
Abstract
Hollow spheres made from titanium dioxide (TiO2) are interesting structures because of their high surface area and low density, combined with semiconducting properties of the TiO2. However, the synthesis is still challenging because of the high reactivity of the titania precursors. Here, we present a simple, reproducible, and scalable way to synthesize TiO2 hollow spheres in the micrometer/sub-micrometer size range comprising three steps: synthesis of polystyrene template particles, growth of TiO2 shells, and calcination to hollow spheres. We investigate the importance of adjusting the seed particle surface functionalization via the appropriate choice of comonomer during the dispersion polymerization. An aging step and a calcination process at low temperatures are mandatory to retain the particle integrity during the seed particle removal. We provide a detailed characterization of each step of this process including electron microscopy, small angle X-ray scattering, and simultaneous thermal analysis.

Graphical abstract
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Environmental pollution is a worldwide pressing issue, which needs to be addressed by society, politicians, and researchers. Whereas “chemistry” certainly contributes in various ways to environmental pollution, it also offers solutions toward a more sustainable future. The awareness of this fact led to the field of “green chemistry.” It was introduced in 1998 by Paul Anastas and John Warner and is based on twelve principles to design chemical products and processes in an environmental friendly way [1,2,3]. It includes less hazardous chemical synthesis and solvents, design for energy efficiency, and catalysis. One particular materials class that is regularly discussed in the context of green chemistry is titanium dioxide. Titanium dioxide (TiO2) can be classified as a green chemical or material because of its photocatalytic activity [4, 5], usage in solar cells [6,7,8] and batteries [9,10,11], low toxicity, and high chemical stability.
TiO2 exists in many different polymorphs, with rutile, brookite, and anatase being the most prominent ones. The main difference of the three polymorphs is their thermodynamic stability. Rutile is the most stable phase in bulk materials and at high temperatures [12, 13]. However, anatase and brookite phases are preferentially formed in small structures in nature as well as during solution-based synthesis [13,14,15]. An amorphous TiO2 phase is also known and typically used as the starting material for transformations into phase pure anatase particles at high temperatures > 100 °C [13, 16, 17].
In addition to the microscopic structure, the mesoscopic shape of the TiO2 material is important for photovoltaic, or photonic applications. Different shapes have been tested as electrodes for photovoltaic applications: thin films, nanoparticle assemblies, inverse opals, and nanotube arrays [18,19,20,21,22,23]. Nanotube arrays achieved photoconversion efficiencies up to 4.9% in solar cells [22] and can also be used for hydrogen storage applications [24]. For optical and photonic applications, typically inverse opal structures are used [12, 25,26,27]. Here, the high refractive index of the different polymorphs of TiO2 is used in the context of structural coloration or efficient scattering.
The different shapes can be achieved via a wide range of synthesis routes [12, 28]. The most common and easy one is the sol-gel method, where a precursor is first hydrolyzed in an acidic or basic environment, followed by polymerization into TiO2. Organic metal compounds or inorganic metal salts are used as precursors. The sol-gel synthesis method leads to a wide variety of structures, from nanoparticles in different sizes and shapes to rod- and tube-like structures. Further methods are hydrothermal or solvothermal synthesis, where the reaction takes place at temperatures above the boiling point of the solvent up to 240 °C in an autoclave, and thus at high pressures [12, 13, 29]. Using this method, it is possible to generate phase pure nanoparticles or nanorods. Chemical or physical vapor deposition processes are further synthesis methods that lead to oriented nanowire arrays [12, 28].
It is also possible to build TiO2 structures in a templated approach. This leads to inverse opals or hollow spheres after removal of the template. To prepare inverse opals, usually, a template structure from assembled polymer particles is infiltrated either with a precursor mixture that undergoes a sol-gel reaction in the pores or with pre-synthesized TiO2 nanoparticles [30]. A different approach was used by Lu et al. who first prepared polymer-TiO2 core-shell particles that were assembled and calcined to get macroporous structures. They were able to prepare phase pure anatase particles at room temperature by using polystyrene particles with grafted poly(styrene sodium sulfonate) chains as template particles. The TiO2 particles were synthesized by a sol-gel process in between the grafted polymer chains by slowly adding a precursor solution. The core-shell particles were assembled by drying the dispersion and calcined in argon to remain the three-dimensional structure [4]. This synthesis is located between an inverse opal and a hollow sphere synthesis. Hollow sphere objects have evolved over the past years as a particularly interesting shape, owing to the material structuring on multiple length scales: shell, particle diameter, and particle ensemble [31,32,33]. The particles have a low density but are large enough to be easily filtered and recycled after a catalysis process. Furthermore, the surface area is large and freely accessible from both sides which may be interesting for solar cells or battery devices.
A wide-spread and general approach toward hollow sphere structures is based on shell growth on top of polymeric seed particles. Three steps need to be controlled for this process: (1) template particle formation, (2) shell growth, and (3) template removal (see Fig. 1).

Overview on the three steps that are needed to prepare TiO2 hollow spheres. First, cationic polystyrene particles are synthesized via dispersion polymerization, using Polyvinylpyrrolidone (PVP) as stabilizer and 2-Methacryloxyethyltrimethylammoniumchloride (MTC) as comonomer. The TiO2 shells are fabricated by a condensation reaction of titanium butoxide (TBT). After an aging step of 24 h, the particles were calcined at 400 °C in air
We briefly outline these steps:
-
1)
The template particles are typically synthesized via emulsifier-free emulsion polymerization or dispersion polymerization. The mechanism of dispersion polymerization has been discussed by Barrett and Arshady and is well known [34, 35]. Generally, size control in dispersion polymerization is achieved by the amount of monomer [36], solvent [35, 37, 38], and stabilizer selection [34, 37, 38], respectively. Cheng et al. [36] used an ethanol/water mixture as solvent and polyvinylpyrrolidone (PVP) as stabilizer. Furthermore, a co-monomer can be used to introduce a specific surface charge.
