solution at 2 wt%. Both oil and aqueous phases were mixed separately under magnetic stirring for 15 min, and then, they were mixed together for 15 min more. Subsequently, the oil in water coarse emulsion, with a solids content of 30%, was sonicated using a Branson Sonifier 450 for 15 min (operating at 8-output control and 70% duty cycle in an ice bath and under magnetic stirring), allowing the formation of a stable miniemulsion. These nanodroplets were polymerized in batch to different extents (73% for PMB1, 0% for PMB2, and 100% for PMB3), before starting the feeding of the rest of MMA and BA content of the formulation. In the case of PMB4, a seeded semibatch emulsion polymerization was also used feeding a preemulsion of POA/MMA/BA to a MMA/BA seed that contained a small amount of cyclodextrin. The use of cyclodextrin allowed the transport of POA through the water phase and its incorporation to the polymer particles by conventional emulsion polymerization. Table 1 presents the formulations for experiments PMB1-3 and Table 2 for experiment PMB4.

Reaction strategies followed for the synthesis of PMB1, PMB2, PMB3, and PMB4 latexes
During the polymerizations, samples were withdrawn from the reactor to measure the unreacted monomer. For this purpose, 1H NMR was used. NMR sample preparation was carried out with 500 μL of latex and 50 μL of deuterated water (D2O), and the 1H-NMR was recorded with a Bruker AVANCE 400 MHz using a Watergate method in order to suppress the signal of water. Integration of the vinyl proton signals of each monomer was performed.
The films cast from the composite latexes were cryosectioned with a Leica EMUC6 cryoultramicrotome at −50 °C, with a Diatome 45° diamond 30 knife. The resulting cryosectioned samples were placed on copper grids covered with Formvar R for transmission electron microscopy (TEM). Transmission electron microscopy (TEM) analysis was carried out with a Tecnai TM G2 20 Twin device at 200 kV (FEI Electron Microscopes). The latexes were diluted with deionized water (with a concentration of 0.05 wt%) placed on copper grids covered with Formvar R and dried at ambient temperature.
The instrument above was also used for high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) of the latex particle morphology. HAADF-STEM is a STEM method, which records inelastically scattered electrons or thermal diffuse scattering (TDS) at high angles using an annular dark-field (ADF) detector (~ 50° to sufficiently high angle).
The hydrophobicity of the films was analyzed by the contact angle produced with a drop of water (10 μl) in an OCA 20 instrument (Dataphysics).
Water sensitivity and barrier properties were assessed by liquid water uptake (WU) test, that consists in monitoring for 15 days the weight gain by circular specimens (diameter = 24 mm, thickness = 2.3 mm) in water, and by water vapor transmission rate (WVTR), in which circular specimens, of the same dimensions as before, follow the cup test described in the ASTM E96.
For assessing corrosion performance, all synthetized latexes were cast on low carbon steel substrate. Previously, steel substrates were degreased with UniClean 251 solution at 70 °C in a shaking bath for 5 min followed by 1 min pickling in HCl solution (1:1). Then, the waterborne latexes were uniformly applied on the steel substrates using a quadruple film applicator (Khushbooscientific). The drying process was carried out using a temperature and humidity chamber (ESPEC SH-641 bench-top type), keeping the relative humidity at RH = 55% and temperature at 23 °C. The thickness of dried systems was determined using a Fischer Dualscope MP20, measuring at 10 different points of each sample, obtaining thicknesses around 75 μm for their electrochemical evaluation.
Films were also assessed in harsh corrosive conditions (5 wt% NaCl salty spray at 35 °C and 99% of RH; ASTM B117) produced by a salt spray chamber (Neurtek SC500). Samples were removed from the chamber after different time of exposure (240, 1200, and 1400 h), and a multichannel potentiostat BIO-LOGIC VMP3 was used to evaluate the corrosion behavior of the systems by EIS measurements. A three-electrode configuration was used in an electrochemical cell: Ag/AgCl saturated with KCl as reference electrode, platinum mesh as a counter electrode, and the different coatings as working electrodes. The electrochemical tests were conducted in 3.5 wt% NaCl solution at room temperature at least by triplicate using an area of 1 cm2. OCP was measured continuously with time although it was interrupted to carry out EIS measurements (once per hour). The frequency range was from 1 to 10 MHz, obtaining 10 points per decade. Frequency scans were carried out by applying ± 10 mV sinusoidal wave perturbation versus OCP.
