
Abstract
This article introduces tri-cationic hemicyanine dye employed as a visible-light photoinitiator of acrylic monomers polymerization. This dye, in combination with borate anions, was found to be a very effective photoinitiating system. The kinetics of trimethylolpropane triacrylate polymerization was studied by a microcalorimetric method. The photoredox pair concentration, the co-initiator structure as well as the light intensity strongly affected the progress of the polymerization, leading, for example, to an increase in the polymerization rate and quantum yield of the process. The efficiency of these photoinitiators was discussed on the basis of the free energy change for electron transfer from a borate anion to an excited hemicyanine dye cation. The ∆G el values were estimated for photoredox pairs containing a series of phenyltrialkylborate anions and one styrylpyridinium dye cation. The relationship between the rate of polymerization and the free energy of activation for electron transfer reaction gives the dependence predicted by the classical theory of electron transfer. The photoinitiating abilities of the selected novel photoredox pairs (BPB61, BPB7, BPB8, and BPB9) are comparable with the photoinitiating efficiency of commercially available photoinitiators.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Radical polymerization is often the preferred mechanism for forming polymers and most commercial polymer materials involve radical chemistry at some stage of their production cycle. The radical reactions can be initiated by various methods. They can be divided into the broad areas of thermolyses, photolyses, and electron transfer reactions [1].
Photoinitiators are compounds that decay on irradiation with UV or visible light. They are used not only in industrial processes involving coatings and surface polymerizations [1] but also find applications involving accurate kinetic measurements.
The main advantage of photoinitiator used in polymerizing systems is the possibility to define exact start- and endpoints of the polymerization process via the duration of the irradiation period. In addition, the rate of (most) photoinitiator decomposition is almost independent of the reaction temperature, but depends strongly on the light intensity. An ideal photoinitiator for a specific polymerization may be defined via the following criteria:
-
1.
The photoinitiator should decompose on irradiation with the (UV) light source. For instance, an absorption should coincide with the radiation wavelength. The monomer(s) used in the specific polymerization process should not absorb light at the selected wavelength.
-
2.
The efficiency of the initiator should be high, preferably close to 1, which says that all radicals generated start a growing chain.
-
3.
At best, there should be only one type of free-radical species that is formed on laser irradiation [1].
According to the mechanism by which initiating radicals are formed, photoinitiators are generally divided into two classes:
-
type I photoinitiators, which undergo a unimolecular bond cleavage upon irradiation to yield free-radicals; and
-
type II photoinitiators, which undergo a bimolecular reaction where the excited state of the photoinitiator interacts with a second molecule (a co-initiator) to generate free radicals [1–3].
Within the type I initiator class, there are several structural variations. The most widely used class are photoinitiators containing a benzoyl group as the effective initiating moiety (acetophenone type).
Type II photoinitiators normally generate radicals by (1) abstracting hydrogen from the environment (e.g., the solvent, an ether or an alcohol) or (2) undergoing photoinduced electron and hydrogen transfer with a co-initiator resulting in radical ions that fragment to generate radicals. The most commonly employed co-initiators for aromatic ketones are tertiary amines, the ionization potential of which is thought to be of crucial importance for the electron transfer process along with steric factors. In the case of the system camphorquinone/amine, it has been shown that the rate of polymerization increases in the order primary < secondary < tertiary amine; the polymerizations are very slow for primary amines and amines lacking an α-hydrogen [2, 3].