-
2)
Shell growth: One major issue of the TiO2 shell growth – in contrast to a silica coating – is the high reactivity of the TiO2 precursors. Barlier et al. examined the condensation reaction in detail [39]. The reaction happens in two steps: the hydrolysis of the titanium precursor and the condensation to the TiO2 network (Scheme 1). Imhof was the first who coated polystyrene (PS) particles with a thin TiO2 layer in a one-step sol-gel approach [40]. Up to now, several more methods have been published based on sol-gel synthesis: varying precursors, solvents, and template particles [36, 41,42,43]. Wang et al. controlled the diffusion of the TiO2 precursor by synthesizing in an ethanol/acetonitrile mixture. With this approach, they were able to get defined shell thicknesses between 8 and 65 nm on 300 nm anionic PS particles [43]. Taniguchi et al. used grafted poly[2-(N,N-dimethylamino)ethyl] methacrylate chains on PS template particles that catalyzed the hydrolysis and condensation of the TiO2 precursor and were able to coat template particles in a size range of 90–450 nm [42]. Cheng et al. were the only ones who controlled the reaction speed by adding the TiO2 precursor dropwise within 30 min instead of one quick addition step. Similar to Imhof et al., they used cationic PS template particles that attracted the TiO2 precursor and lead to shell growth [36].

Condensation reaction of titanium butoxide in water
-
3)
There are two options to remove the template particles: dissolution and calcination. Toluene [40] or THF [41] are commonly used to dissolve non-crosslinked polymer cores via repeated centrifugation and redispersion. This requires a certain degree of porosity and pore sizes in the coated shell to allow for sufficient mass transport. Cheng et al. removed the core directly after the synthesis while heating the core-shell particles in an ethanol-ammonia mixture [36]. Calcination, however, is the more widespread strategy to remove the template [40,41,42,43,44]. An inherent side-effect of the thermal decomposition is the concomitant phase transition of the amorphous TiO2 shell into its anatase form. Therefore, the selection of the right temperature profile and calcination atmosphere is of main importance for the stability of the final hollow particles. It is common to simply heat the samples in air between 500 and 600 °C, followed by an isothermal step of 2 to 3 h [40, 42, 43]. Lu et al. found that their structures collapsed when using this simple approach. That is why they first pyrolised their structures in an inert atmosphere at 500 °C, followed by a calcination step in air to remove the carbon that stabilized the structure [4]. Schroden et al. solved the stability problem by applying a more complex heating ramp. Generally, they used very slow heating rates of 2 K/min and heated the sample first to 300 °C for 2 h, followed by a second heating step to 400 °C for 2 h. With this profile, they were able to get stable inverse opal structures without using an inert atmosphere during the thermal treatment [25].
We build upon these existing methods and provide an approach toward highly uniform TiO2 hollow spheres with a scalable and simple synthesis route. We used dispersion polymerization to prepare monodisperse polystyrene particles in a size range of 700 nm to 1.3 μm. Our method extends the range of accessible particle sizes known from emulsifier-free emulsion polymerization considerably, where an upper limit of 600–800 nm is known [45]. Furthermore, few purification steps are needed in our protocol, which improves the efficiency and yield of the synthesis. Using a combined mass loss – differential scanning calorimetry – infrared analysis, we also provide a better understanding of the calcination mechanism.
Materials and methods
Materials
2-Methacryloxyethyltrimethylammoniumchloride (MTC, Sigma-Aldrich GmbH, 75% soln. in water), ethanol abs. (Sigma-Aldrich GmbH, ≥ 99.8%), polyvinylpyrrolidone (PVP, Sigma-Aldrich GmbH, 40,000 g/mol), styrene (Sigma-Aldrich GmbH, > 99%), titanium butoxide (TBT, Sigma Aldrich GmbH, 97%) were used as received. Millipore water was taken from a Millipore Direct Q3UV unit (Merck Millipore). 2,2′-Azobis(2-methylpropionitril) (AIBN, Sigma-Aldrich GmbH) was recrystallized from ethanol before use.
Synthesis of PS particles
3 g PVP (40,000 g/mol) were dissolved in 10 mL ethanol via ultrasonication. The PVP solution, 46 mL of ethanol, 10 mL ultrapure water, half of the styrene (see Table 1), and 300 mg AIBN were added to a 250 mL three-necked flask equipped with a reflux condenser and a gas inlet. The solution was degassed while stirring with an egg-shaped stirring bar with a speed of 150 rpm. After 30 min, the mixture was slowly heated to the reaction temperature of 70 °C by turning the hot plate on. Fifty-six milliliters of ethanol, the second half of the styrene, and the MTC were premixed in an Erlenmeyer flask and added after 90 min. The reaction was carried out overnight stirring continuously with a speed of 150 rpm under a slight argon flow. The polymerization was stopped by exposing the dispersion to ambient air and filtrated using a 125 μm nylon filter sieve. The concentration of the particles was determined gravimetrically. For the calculation of the conversion of the particles, the concentration was divided by the theoretical concentration at 100% conversion.
Synthesis of TiO2 shells
The synthesis was performed at room temperature. 6.3 mL PS dispersion and 37 mL ethanol were added to an Erlenmeyer flask equipped with a septum. The dispersion was stirred at 350 rpm using a magnetic stirrer bar during the degassing and TBT addition steps. The dispersion was degassed for 10 min with argon. 0.8 mL TBT was mixed with 3.2 mL ethanol and added within 30 min using a syringe pump. After the addition, the dispersion was allowed to age for 24 h without stirring. This aging step is essential to obtain core-shell particles of sufficient mechanical robustness to allow for the final calcination procedure. Particles were washed three times with ethanol for purification.