X-ray photoelectron spectroscopy (XPS) was conducted in a SPECS system (Berlin, Germany) equipped with a Phoibos 150 1D-DLD analyzer and a monochromatic radiation source Al Kα (1486.7 eV). First, a wide scan was carried out in order to determine the elements present on the surface (step energy 1 eV, dwell time 0.1 s, pass energy 80 eV). Then, detailed scans were carried out in order to analyze the detected elements in detail (step energy 0.08 eV, dwell time 0.1 s, pass energy 30 eV) with an electron exit angle of 90°. The spectrophotometer was previously calibrated with Ag (Ag 3d5/2, 368.26 eV).
Results and discussion
Kinetics and cumulative composition of the copolymers
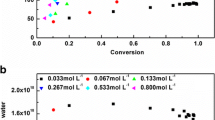
The four polymerization strategies employed in this work successfully produced latexes with final solid content of 50% (final monomer conversions higher than 99% were obtained in all the cases as shown in the Supporting Information, Fig. S2). The evolution of the conversion of each monomer was calculated by monitoring the unreacted monomer by NMR in samples withdrawn from the reactor (a representative NMR spectra of a sample taken in the polymerization to produce latex PMB1 is shown in Fig. S1 in the Supporting Information (SI) with the assignment of the double bond peaks of each monomer). Figure 2 displays the concentration of unreacted monomer in the reactor and the cumulative copolymer composition of the polymer for each experiment. Note that for experiment PMB3, only the gravimetric overall instantaneous conversion was measured because POA was polymerized in batch to full conversion (see Fig. S2 in the supporting information).

Time evolution of the unreacted concentration of comonomers (left), and cumulative copolymer composition (right) for experiments PMB1 (top). PMB2 (center) and PMB4 (bottom)
Figure 2 displays substantial differences in the built-up of the copolymers in each experiment. Thus, in PMB1 and PMB2, there is a clear drift in the composition of the chains; namely, the initial copolymers are rich in POA, and along the reaction, the chains are becoming richer in MMA/BA. Note that in PMB1, initially the copolymers are rich in MMA but quickly shift to rich in POA once the batch time is over and the MMA is almost depleted.
Although there are differences between PMB1 and PMB2, the most important difference is with respect to experiment PMB4. In this experiment, the composition remained constant during the whole process and close to the feed composition (POA/MMA/BA = 30/40/30 by weight) because true starved conditions were achieved (see Fig. 2 left and Fig. S2). This difference in the composition of the chains formed during the experiment will have implications on the morphologies of the polymer particles as it will be discussed below.
The intensity-average particle size and the dispersity of the distribution measured by DLS for the four experiments is displayed in Table 3.
Latexes produced using a miniemulsion that contained POA monomer (PMB1-3) yield latexes with large particles because the nanodroplets obtained in the miniemulsification were large too (400–500 nm). The reasons behind the large nanodroplet size are probably the high viscosity of POA, its high hydrophobicity (which lowers the emulsifier efficiency), and the not enough energy used in the miniemusification step [35]. No attempt was done to reduce the nanodroplet size in this work. On the other hand, the particle size obtained in PMB4 experiment was substantially lower because the seed did not contain POA (a MMA/BA latex with a particle size of 65 nm was used as seed). POA, MMA, and BA were fed in a preemulsion, and the particle growth followed the expected trend in absence of secondary nucleations and coagulation and yield a latex with 203 nm and very narrow dispersity.
Particle morphology
Despite the overall copolymer composition (POA/MMA/BA = 10/57/33 mol basis) was the same in all the polymerization strategies, latex particle morphology was different in each process. Figure 3 presents the TEM (STEM) micrographs of the particles for PMB1–4.

HAADF-STEM (a, b, and d) and TEM (c) micrographs of the polymer particles produced which each polymerization strategy. In HAADF-STEM images, bright regions correspond to electronically dense regions (i.e., F atom containing regions) as opposed to TEM (Figure 4a is reprinted with permission from reference [16])
The morphology of experiment PMB1 is a core–shell-shell with a core composed by terpolymer chains rich in MMA/BA (67%), an inner shell composed by terpolymer chains rich in POA (70%), and the external shell composed by terpolymer chains rich in MMA/BA (73%). The exact compositions of each of the phases were determined by nanoFTIR as described in a recent work, and the reader is referred to there for details [16]. For the purposes of this work, it suffices to say that the morphology achieved was clearly a non-equilibrium morphology because the inner shell is substantially more hydrophobic than the core, and hence, it was expected to be in the core. The latex particles of PMB2 present bright domains (rich in POA) distributed in the particle but only two phases can be detected. The polymerization process of PMB2 is similar to that of PMB1, but the initial miniemulsion only contained POA and all the MMA/BA was fed to the POA nanodroplets. As shown in Fig. 1 the evolution of the unreacted monomers and cumulative copolymers are similar, but in the initial stages of the reaction in experiment PMB2, the polymer is poorer in MMA/BA (because there was not MMA and BA in the initial nanodroplets loaded to the reactor). This difference in the composition of the chains yielded distinct morphologies in each case. It is out of the scope of this article to unveil the dynamic of the particle morphology production in these experiments.