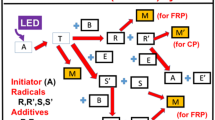
Another example of type II photoinitiators which generate primary radical via the electron transfer process are cyanine borates. Irradiation of the ion pair with visible light generates the excited singlet state of the cyanine, which can undergo three possible reactions: fluorescence, photoisomerization, or electron transfer to form the cyanine and boranyl radicals. The cyanine–boranyl radical pair may undergo back electron transfer to regenerate the substrate or undergo cleavage of a carbon–boron bond to form an alkyl radical and triphenylboron. The cyanine-alkyl radical pair formed in the latter process might: (1) undergo electron transfer to form cyanine cation and an alkyl anion; (2) couple to form an alkylated cyanine; (3) or to diffuse from the initial solvent cage, giving free radicals.[3]
Generally, in the case of a donor–acceptor photoinitiating system, the polymerization chain reaction starts as a result of photoinduced intermolecular electron transfer (PET), which is a nonclassical, endothermic energy-transfer process. Marcus [4] provided a simple approach allowing to predict the kinetics of the electron-transfer process, using thermodynamic and spectroscopic parameters characterizing both an electron donor and an electron acceptor. His theory assumes that bimolecular electron transfer occurs in three stages: (1) the formation of the precursor complex, (2) the electron transfer, and (3) the change in the organization of the solvent cage in which electron transfer primary products exist. It is commonly assumed that solvent reorganization will dominate electron transfer kinetically. On the basis of the thermodynamics of the electron exchange, it is possible quantitatively to predict a relationship between the rate of the free radical formation and the free energy of activation for the electron-transfer process. Essentially, one can test a possibility of the application of the Marcus theory for the prediction of the photoinitiated polymerization rate by (1) the use of one electron acceptor and a series of electron donors and (2) the use of a series of dyes and only one type of electron donor. The first method usually yields free radicals with different reactivities, [5, 6] and this might have an impact on the final rate of photoinitiated polymerization. The use of the photoredox pairs under the study represents the first method. Besides the thermodynamics of the electron-transfer process, one should take into account the fact that the photoinitiation ability of the donor–acceptor pairs depend on the nature of the reacting partners [2, 6].
In this paper, the synthesis and kinetic studies of photoinitiating systems that consist of tri-cationic styrylpyridinium dye cation [1,1′,1″-(benzene-1,3,5-triyltrimethanetriyl)-tris(N,N-dibutylaminostyrylpyridinium bromide)—BP], which act as light absorber, and different electron donors (borate anions) are described. The photoinitiators form the ground-state ion pairs.
Experimental
Materials
Monomer, 2-ethyl-2-(hydroxymethyl)-1,3-propanediol triacrylate (TMPTA), all chemicals and solvents were purchased from Aldrich Chemical Co. and were used without further purification. The tri-cationic styrylpyridinium phenyltrialkylborate salts applied as visible-light initiators were synthesized in my laboratory. Structures of the dye and borate salts are collected in Scheme 1. The 5,7-diiodo-3-butoxy-6-fluorone (DIBF) and titanocene (Ti(C6F5)2(C5H4)2; T) were applied as light absorber and N-phenylglycine was used as co-initiator for comparison, respectively. They were commercially available.
Techniques
Spectral measurements
The 1H NMR (200 MHz) spectra were recorded with the use of a Varian Gemini 200 NMR spectrometer (Palo Alto, CA, USA). Dimethylsulfoxide (DMSO-d6) was used as the solvent and tetramethylsilane as internal standard.
Absorption and emission spectra in N,N-dimethylformamide (DMF), acetonitrile (AcCN), and methanol (MeOH) were recorded at room temperature using a Shimadzu UV–vis Multispec-1501 spectrophotometer and a Hitachi F-4500 spectrofluorimeter, respectively.
The fluorescence lifetimes were measured using an Edinburgh Instruments FLS920P Spectrometers. The apparatus utilizes for the excitation a picosecond diode laser generating pulses of about 55 ps at 466 nm. Short laser pulses in combination with a fast microchannel plate photodetector and ultrafast electronics make a successful analysis of fluorescence decay signals with resolution in the range of single picoseconds possible.
The fluorescence quantum yield (ϕ dye) of the dye in organic solvents were determined with respect to Rhodamine B as the standard. The quantum yield of Rhodamine B in ethanol is 0.55 using excitation wavelength 530 nm [7]. The quantum yield was calculated using Eq. (1)
where:
ϕref is the fluorescence quantum yield of reference (Rhodamine B) in ethanol, A dye and A ref are the absorbances of the dye and reference samples at the excitation wavelengths (530 nm), I dye and I ref are the areas arbitrary units of the corrected fluorescence spectra for the dyes and reference samples, n dye and n ref are the refractive indices of the solvents used for the dyes and the reference, respectively.