Synthesis of hollow TiO2 particles
The particles were freeze-dried in an 80:20 vol% ethanol-water mixture. The PS core was removed by calcination in air. A modified temperature profile of Schroden et al. [25] was used. The samples were heated to 300 °C with a heating rate of 2 K/min, followed by an isothermal step of 2 h. The samples were then heated to 400 °C with a heating rate of 2 K/min, followed by an isothermal step of 12 h. Finally, the samples were cooled down to room temperature for 5 h.
Characterization methods
Scanning electron microscopy (SEM) and scanning transmission electron microscopy (STEM) were performed using a Zeiss Ultraplus instrument using acceleration voltages of 3 kV or 10 kV. InLens, Everhard-Thornley, and STEM detector were used. Core-shell particles were calcined directly on a silicon wafer or SiO2 TEM grid (Plano GmbH).
The diameter of the PS template particles was evaluated using the MATLAB circle detection function (see S1). The search parameters were optimized manually.
Zeta potential was measured using Zetasizer Nano-ZS (Malvern Panalytical). Three measurements consisting of 10–100 runs were performed. The particles were diluted in ethanol without further purification. No additional substances were added to adjust the pH and background salt concentration.
Transmission electron microscopy (TEM) measurements were performed with a JEOL JEM-2200FS field emission energy filtering transmission electron microscope (FE-EFTEM) operated at an acceleration voltage of 200 kV. Zero-loss filtered micrographs (∆E ~ 0 eV) were recorded with a bottom-mounted CMOS camera system (OneView, Gatan) and processed with DM 3.3 image processing software (Gatan). Tilt series and tomography reconstructions were performed with SerialEM and IMOD software packages, supporting the entire tomography workflow, from data acquisition to image processing and modeling. The software was developed by David Mastronarde at the Boulder Laboratory for 3D Electron Microscopy (Boulder, Colorado, USA). Videos of the tilt-series of hollow TiO2 particles were exported from ImageJ distribution Fiji [46].
Small-angle X-ray scattering (SAXS) measurements were performed on freeze-dried samples in 1 mm glass capillaries (Hilgenberg, code 4007610, Germany) at room temperature. The measurements were performed in a transmission geometry using a Double Ganesha AIR system (SAXSLAB). A rotating copper anode (MicroMax 007HF, Rigaku Corporation) is the X-ray source of this system. Data was recorded using a position-sensitive detector (PILATUS 300 K, Dectris). Different detector positions were used to cover scattering vectors q between 0.0024 and 0.2 Å−1. The radially averaged data were normalized to the incident beam and sample thickness. Calculations were done using the software SASFIT (version 0.94.1, Kohlbrecher and Bressler) [47] or SasView (version 4.2) [48] or Scatter (version 2.5) [49].
X-ray powder diffraction patterns for the core-shell and hollow spheres were recorded in Bragg-Brentano-geometry on an Empyrean diffractometer (PANalytical B.V.; the Netherlands) using Cu-Kα radiation (λ = 1.54187 Å).
Simultaneous thermal analysis (STA) measurements were performed on a STA 449 F3 Jupiter (Netzsch) equipped with a Bruker Alpha II IR spectrometer using the same temperature ramp that was used for calcination. A DSC/TG OctoS sample holder and PtRh20 crucibles with lids were used. An airflow of 50 mL/min was adjusted for the measurement. Differential scanning calorimetry (DSC) measurements show an increase in the baseline at the heating step from 400 to 700 °C, which is caused by the baseline calibration. IR measurements were divided by a reference measurement that was taken before the sample measurement started. Due to fluctuations of the baseline over the measurement time, this led to transmission values above 100% for water bands (3750 cm−1 and 1500 cm−1) and CO2 bands (2250 cm−1). Furthermore, a rubber band baseline correction was performed to cancel out an overall intensity shift that was caused by the increasing temperature of the measured gas.
Results and discussion
Synthesis of polystyrene template particles
An overview on the explicit particle recipes is shown in Table 1. To control the particle size, the amount of styrene was increased from 6 to 22 mL. As can be seen in Fig. 2a, the particle diameter can be adjusted linearly with the amount of added styrene. The amount of initiator 2,2′-Azobis(2-methylpropionitril) (AIBN) does not influence the final particle size. This can be inferred from particles A–E with 0.3 g of AIBN, and particles F–J with 0.15 g AIBN. Further, we find no influence of the initiator concentration on the conversion of the synthesis within this range. The overall conversion was determined to be 80 to 90% for all syntheses (see Table 1).

Diameter of particles from dispersion polymerization dependent on the amount of styrene and the amount of initiator AIBN (a). Corresponding SEM images of the PS particles (b) top row with a high and bottom row with a low initiator concentration. The styrene volume increases from left to right
Electrostatic stabilization is introduced by the addition of the comonomer 2-Methacryloxyethyltrimethylammoniumchloride (MTC). This introduces a positive charge to the particle surface. For particles A–J, we kept the molar ratio between monomer and comonomer constant with a ratio of 0.8 mol%. This ratio results in a zeta potential of about + 40 mV for all particles. A ratio between monomer and comonomer in the range of 0.8 mol% (particles A–J) up to 1.2 mol% (particles Y) leads to well-functionalized, stable colloids. This is demonstrated by the synthesis of particles X–Z with different amounts of MTC, while styrene and AIBN concentration were kept constant (Fig. 3). Particle X was fabricated without MTC, resulting in a zeta potential ~ 0 mV. Nevertheless, owing to the use of PVP as a steric stabilizer, the dispersion is still stable. Without MTC, the particles exhibit a very smooth surface. Increasing the amount of MTC to 170 μl lead to a rougher surface, while the particle shape remained spherical. Further increasing the amount of MTC to 300 μl lead to aggregated and deformed particles. The particle aggregation is accompanied by a reduction in the overall conversion. The particle diameter decreased by 200 nm from particles X to Y. A potential reason for this deviation from the expected particle diameter is the better solubility of the PS oligomers due to the copolymerization with MTC. This can reduce the tendency for newly formed oligomers to precipitate onto the existing nuclei. As a consequence, the particle growth is less compared to the comonomer free synthesis. Furthermore, newly formed, small nuclei may aggregate on larger particles, resulting in an increasing particle roughness. Overall, using dispersion polymerization, it is possible to prepare polymer particles with standard deviations that are less or equal 5% of the diameter and are, therefore, highly monodisperse (see SI Fig. 2). It provides access to a complementary size range with particle sizes around 1 μm. We want to stress the simplicity of these recipes, where all chemicals, except for AIBN were used without any additional purification.