The polymerization process to obtain latex PMB3 is equal to PMB2, but in this case, the addition of the MMA/BA monomer preemulsion stream started once the POA nanodroplets were fully converted to polyPOA homopolymer. In this case, the MMA/BA copolymer produced in the second stage formed a shell in the POA core as can be seen in the TEM micrograph (according to the evolution of the instantaneous conversion (see SI), true starved conditions were achieved and the composition of the shell was equal to the composition of the feed employed). The morphology is core–shell, but it is worth to note that plenty of small particles have also formed during the second stage (secondary nucleation).
Latex PMB4 presents a homogeneous morphology; namely, only a single bright phase can be distinguished, which according to the polymerization strategy indicated that chains with similar composition have been formed along the process, and hence, there is no noticeable phase separation. In this strategy, cyclodextrin was added in the initial charge (together with a small amount of seed polymer) and the three monomers were slowly fed to the reactor in a preemulsion under starved conditions. This result suggests that cyclodextrin has allowed to incorporate POA monomer together with the other two comonomers correctly to the particles yielding homogeneous copolymers with the composition close to the feed composition (POA/MMA/BA = 10/57/33 mol basis). This is in good agreement with the data of the cumulative copolymer composition of Fig. 1.
Film morphology
Films were cast from the latexes and thin cross-sections analyzed by TEM and SEM. AFM analysis of the air-film interface (top) was carried out, and the roughness of the surfaces measured using the geometric roughness (Rg).
Figure 4 presents the TEM micrographs for the four latex film cross-sections. Very different film morphologies were obtained as shown by the TEM micrographs. PMB1 film presented a complex morphology, which was attributed to the core–shell-shell morphology of the particles discussed above. The morphology of the particles was maintained during film formation. In the case of PMB2, the particle morphology was not as clear as the one of PMB1, but the phase separation between POA richer and MMA/BA richer domains could still be noticed by differences in the electron density along the film. On the other hand, PMB3 film presented a continuous MMA/BA phase (bright), in which POA rich domains (black) could be clearly identified. Finally, PMB4 film presented a much more homogeneous morphology in which the differences between POA rich and POA poor domains could not be noticed, because as confirmed by the cumulative copolymer composition measured by NMR, all the chains have similar composition, and hence, no phase separation occurred and morphology was homogeneous.

TEM micrographs of cross-sectioned films cast from PMB1, PMB2, PMB3, and PMB4
Figure 5 presents SEM images of the cross-sections of the films cast from latexes PMB1-4. Figure 5a shows a film (for PMB1) in which the polymer particles are not fully coalesced. This is likely caused by the lack of coalescence of the inner shell of the particles (as observed in the TEM micrograph, the core–shell-shell morphology was preserved in the film) that is incompatible with the polymers in the core and the outer shell and hence yields a non-coalesced film. Figure 5b and c (for PMB2 and PMB3 latexes, respectively) show similar film morphologies. In both films, a coherent film corresponding to the MMA/BA-rich polymer was formed and phase separated spots (bright clusters), corresponding to rich POA copolymers for PMB2 and pure POA homopolymer in PMB3, can be distinguished. These clusters are homogeneously distributed in the MMA/BA rich matrix. On the other hand, PMB4 film is homogeneous in good agreement with the observations in the TEM images of the film and also of the polymer particles.

Height and phase AFM images of the air-film interfaces are presented in Fig. 6, and the corresponding average roughness (Rg) are also embedded in the figure.

AFM height (left) and phase (right) images of the air-film substrate and the average roughness of the films PMB1–4
The height images show that films PMB2 and PMB4 both have more homogenous surface than PMB1 and PMB3 that show phase separation of soft and hard phases. PMB4 latexes yield small particles and with homogeneous copolymer composition of the chains, and this is clearly seen in the air-film surface as compared with PMB2 that yielded larger particles with some composition drift which apparently was not enough as to show phase separation in the surface. PMB1 process produced also large particles, but the miniemulsion rich in POA was polymerized in batch yielding at the beginning copolymers rich in POA and then copolymers that were richer in the MMA/BA fed to the reactor. As shown in Figs. 3–5, the morphology was complex; a core–shell-shell morphology was achieved. The air-film interface presents hard domains (bright) dispersed in a softer matrix (dark). Admittedly, we are not able to interpret this surface with the knowledge of the morphologies presented in Figs. 3–5. On the other hand, PMB3 presents phase separation on the surface. The process yielded pure POA polymer and copolymers of MMA/BA that during film formation phase separated (see Figs. 4 and 5). According to the AFM image, part of the soft material (POA phase) migrated to the air-film interface. The roughness of the films was higher for the latexes PMB1 and PMB2 and lower for PMB3 and PMB4.
Film properties
DSC measurements of the films (see Fig. 7) confirm that the most homogenous polymer corresponds to that produced in PMB4. A single narrow peak at around 19 °C is observed, which is in fair agreement with terpolymers of POA/MMA/BA = 30/40/30 (weight basis) that according to the Fox equation will have a Tg of 12 °C. In films PMB1 and PMB3, two transitions can be distinguished, whereas in PMB2, a single broad transition is observed. The two Tg in PMB1 and PMB3 correspond to the two incompatible phases present in the particles, namely, one rich in POA (lower Tg) and one rich in the MMA/BA comonomers (higher Tg). The nanoFTIR analysis done for the PMB1 sample [16] indicated the presence of three phases two of them rich in MMA/BA (core and outer shell) comonomers and one rich in POA monomer (inner shell). The morphology of PMB3 presents two phases that correspond to pure POA homopolymer and a copolymer of MMA/BA synthesized under starved conditions. Therefore, the lower transition temperature (at 0.53 °C) should correspond to polyPOA (whose Tg was measured as −6 °C), and the higher temperature one (at 27 °C) should correspond to the MMA/BA copolymer (according to the Fox equation at the ratio MMA/BA = 57/43 the Tg should be 21 °C). These transition temperatures for PMB1 are both shifted to higher (3.40 °C) and lower (22 °C) values as compared with the two phases in PMB3, respectively, because each phase is not pure as in PMB3. In other words, in PMB1, the POA-rich phase contains MMA/BA comonomers in the chains increasing the Tg and the reverse occurs for the MMA/BA-rich phase that contains POA comonomer.

First heating DSC curves of the PBM1-4 films
PMB2 presents a single broad transition spanning from −10 to 30 °C with a maximum at around 17 °C. This means that in this strategy, terpolymers with varying compositions were obtained throughout the process although a large fraction of the chains presents a composition that is close to the formulation composition (POA/MMA/BA = 30/40/30 mol basis). This Tg and the Tg of the homogeneous polymer achieved in strategy PMB4 are similar. It is worth noting that this strategy resembles the pseudo-optimal strategies developed by Arzamendi and Asua [37] to produce homogeneous copolymer when monomers with different reactivity ratios are copolymerized. As discussed by the authors, depending on the constant feeding rate used, higher or lower composition drift is obtained with this strategy.
In the last two decades, large efforts have been devoted to the synthesis of core–shell morphology fluorinated acrylate latexes with the aim to create coatings with reduced surface energy and (super)hydrophobic properties. It is claimed that due to the core–shell morphology exhibited by the polymer particles with the FMA monomers located in the shell, the films made out of these latexes have a tendency to enrich the air-film interface with fluoroalkyl groups [6, 7, 10, 13, 15, 20,21,22, 26]. Furthermore, in some cases, a gradient of fluorine atoms from air-film to film-substrate interfaces is also reported [6, 20, 21, 24]. This accumulation of the fluorine atoms at the air-film interface is determined by analyzing the surfaces with X-ray photoelectron spectroscopy (XPS), AFM, and measuring contact angles (CA), which are also affected by the roughness of the film and hence can be misleading. In this work, XPS was used to measure the fluorine content at the air-film and film-substrate of the films made from the latexes PMB1-4 that present different morphologies (see Fig. 3 for TEM of the polymer particles, and Fig. 6 for AFMs of the air-film surface). The XPS data is summarized in Table 4.