SEM and TEM images of particles X–Z, the corresponding PS@TiO2 core-shell particles, and hollow TiO2 particles, as well as TEM tomography reconstructions of parts of a slice of the hollow particles
Synthesis of TiO2 shells
The PS particles were used as seed templates without purification – not even centrifugation after the dispersion polymerization was employed. This procedure is similar to the scalable synthesis of PS@SiO2 core-shell and SiO2 hollow spheres [50], which provided access to gram-scale amounts of hollow silica spheres. An ethanolic solution (V = 4 mL) of the precursor titanium butoxide (TBT) was added with a concentration of 0.6 molL−1 to the ethanolic particle dispersion using a syringe pump within 30 min.
We first investigated the influence of the template particle surface functionalization on the TiO2 immobilization and shell formation.
Figure 3 demonstrates the necessity to adjust the cationic surface functionalization. Without the addition of MTC granular TiO2 nuclei are immobilized on the polymer surface. For both cases of added MTC (particles Y and Z, respectively) an increasingly smooth shell was observed. The granular appearance of the TiO2 shell is already apparent in the amorphous shell directly after the TiO2 condensation. This can be inferred from the SEM images of the core-shell structures (Fig. 3, second column), where white speckles cover the previously smooth surface. The presence of MTC at the particle surface apparently influences the nucleation and growth mechanism, which we assign to the altered electrostatic environment. Removing the template core by calcination preserves this granularity, which is shown in TEM and TEM tomography images (Fig. 3 right panels). The spherical shape of the hollow sphere is also preserved, which is apparent from the TEM tilt series (see SI gif files). The highest amount of MTC resulted in the most compact and least granular shells. Nevertheless, the TiO2 coated structures were clustered due to the already clustered particles in the PS seed dispersion. Consequently, a balanced adjustment of the particle surface functionality via MTC is crucial for the colloidal stability and successful coating step.
Calcination process
Calcination is the final step to obtain hollow TiO2 particles (Fig. 3 right panel). We want to stress that the core-shell particles can only be transformed into hollow spheres successfully after aging the core-shell dispersion for 24 h prior to purification. This aging process apparently improves the formation of a pre-condensed TiO2 network and proper covalent connectivity among the granular nuclei in the shell. The shell resilience to the thermal decomposition process is improved. Furthermore, the calcination process itself is very important. We used an adapted temperature profile of Schroden et al. [25], where the template removal is achieved in a two-step process. The first calcination step is undertaken just at the onset of PS decomposition at a relatively low temperature (300 °C). The complete degradation of the PS is then achieved by a second step at 400 °C. To gain a deeper understanding of the calcination process, combined TGA, DSC, and IR measurements have been performed in air and nitrogen (see Fig. 4 a–d and Fig. S4).

Combined STA (a), DSC (b), and IR measurements to investigate the calcination process of pure PS particles (c) and PS@TiO2 particles (d). Results of SAXS measurements of PS, PS@TiO2, and TiO2 hollow spheres (e) and XRD measurements of the PS@TiO2 and hollow TiO2 spheres (f). For all measurements, particles Y have been used
The pure template particles (polystyrene) show three prominent steps. These are directly related to the applied temperature profile. The degradation starts at the end of the first heating ramp (2 K/min) (1). Already 80% of polystyrene decompose in the form of CO2 during the isothermal conditions (300 °C), which took 2 h. The PS decomposition is strongly accelerated during the second heat ramp (2 K/min) to 400 °C (2). Within 300 mins another 13% of PS are decomposed to CO2, whereas the residues remain stable in air during the 5 h isothermal heating step at 400 °C. The last 5% of material is fully decomposed during the last heating ramp up to 700 °C (3). The IR spectra (Fig. 4c) only indicated CO2 as the decomposition product. Single IR spectra of the three steps can be found in the supporting information. Each of the decomposition steps was accompanied by an exothermal event in the DSC signal. This indicates the oxidative decomposition of the polystyrene backbone [51, 52].
The calcination of the core-shell particles shows a couple of interesting deviations from the pure PS decomposition. The degradation starts at the same time/temperature as the pure polystyrene particles (1). Since the IR spectra show exclusively CO2 bands (see Fig. 4d), it is reasonable to assume that only the polymer decomposed at this point. The PS decomposition, however, is significantly slower compared to the neat seed particles. This could be caused by the limited mass transport to the PS core owing to the presence of the TiO2 shell. Only 40% mass has been lost by the end of the first isothermal annealing step at 300 °C. The DSC signal reveals a second event occurring during the isothermal part at 300 °C (2). Xie et al. saw a similar exothermic peak in their differential thermal analysis measurements on TiO2 particles from different phases at 280 °C [53]. They found that this peak is due to the loss of water absorbed at the TiO2 particle surface. Therefore, the peak could be linked to a condensation process in the TiO2 shell and the release of the enclosed water molecules. However, we cannot unambiguously determine the onset of water loss in the IR spectra because the amount of released water is very low and cannot be separated from the background water bands at 3750 cm−1 and 1500 cm−1. The second heating ramp lead to a third prominent step at 400 °C (3). In this case, the degradation product is also CO2. In contrast to the pure PS particles, the last heating step to 700 °C results in a very small mass loss of only 2%. Since the corresponding IR data show no trace of CO2, all PS must already be decomposed at the end of the second isothermal step, and a further condensation reaction of the TiO2 is assumed. Calculations show that 34% material should be left over, which agrees well with the experimental data of 33%. We conclude that the TiO2 shell aids the decomposition reaction of the polymer even though the mass transfer is reduced. In our case, it is not possible to see the transformation of the amorphous to the anatase phase in the DSC curves. Xi et al. and Li et al. saw this event happening at temperatures above 400 °C [53, 54]. Therefore, the effect is likely to be superimposed by the exothermic degradation peak of PS.