The fluorine content (mol basis) of a terpolymer with the composition used in the formulation of the latexes is 6.5% (neglecting the presence of surfactant, initiator, and buffer used in the formulation). On the other hand, the fluorine content of the pure POA polymer is 39.4%. As discussed above, the only latex produced with homogenous composition and equal to that of the feed was PMB4. Interestingly, the F content of the top and bottom surfaces of PMB4 film shows values that are really close to the expected F content of the terpolymer with the feed composition. This result is in excellent agreement with the kinetics and morphology of the particles and films presented above. Contrary to what is reported in the literature, there is no noticeable enrichment of F atoms in the air-film interface.
Film PMB2 shows also values that are close to the average value, but in this case, the air-film F content is larger than the average value and film-substrate is lower suggesting some segregation of F richer chains in the air-film interface. The composition drift of the chains formed in this strategy was not large as reported in Fig. 1 and also confirmed in the DSC (Fig. 7). The AFM also shows a relatively homogeneous air-film interface, similar to that of PMB4 but with larger particles.
Films PMB1 and PMB3 present the highest fluorine contents at both interfaces. The polymerization strategy to produce latex PMB1 led to substantial composition drift in the copolymer chains (rich copolymers of POA and rich copolymers in MMA/BA that produced a core–shell-shell morphology where three distinct phases could be recognized in the TEM analysis). The air-film interface of PMB1 presented visible soft domains, which should correspond to POA rich chains. However, the SEM and TEM images of cross-sections of the film display that the morphology of the particles was preserved; namely, that the inner shell that is composed by copolymer chains rich in POA did not phase separate upon film formation. Consequently, the high fluorine content measured at the air-film interphase and film-substrate interphase should be a consequence of the arrangements of the POA monomer units present in the copolymer chains of the outer shell. Admittedly, we do not have an explanation why the film-substrate presents a higher F content than the air-film interface and neither why the F content at the film-substrate is that close to pure POA.
PMB3 latex is composed by two populations of particles. The large particles are core–shell particles with the core made out of pure POA and a shell of the MMA/BA copolymer. The small ones are composed by pure MMA/BA copolymer chains. The TEM images of the film cross-sections show POA domains dispersed in the MMA/BA matrix generated by the coalescence of the small MMA/BA nanoparticles. The AFM image of the air-film interface shows large soft domains that could have been generated by migration and phase separation of the POA domains, but these large domains (several microns) are not seen in the TEM and SEM images. The high F content values measured at the air-film and film-substrate agrees with the phase separation observed in the AFM, but why this phase separation only occurs at the interface is not understood yet.
For PMB1 and PMB3, the bottom interface is richer in F content than the top, contrary to what it has been reported in the literature that showed a preferential enrichment of F atoms at the air-film interface.
Water uptake, water vapor transmission rate (VVTR), and contact angles (in the air-film interface) were also measured for the four films. For comparison purposes, the results of a non-fluorinated acrylate latex (MB film) with a composition MMA/BA = 50/50 and produced by seeded semibatch emulsion polymerization are also shown in Table 5.
The incorporation of the fluorinated monomer led to higher surface contact angle than the one presented by the pure acrylic film (MB). Among the fluorinated films, PMB1 presented the higher contact angle which should be related with the balance between high roughness (Rg = 18.1 nm) and high fluorine content (20.1%) on the surface. The rest of the films presented contact angles in the range 90–105°.
Water uptake values were more scattered. In fact, PMB1 film showed a decreased water uptake compared to MB, but PMB4 presented much higher water uptake values, whilst PMB3 was similar to MB. The high value of PMB4 can be likely attributed to the presence of small amounts of cyclodextrin. On the other hand, the integrity of PMB2 did not resist the immersion in water. As a result, the water resistance measurements provided conflicting results in terms of the effect of the incorporation of the fluorinated monomer.
WVTR were lower for the fluorinated latexes with the exception of PMB1 that presented a notably much higher value, likely because as shown in the SEM micrograph, the film did not completely coalesce and created pores from which water vapor molecules travel [38].
Corrosion performance of the films
In order to evaluate the anticorrosion properties of these films, each latex was cast onto steel substrate to promote the in situ phosphatization of the metal/film interface (optimized conditions are at 23 °C and 55% RH) [2] and afterwards, films of approximately 75 μm of thickness, were tested by salt spray (using 5 wt% NaCl to create a fog under saturated humidity). In all the cases, the in situ phosphatization was produced, as confirmed by the yellowish coloration that appeared on each steel substrate after the film formation. However, despite the beneficial effect of the in situ phosphatization layer on the anticorrosion properties of waterborne coatings [2], only PMB3 film provided corrosion protection superior to non-fluorinated acrylate latexes synthesized using phosphate polymerizable surfactants [2, 39]. Notably, this film presents the best balance between water uptake and water vapor permeation rate of the four films.
Figure 8a and b present the EIS spectra (both Bode plots) of the PMB3 film after different time of exposure to the salt spray chamber. It is remarkable that this film withstand up to 1200 h without suffering any noticeable corrosion effect. In fact, it is confirmed by the capacitive behavior shown by the coating according to both Bode plots. The impedance modulus (|Z|) obtained at low frequency (from 1 to 10−2 Hz) reached a plateau within the range of 109–1010 Ω cm2, whilst the phase angle remained in the range of −70/ −90° (although there are some scatter points due to noise at frequencies below 102 Hz). However, if the exposure is increased up to 1400 h, the impedance (|Z| at 10–2 Hz) drastically decreased to values around 106 Ω cm2. Figure 8c displays the image of the sample after 1400 h of exposure where clearly a heterogeneous surface that may be affected by the corrosion process according to the EIS results can be seen.

EIS (impedance a and phase b) spectra after different immersion times in salt-spray chamber and c photograph of the sample immersed in the chamber after 1400 h of exposure
Conclusions
The incorporation of perfluoro octyl acrylate (POA) in the formulation of waterborne acrylic coatings for corrosion protection has been carried out following four different approaches (from PMB1 to PMB4). Due to the insolubility of the POA monomer in water, miniemulsion polymerization was used in the first three strategies to incorporate the FMA monomer to the copolymer latex. Thus, in PMB1, nanodroplets containing the whole POA and a fraction of MMA/BA were first polymerized in batch, and then, the remaining MMA/BA was fed to the reactor. In PMB2, pure nanodroplets of POA were loaded in the reactor and the polymerization was started at the same time that the feeding of the MMA/BA comonomers. In PMB3, pure nanodroplets of POA were fully polymerized in batch to produce a seed of polyPOA and a MMA/BA preemulsion was fed to the seed at constant flow rate. In contrast to these strategies that used miniemulsion polymerization, PMB4 was carried out using a conventional seeded semibatch emulsion polymerization. The seed contained a small amount of cyclodextrin that acted as phase transfer catalyst to help transporting the POA to the polymer particles. The comonomer mixture (POA/MMA/BA) was fed to the reactor as a preemulsion at constant flow rate.
Distinct particle sizes and morphologies were achieved. The processes using miniemulsion polymerization yielded latexes with large average particle sizes (> 400 nm) and PMB4 yielded a latex with smaller particles (around 200 nm). Morphologies were multiphasic for PMB1-PMB3, whereas homogeneous particles were achieved for PMB4 in agreement with the composition of the chains formed in each process.
The morphologies of the films were also distinct. The particle morphology was preserved in the film for PMB1 and PMB4. For PMB2 and PMB3, phase separation occurred during film formation (and also during polymerization) and domains rich in POA (pure POA for PMB3) were formed. AFM, XPS, and contact angle measurements of the air-film interface were in reasonable agreement with the achieved morphologies. We did not find a noticeable enrichment of F atoms in the air-film interface as reported by other authors.
All the latexes showed the ability to in situ phosphatize the steel substrate when they were applied on the steel specimens, but only the coating based on PMB3 latex was able to provide full corrosion protection against the harsh corrosive conditions. Notably, the PMB3 film maintained its corrosion resistance for 1200 h. These results highlight the strong influence of the particle and film morphology on the anticorrosion performance and the key role that the chosen reaction strategy plays on the design of efficient waterborne coatings for anticorrosion application.