These results show the importance of the right temperature profile for the calcination process. The overall particle shrinkage can be estimated by the mass loss of the PS particle at the first heating step. The mass loss translates into a volume shrinkage of the template particle. Assuming an isotropic shrinkage of the template particle, the diameter of the shrunk particle can be recalculated. The shrunk particle size then corresponds to the final hollow-core diameter. Starting with an initial particle with a diameter of 621 nm and considering a mass loss of 40%, this would result in a shrunk particle diameter of 520 nm. This is in reasonable agreement with the experimental data, where the hollow core has a size of ~ 500 nm. Calcinations in inert atmosphere, where the template particle is intact much longer, show less shrinkage of the hollow spheres (see SI Fig. 4). This observation strengthens the interpretation that the second peak in the DSC measurements is connected to the formation of the final and stable TiO2 structure, and therefore to the shell condensation process. Calcination profiles, which omit the mild calcination at 300 °C or which feature too fast heating ramps, sacrifice the shell integrity and result in collapsed structures (see SI Fig. 5).

SEM images of PS@TiO2 core-shell particles of different sizes (A–E) and corresponding TiO2 hollow spheres. The last row shows STEM images of single TiO2 hollow spheres
The structures of the polystyrene, core-shell, and hollow particles Y (see Fig. 4e) were further characterized by SAXS and SEM/TEM. The measured SAXS data exhibit only weak features, which prevent a thorough fitting analysis. Furthermore, PS seed particles are too large to identify the radius in the experimentally reachable q range. Thus, based on the TEM result, we calculated the form factor of homogeneous spheres with a diameter of 621 nm and compared it to our experimental data – both agree well. The model has a Gaussian size distribution with a standard derivation of 10%, which is slightly higher compared to the SEM images due to instrumental smearing effects. The measurement of the PS@TiO2 core-shell particles is shown in Fig. 4e (green symbols). The scattering of PS@SiO2 can be described by the model of a homogeneous core-homogeneous shell [50]. The sharp and well defined boundary between core and (monodisperse) shell leads to significant oscillations in the scattering data. In contrast to PS@SiO2 the shell of PS@TiO2 is less dense and highly particulate (see Fig. 3). As consequence of such a fractal-like shell morphology, the corresponding form factor scattering miss such pronounced oscillations. The main features are a q−4 scaling at intermediate q (ca. 0.008–0.05 Å−1) and a q−2 power law for q > 0.057 Å−1. A very weak oscillation around 0.017 Å−1 hints toward the expected dimension of the shell thickness of about 30–40 nm before calcination. The q−2 power law for q > 0.057 Å−1 is indicative of strong scatters with a mainly 2D-structure. This is in contrast to scattering patterns of similar (silica-based) core-shell systems [55, 56]. These systems exhibit a pronounced form factor and no q−2 scaling law at high q. The deviation of our system from these findings can be understood by the scattering contrast situation (PS ≈ 9.51∙10−6 Å−2, TiO2 ≈ 31.8∙10−6 Å−2, no solvent) and the lower bending curvature due to the large template particle diameter. At high q, the q−2 term seems to simply add up to the scattering of a pure PS sphere (q−4) underlining the fact that the shell is particulate. For comparison, the scattering of homogeneous core-homogeneous shell spheres is given (d = 621 nm, 10% Gaussian distribution and dshell = 35 nm, 20% Gaussian distribution; green dotted line PS@TiO2). During the calcination process, the amorphous TiO2 shells undergo a transformation into anatase phase (Fig. 4f). During this process the overall size of the particle shrinks. The experimental SAXS scattering pattern of these hollow spheres exhibits a minimum at q ≈ 0.026 Å−1. The corresponding correlation length of about 24 nm agrees well with the thickness of the TiO2 shell obtained from TEM analysis. The q−4 behavior at intermediate and high q reflects the contrast situation for hollow spheres. This is corroborated by the calculation of homogenous hollow spheres (blue dotted line; dinner = 621 nm, 10% Gaussian with zero contrast, dshell = 24 nm, 25% Gaussian with contrast 31.8∙10−6 Å−2). The q−2 power law at low q is attributed to the particulate shell, since rough surfaces can be considered as fractals.
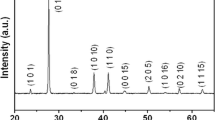
As stated above, the TiO2 shell undergoes a phase transition during the calcination procedure. This can be seen in X-ray diffraction measurements in Fig. 4f. The core-shell particles (green line) do not show any features except for an amorphous halo. This pattern is caused by the TiO2 and the amorphous polymer core. After calcination, distinct peaks are visible (blue line). The inset shows the normalized data that agree very well with the expected diffraction pattern of anatase.