References
Pilcher G (2002) Meeting the challenge of radical change: coatings R&D as we enter the 21st century. 187:1–16
Chimenti S, Vega JM, García-Lecina E et al (2019) In-situ phosphatization and enhanced corrosion properties of films made of phosphate functionalized nanoparticles. React Funct Polym 143:104334. https://doi.org/10.1016/j.reactfunctpolym.2019.104334
Thomas RR, Lloyd KG, Stika KM et al (2000) Low free energy surfaces using blends of fluorinated acrylic copolymer and hydrocarbon acrylic copolymer latexes. Macromolecules 33:8828–8841. https://doi.org/10.1021/ma000221p
Ha JW, Park IJ, Lee SB (2005) Hydrophobicity and sliding behavior of liquid droplets on the fluorinated latex films. Macromolecules 38:736–744. https://doi.org/10.1021/ma0488764
Hao L, An Q, Xu W, Huang L (2012) Synthesis, film morphology and hydrophobicity of novel fluorinated polyacrylate emulsion and solution on silicon wafer. Colloids Surfaces A Physicochem Eng Asp 396:83–89. https://doi.org/10.1016/j.colsurfa.2011.12.045
Cheng Y, Wang Z (2013) Fluorinated poly(isobornyl methacrylate-co-butyl acrylate) core-shell latex nanoparticles: synthesis, morphology and wettability of films. Polymer (Guildf) 54:3047–3054. https://doi.org/10.1016/j.polymer.2013.03.029
Xu W, An Q, Hao L et al (2014) Synthesis of self-crosslinking fluorinated polyacrylate soap-free latex and its waterproofing application on cotton fabrics. Fibers Polym 15:457–464. https://doi.org/10.1007/s12221-014-0457-8
Zhang Z, Wang P (2015) Fluorinated latex particles prepared by semibatch miniemulsion polymerization and its film surface properties. J Dispers Sci Technol 36:291–297. https://doi.org/10.1080/01932691.2014.903807
Yang W, Zhu L, Chen Y (2015) Spherical and core-shell fluorinated polyacrylate latex particles: preparation and characterization. Colloid Polym Sci 293:2349–2357. https://doi.org/10.1007/s00396-015-3583-y
Wang Z, Wang Z (2015) Synthesis of cross-linkable fluorinated core-shell latex nanoparticles and the hydrophobic stability of films. Polymer (Guildf) 74:216–223. https://doi.org/10.1016/j.polymer.2015.08.012
Lü T, Qi D, Zhang D et al (2016) Fabrication of self-cross-linking fluorinated polyacrylate latex particles with core-shell structure and film properties. React Funct Polym 104:9–14. https://doi.org/10.1016/j.reactfunctpolym.2016.04.020
Gu Z, Cheng J, Zhang M et al (2017) Effect of groups at α-position and side-chain structure of comonomers on surface free energy and surface reorganization of fluorinated methacrylate copolymer. Polymer (Guildf) 114:79–87. https://doi.org/10.1016/j.polymer.2017.02.073
Li J, Zhong S, Chen Z et al (2018) Fabrication and properties of polysilsesquioxane-based trilayer core–shell structure latex coatings with fluorinated polyacrylate and silica nanocomposite as the shell layer. J Coatings Technol Res 15:1077–1088. https://doi.org/10.1007/s11998-018-0044-9
Wu G, Li W, Ni W et al (2019) Preparation of nonspherical fluorinated acrylate polymer particles by a “surface tension controlling” method and their applications in light-diffusing films. Macromol Mater Eng 304:1–12. https://doi.org/10.1002/mame.201900174
Zhang C, Chen Y (2005) Investigation of fluorinated polyacrylate latex with core-shell structure. Polym Int 54:1027–1033. https://doi.org/10.1002/pi.1803
Goikoetxea M, Amenabar I, Chimenti S et al (2021) Cross-sectional chemical nanoimaging of composite polymer nanoparticles by infrared nanospectroscopy. Macromolecules 54:995–1005. https://doi.org/10.1021/acs.macromol.0c02287
López AB, De La Cal JC, Asua JM (2016) Highly hydrophobic coatings from waterborne latexes. Langmuir 32:7459–7466. https://doi.org/10.1021/acs.langmuir.6b01072
López AB, de la Cal JC, Asua JM (2017) From fractal polymer dispersions to mechanically resistant waterborne superhydrophobic coatings. Polymer (Guildf) 124:12–19. https://doi.org/10.1016/j.polymer.2017.07.043
López AB, De La Cal JC, Asua JM (2016) Controlling film topography to form highly hydrophobic waterborne coatings. Soft Matter 12:7005–7011. https://doi.org/10.1039/c6sm01081d
Cui X, Zhong S, Gao Y, Wang H (2008) Preparation and characterization of emulsifier-free core-shell interpenetrating polymer network-fluorinated polyacrylate latex particles. Colloids Surfaces A Physicochem Eng Asp 324:14–21. https://doi.org/10.1016/j.colsurfa.2008.03.018
Xiao X, Wang Y (2009) Emulsion copolymerization of fluorinated acrylate in the presence of a polymerizable emulsifier. Colloids Surfaces A Physicochem Eng Asp 348:151–156. https://doi.org/10.1016/j.colsurfa.2009.07.006
Wang J, Zeng XR, Li HQ (2010) Preparation and characterization of soap-free fluorine-containing acrylate latex. J Coatings Technol Res 7:469–476. https://doi.org/10.1007/s11998-009-9199-8
He L, Liang J, Zhao X et al (2010) Preparation and comparative evaluation of well-defined fluorinated acrylic copolymer latex and solution for ancient stone protection. Prog Org Coatings 69:352–358. https://doi.org/10.1016/j.porgcoat.2010.07.008
Yang C, Castelvetro V, Scalarone D et al (2011) Three different β-cyclodextrins direct the emulsion copolymerization of a highly fluorinated methacrylate toward distinctive nanostructured particle morphologies. J Polym Sci Part A Polym Chem 49:4518–4530. https://doi.org/10.1002/pola.24921
Chen LJ, Wu FQ (2011) Structure and properties of novel fluorinated polyacrylate latex prepared with reactive surfactant. Polym Sci - Ser B 53:606–611. https://doi.org/10.1134/S1560090411120013
Bai R, Qiu T, Duan M et al (2012) Synthesis and characterization of core-shell polysilsesquioxane-poly(styrene-butyl acrylate-fluorinated acrylate) hybrid latex particles. Colloids Surfaces A Physicochem Eng Asp 396:251–257. https://doi.org/10.1016/j.colsurfa.2012.01.003
Ganesh VA, Raut HK, Nair AS, Ramakrishna S (2011) A review on self-cleaning coatings. J Mater Chem 21:16304–16322. https://doi.org/10.1039/c1jm12523k
Yao W, Li Y, Huang X (2014) Fluorinated poly(meth)acrylate: synthesis and properties. Polymer (Guildf) 55:6197–6211. https://doi.org/10.1016/j.polymer.2014.09.036
Darmanin T, Guittard F (2014) Recent advances in the potential applications of bioinspired superhydrophobic materials. J Mater Chem A 2:16319–16359. https://doi.org/10.1039/c4ta02071e
Sun Y, Zhao X, Liu R et al (2018) Synthesis and characterization of fluorinated polyacrylate as water and oil repellent and soil release finishing agent for polyester fabric. Prog Org Coatings 123:306–313. https://doi.org/10.1016/j.porgcoat.2018.07.013
Lopez AB, Bohórquez SJ, Meeuwisse M et al (2018) Self-matting waterborne fluoropolymers Prog Org Coatings 116:57–69. https://doi.org/10.1016/j.porgcoat.2017.12.003
Volkov VV, Plat NA (1992) Aggregation state and mesophase structure of comb-shaped polymers with fluorocarbon side groups 33:1316–1320
Shimizu T, Tanaka Y, Kutsumizu S, Yano S (1996) Ordered structure of poly(1H,1H-fluoroalkyl R-fluoroacrylate)s. Macromolecules 29:159–164
Liang J, Ling H, Zheng Y (2009) Synthesis and property investigation of three core-shell fluoroacrylate copolymer latexes. J Appl Polym Sci 112:1615–1621
Asua JM (2002) Miniemulsion polymerization. Prog Polym Sci 27:1283–1346. https://doi.org/10.1016/S0079-6700(02)00010-2
Lau W (2002) Emulsion polymerization of hydrophobic monomers. Macromol Symp 182:283–289
Arzamendi G, Asua JM Monomer addition policies for copolymer composition control in semicontinuous emulsion copolymerization
Vega JM, Chimenti S, García-Lecina E et al (2020) Impact of the in-situ phosphatization on the corrosion resistance of steel coated with fluorinated waterborne binders assessed by SKP and EIS. Prog Org Coatings 148. https://doi.org/10.1016/j.porgcoat.2020.105706
Chimenti S, Vega JM, Lecina EG et al (2019) Combined effect of crystalline nanodomains and in situ phosphatization on the anticorrosion properties of waterborne composite latex films. Ind Eng Chem Res 58:21022–21030. https://doi.org/10.1021/acs.iecr.9b02233
Acknowledgements
The authors would like to thank the financial support received from Basque Goverment (ELKARTEK KK-2017/00096, KK-2018/00108 and IT-999-16), and from the Spanish Goverment (MINECO CTQ -2017-87841-R) and MICINN (PDC2021-121416-100). The authors would like also to thank for microscopy analysis and human support provided by SGKiker of UPV/EHU and European funding (ERDF and ESF).
Funding
Open Access funding provided thanks to the CRUE-CSIC agreement with Springer Nature.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chimenti, S., Vega, J.M., Paulis, M. et al. Strategies to incorporate a fluorinated acrylate monomer into polymer particles: from particle morphology to film morphology and anticorrosion properties. Colloid Polym Sci 300, 429–443 (2022). https://doi.org/10.1007/s00396-022-04943-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00396-022-04943-9