Size series of TiO2 hollow particles
We now want to highlight the robustness of our synthetic protocol. Therefore, TiO2 shells have been synthesized on PS particles A–E. Setting the MTC/Styrene ratio to 0.8mol% during the seed synthesis and the amount of TBT to 5.5∙10−4 to 6.5∙10−4 mol/m2 particle surface during the coating step, it is possible to fabricate TiO2 core-shell particles without secondary nucleation or particle clustering (Fig. 5). When using 800 μL of TBT, shell thicknesses between 45 and 60 nm could be achieved. Stable and smooth TiO2 shells could be immobilized on the PS seed particles. Also, the template particles themselves remain colloidally stable and are coated as individual objects. One may expect that the shell thickness decreases with increasing template particle diameter when employing the same amount of TBT precursor. This, however, would only be true, if the same particle concentration was used. Owing to our scalable process, we directly use the as-synthesized polymer dispersion, where both, particle diameter and particle concentration vary with the initial monomer concentration. Both parameters cancel each other out leading to comparable shell thicknesses for each batch.
Calcination of the particles in air leads to an isotropic shrinkage up to 20% compared to the core-shell size (see STEM, Fig. 5). For particles > 700 nm we find no systematic variation of the degree of shrinkage to the particle size. As a consequence, the TiO2 hollow spheres B, C, and D all have the same particle diameter of ~ 900 nm; particle E is slightly larger with 1025 nm. Yet, despite the considerable shrinkage, the particles retain their spherical shape and shrink in an isotropic fashion. This is even more remarkable as the ratio t/D between shell thickness (t) and particle diameter (D) is very small. t/D ranges from 4% for the smallest hollow spheres to 2% for the largest one. With decreasing t/R ratio the mechanical stability of the TiO2 hollow spheres decreases, which is also known for their silica shell counterparts [57]. We, consequently, observed an increased portion of fractured or buckled hollow spheres from particles C to E.
Conclusions
Our contribution addresses several important aspects in the field of templated hollow sphere synthesis. We firstly introduced dispersion polymerization as a suitable alternative to established emulsion polymerization techniques for synthesizing template particles in the 500 to 1300 nm size regime. The template particles can be functionalized with comonomers to control the particle surface charge. These template beads can be used without additional purification steps for the synthesis of homogeneous TiO2 shells. This strategy allows for a scalable synthesis of well-coated TiO2 core-shell particles. However, it is important to control the amount of comonomer used during the dispersion polymerization. This affects the stability of the colloidal particles and the granularity of the TiO2 shell. We thoroughly investigated the calcination procedure yielding the hollow particles. The usage of an isothermal step at the onset of polystyrene decomposition is crucial for retaining the hollow particle shape. Our presented method could be applied to a range of template particles with different sizes. Overall, this facile, reproducible and scalable method, creates well-defined TiO2 core-shell or hollow particles that can be used in applications, where the properties of TiO2 are beneficial.
References
Anastas P, Eghbali N (2010) Green chemistry: principles and practice. Chem Soc Rev 39(1):301–312. https://doi.org/10.1039/B918763B
Sheldon RA, Arends I, Hanefeld U (2007) Green chemistry and catalysis. Wiley. https://doi.org/10.1002/9783527611003
Anastas PT (2007) Introduction: green chemistry. Chem Rev 107(6):2167–2168. https://doi.org/10.1021/cr0783784
Lu Y, Hoffmann M, Yelamanchili RS, Terrenoire A, Schrinner M, Drechsler M, Möller MW, Breu J, Ballauff M (2009) Well-defined crystalline TiO2 nanoparticles generated and immobilized on a colloidal nanoreactor. Macromol Chem Phys 210(5):377–386. https://doi.org/10.1002/macp.200800608
Kisch H (2015) Semiconductor photocatalysis: principles and applications. Wiley. https://doi.org/10.1002/9783527673315
O'Regan B, Grätzel M (1991) A low-cost, high-efficiency solar cell based on dye-sensitized colloidal TiO2 films. Nature 353(6346):737–740. https://doi.org/10.1038/353737a0
Varghese OK, Paulose M, Grimes CA (2009) Long vertically aligned titania nanotubes on transparent conducting oxide for highly efficient solar cells. Nature Nanotech 4(9):592. https://doi.org/10.1038/nnano.2009.226
Phani G, Tulloch G, Vittorio D, Skryabin I (2001) Titania solar cells: new photovoltaic technology. Renew Energy 22(1):303–309. https://doi.org/10.1016/S0960-1481(00)00059-8
Fu LJ, Zhang T, Cao Q, Zhang HP, Wu YP (2007) Preparation and characterization of three-dimensionally ordered mesoporous titania microparticles as anode material for lithium ion battery. Electrochem Commun 9(8):2140–2144. https://doi.org/10.1016/j.elecom.2007.06.009
Ortiz GF, Hanzu I, Djenizian T, Lavela P, Tirado JL, Knauth P (2009) Alternative Li-ion battery electrode based on self-organized titania nanotubes. Chem Mater 21(1):63–67. https://doi.org/10.1021/cm801670u
Kim K-T, Ali G, Chung KY, Yoon CS, Yashiro H, Sun Y-K, Lu J, Amine K, Myung S-T (2014) Anatase titania nanorods as an intercalation anode material for rechargeable sodium batteries. Nano Lett 14(2):416–422. https://doi.org/10.1021/nl402747x
Chen X, Mao SS (2007) Titanium dioxide nanomaterials: synthesis, properties, modifications, and applications. Chem Rev 107(7):2891–2959. https://doi.org/10.1021/cr0500535
Reyes-Coronado D, Rodriguez-Gattorno G, Espinosa-Pesqueira ME, Cab C, de Coss R, Oskam G (2008) Phase-pure TiO(2) nanoparticles: anatase, brookite and rutile. Nanotech 19(14):145605. https://doi.org/10.1088/0957-4484/19/14/145605
Zaban A, Aruna ST, Tirosh S, Gregg BA, Mastai Y (2000) The effect of the preparation condition of TiO2 colloids on their surface structures. J Phys Chem B 104(17):4130–4133. https://doi.org/10.1021/jp993198m
Banfield JF, Bischoff BL, Anderson MA (1993) TiO2 accessory minerals: coarsening, and transformation kinetics in pure and doped synthetic nanocrystalline materials. Chem Geol 110(1):211–231. https://doi.org/10.1016/0009-2541(93)90255-H
Yanagisawa K, Ovenstone J (1999) Crystallization of anatase from amorphous titania using the hydrothermal technique: effects of starting material and temperature. J Phys Chem B 103(37):7781–7787. https://doi.org/10.1021/jp990521c
Yin H, Wada Y, Kitamura T, Kambe S, Murasawa S, Mori H, Sakata T, Yanagida S (2001) Hydrothermal synthesis of nanosized anatase and rutile TiO2 using amorphous phase TiO2. J Mater Chem 11(6):1694–1703. https://doi.org/10.1039/B008974P
Shankar K, Bandara J, Paulose M, Wietasch H, Varghese OK, Mor GK, LaTempa TJ, Thelakkat M, Grimes CA (2008) Highly efficient solar cells using TiO2 nanotube arrays sensitized with a donor-antenna dye. Nano Lett 8(6):1654–1659. https://doi.org/10.1021/nl080421v
Paulose M, Shankar K, Varghese OK, Mor GK, Hardin B, Grimes CA (2006) Backside illuminated dye-sensitized solar cells based on titania nanotube array electrodes. Nanotech 17(5):1446–1448. https://doi.org/10.1088/0957-4484/17/5/046
Zukalová M, Zukal A, Kavan L, Nazeeruddin MK, Liska P, Grätzel M (2005) Organized mesoporous TiO2 films exhibiting greatly enhanced performance in dye-sensitized solar cells. Nano Lett 5(9):1789–1792. https://doi.org/10.1021/nl051401l
Somani PR, Dionigi C, Murgia M, Palles D, Nozar P, Ruani G (2005) Solid-state dye PV cells using inverse opal TiO2 films. Sol Energy Mater Sol Cells 87(1):513–519. https://doi.org/10.1016/j.solmat.2004.07.037
Adachi M, Murata Y, Okada I, Yoshikawa S (2003) Formation of titania nanotubes and applications for dye-sensitized solar cells. J Electrochem Soc 150(8):G488–G493. https://doi.org/10.1149/1.1589763
Ohsaki Y, Masaki N, Kitamura T, Wada Y, Okamoto T, Sekino T, Niihara K, Yanagida S (2005) Dye-sensitized TiO2 nanotube solar cells: fabrication and electronic characterization. PCCP 7(24):4157–4163. https://doi.org/10.1039/B511016E
Lim SH, Luo J, Zhong Z, Ji W, Lin J (2005) Room-temperature hydrogen uptake by TiO2 nanotubes. Inorg Chem 44(12):4124–4126. https://doi.org/10.1021/ic0501723
Schroden RC, Al-Daous M, Blanford CF, Stein A (2002) Optical properties of inverse opal photonic crystals. Chem Mater 14(8):3305–3315. https://doi.org/10.1021/cm020100z
Retsch M, Jonas U (2013) Hierarchically structured, double-periodic inverse composite opals. Adv Funct Mater 23(43):5381–5389. https://doi.org/10.1002/adfm.201300803
Stein A, Wilson BE, Rudisill SG (2013) Design and functionality of colloidal-crystal-templated materials--chemical applications of inverse opals. Chem Soc Rev 42(7):2763–2803. https://doi.org/10.1039/c2cs35317b
Noman MT, Ashraf MA, Ali A (2019) Synthesis and applications of nano-TiO2: a review. Environ Sci Pollut Res 26(4):3262–3291. https://doi.org/10.1007/s11356-018-3884-z
Rabenau A (1985) The role of hydrothermal synthesis in preparative chemistry. Angew Chem Int Ed Eng 24(12):1026–1040. https://doi.org/10.1002/anie.198510261
Cho C-Y, Moon JH (2012) Hierarchical twin-scale inverse opal TiO2 electrodes for dye-sensitized solar cells. Langmuir 28(25):9372–9377. https://doi.org/10.1021/la3014656
Wang X, Feng J, Bai Y, Zhang Q, Yin Y (2016) Synthesis, properties, and applications of hollow micro-/nanostructures. Chem Rev 116(18):10983–11060. https://doi.org/10.1021/acs.chemrev.5b00731
Chen M, Ye C, Zhou S, Wu L (2013) Recent advances in applications and performance of inorganic hollow spheres in devices. Adv Mater 25(37):5343–5351. https://doi.org/10.1002/adma.201301911
Zhou L, Zhuang Z, Zhao H, Lin M, Zhao D, Mai L (2017) Intricate hollow structures: controlled synthesis and applications in energy storage and conversion. Adv Mater 29(20):1602914. https://doi.org/10.1002/adma.201602914
Arshady R (1992) Suspension, emulsion, and dispersion polymerization: a methodological survey. Colloid Polym Sci 270(8):717–732. https://doi.org/10.1007/bf00776142
Barrett KEJ (1973) Dispersion polymerisation in organic media. Br Polym J 5(4):259–271. https://doi.org/10.1002/pi.4980050403
Cheng X, Chen M, Wu L, Gu G (2006) Novel and facile method for the preparation of monodispersed titania hollow spheres. Langmuir 22(8):3858–3863. https://doi.org/10.1021/la0534221
Kawaguchi S, Ito K (2005) Advances in polymer science. polymer particles, vol 175. Advances in polymer science, pp 299–328. https://doi.org/10.1007/b100118
Paine AJ, Luymes W, McNulty J (1990) Dispersion polymerization of styrene in polar solvents. 6. Influence of reaction parameters on particle size and molecular weight in poly(N-vinylpyrrolidone)-stabilized reactions. Macromolecules 23(12):3104–3109. https://doi.org/10.1021/ma00214a012
Barlier V, Bounor-Legaré V, Boiteux G, Davenas J, Léonard D (2008) Hydrolysis–condensation reactions of titanium alkoxides in thin films: a study of the steric hindrance effect by X-ray photoelectron spectroscopy. Appl Surf Sci 254(17):5408–5412. https://doi.org/10.1016/j.apsusc.2008.02.076
Imhof A (2001) Preparation and characterization of titania-coated polystyrene spheres and hollow titania shells. Langmuir 17(12):3579–3585. https://doi.org/10.1021/la001604j
Agrawal M, Pich A, Zafeiropoulos NE, Stamm M (2008) Fabrication of hollow titania microspheres with tailored shell thickness. Colloid Polym Sci 286(5):593–601. https://doi.org/10.1007/s00396-007-1833-3
Taniguchi T, Murakami F, Kasuya M, Kojima T, Kohri M, Saito K, Nakahira T (2013) Preparation of titania hollow particles with independently controlled void size and shell thickness by catalytic templating core–shell polymer particles. Colloid Polym Sci 291(1):215–222. https://doi.org/10.1007/s00396-012-2658-2
Wang P, Chen D, Tang F-Q (2006) Preparation of Titania-coated polystyrene particles in mixed solvents by Ammonia catalysis. Langmuir 22(10):4832–4835. https://doi.org/10.1021/la060112p
Yelamanchili RS, Lu Y, Lunkenbein T, Miyajima N, Yan L-T, Ballauff M, Breu J (2009) Shaping colloidal rutile into thermally stable and porous Mesoscopic Titania balls. Small 5(11):1326–1333. https://doi.org/10.1002/smll.200801298
Ruckdeschel P (2017) Transport phenomena in silica hollow spheres and hybrid materials. University of Bayreuth, Bayreuth
Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez J-Y, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A (2012) Fiji: an open-source platform for biological-image analysis. Nat Methods 9(7):676–682. https://doi.org/10.1038/nmeth.2019
Bressler I, Kohlbrecher J, Thunemann AF (2015) SASfit: a tool for small-angle scattering data analysis using a library of analytical expressions. J Appl Crystallogr 48(5):1587–1598. https://doi.org/10.1107/S1600576715016544
Doucet M, Cho, Jae Hie, Alina, Gervaise, Bakker, Jurrian, Bouwman, Wim, Butler, Paul, Washington, Adam (2019) SasView version 4.2.2. Zenodo
Förster S, Fischer S, Zielske K, Schellbach C, Sztucki M, Lindner P, Perlich J (2011) Calculation of scattering-patterns of ordered nano- and mesoscale materials. Adv Colloid Interf Sci 163(1):53–83. https://doi.org/10.1016/j.cis.2010.12.003
Ruckdeschel P, Dulle M, Honold T, Förster S, Karg M, Retsch M (2016) Monodisperse hollow silica spheres: an in-depth scattering analysis. Nano Res 9(5):1366–1376. https://doi.org/10.1007/s12274-016-1032-y
Kannan P, Biernacki JJ, Visco DP, Lambert W (2009) Kinetics of thermal decomposition of expandable polystyrene in different gaseous environments. J Anal Appl Pyrolysis 84(2):139–144. https://doi.org/10.1016/j.jaap.2009.01.003
Malhotra SL, Hesse J, Blanchard L-P (1975) Thermal decomposition of polystyrene. Polymer 16(2):81–93. https://doi.org/10.1016/0032-3861(75)90133-0
Xie H, Zhang Q, Xi T, Wang J, Liu Y (2002) Thermal analysis on nanosized TiO2 prepared by hydrolysis. Thermochim Acta 381(1):45–48. https://doi.org/10.1016/S0040-6031(01)00642-6
Li D, Chen S, Wang D, Li Y, Shao W, Long Y, Liu Z, Ringer SP (2010) Thermo-analysis of nanocrystalline TiO2 ceramics during the whole sintering process using differential scanning calorimetry. Ceram Int 36(2):827–829. https://doi.org/10.1016/j.ceramint.2009.10.004
Balmer JA, Mykhaylyk OO, Schmid A, Armes SP, Fairclough JPA, Ryan AJ (2011) Characterization of polymer-silica nanocomposite particles with core–shell morphologies using Monte Carlo simulations and small angle X-ray scattering. Langmuir 27(13):8075–8089. https://doi.org/10.1021/la201319h
Fielding LA, Mykhaylyk OO, Schmid A, Pontoni D, Armes SP, Fowler PW (2014) Visible Mie scattering from hollow silica particles with particulate shells. Chem Mater 26(2):1270–1277. https://doi.org/10.1021/cm4039347
Yin J, Retsch M, Lee J-H, Thomas EL, Boyce MC (2011) Mechanics of nanoindentation on a monolayer of colloidal hollow nanoparticles. Langmuir 27(17):10492–10500. https://doi.org/10.1021/la2018117
Acknowledgments
We thank Stefan Rettinger for the help with STA experiments and the Bavarian Polymer Institute, especially Martina Heider, for helping with SEM and STEM measurements. Prof. André Gröschel is acknowledged for the help regarding the TEM reconstruction.
Funding
Open Access funding provided by Projekt DEAL. This project was funded by the German Research Foundation (DFG RE3550/2–1). Additional support was provided by ERC Starting Grant VISIRday under Grant No. 714968. This work benefited from the use of the SasView application, originally developed under NSF Award DMR-0520547. SasView also contains code developed with funding from EU Horizon 2020 programme under the SINE2020 project Grant No. 654000.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lechner, A.M., Feller, T., Song, Q. et al. Scalable synthesis of smooth PS@TiO2 core-shell and TiO2 hollow spheres in the (sub) micron size range: understanding synthesis and calcination parameters. Colloid Polym Sci 298, 867–878 (2020). https://doi.org/10.1007/s00396-020-04626-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00396-020-04626-3