Electrochemical measurements
The reduction potential of the dye and the oxidation potentials of the borate salts were measured by cyclic voltammetry using an Electroanalytical Cypress System Model CS-1090. The typical three-electrode setup was employed for electrochemical measurements. The electrolyte was 0.1 M tertrabutylammonium perchlorate, which was purged with argon prior to a measurement. Platinum 1-mm electrode was applied as working electrode and platinum and Ag/AgCl was used as auxiliary and reference electrodes, respectively.
Polymerization measurements
The kinetics of free radical polymerization were studied using a polymerization solution composed of 1 mL of 1-methyl-2-pyrrolidinone (MP) and 9 mL of TMPTA. The hemicyanine borate salts (photoinitiator) concentration was 1 × 10−3 M. A reference formulation contained hemicyanine bromide (dye without an electron donor) instead of hemicyanine borate salt (photoinitiator). The measurements were carried out at an ambient temperature and the polymerizing mixture was not deaerated before curing.
The kinetics of free radical polymerization were measured based on the measurements of the rate of the heat evolution during polymerization in thin film cured sample (0.035 ± 0.002 g). The measurements were performed by measuring photopolymerization exotherms using photo-DSC apparatus constructed on the basis of a TA Instruments DSC 2010 Differential Scanning Calorimeter. Irradiation of the polymerization mixture was carried out using the emission (line at 514 nm) of an argon ion laser Model Melles Griot 43 series with intensity of light of 100 mW/cm2. The light intensity was measured by a Coherent Model Fieldmaster power meter.
The rate of polymerization (R p) was calculated using the formula (2) where dH/dt is maximal heat flow during reaction and ΔH p theor is the theoretical enthalpy for complete conversion of acrylates’ double bonds. ΔH p theor for acrylic double bond is equal 78.2 kJ/mol [8].
The reaction heat liberated in the polymerization was directly proportional to the number of vinyl groups reacting in the system. By integrating the area under the exothermic peak, the conversion of the vinyl groups (C) or the extent of reaction could be determined according to Eq. 3.
where ΔH t is the reaction heat evolved at time t, M is the molar mass of the monomer, m is the mass of the sample, and n is the number of double bonds per monomer molecule.
The quantum yield of polymerization Φ p was defined as the number of polymerized double bonds per absorbed photon [9, 10].
Synthesis
Phenyltrialkylborate tetramethylammonium salts listed in Scheme 1 were prepared in my laboratory with modified methods described by Neckers et al. [11, 12]. The detailed synthetic procedures as well as the basis of analytical results and spectral evidences of the tetramethylammonium phenyltrialkylborates the readers can find in my previous paper [13].


Structures of the sensitizer and co-initiators
BP was synthesised by the reaction of 1,3,5-tris(bromomethyl)benzene with three equivalents of 4-picoline followed by Knoevenagel condensation with N,N-dimethylaminobenzaldehyde according to the procedure described in literature [7, 14].
1,1′,1″-(Benzene-1,3,5-triyltrimethanetriyl)-tris(4-methylpyridinium bromide)
A mixture of 1,3,5-tris(bromomethyl)benzene, (5.2 g, 14 mmol) and 4-picoline (4.2 g, 45 mmol) in EtOH (50 mL) was refluxed for 8 h. After cooling to 25 °C, the solvent was removed in vacuo giving a residue, which was washed with Et2O to afford the 4-picolinium salt as white solid: 8.5 g, m.p. 265–269 °C; 1H NMR (DMSO-d6, δ ppm) 2.622 (s, 9H, CH3), 5.803 (s, 6H, CH2), 7.656 (s, 3H, ArH), 8.015–8.044 (d, 6H, J = 5.8 Hz, ArH), 9.028–9.059 (d, 6H, J = 6.2 Hz, ArH).
1,1′,1″-(Benzene-1,3,5-triyltrimethanetriyl)-tris(N,N-dibutylaminostyrylpyridinium bromide)
The N,N-dibutylaminobenzaldehyde (1.05 g, 4.5 mmol) in EtOH (20 mL) and two to three drops of piperidine, as catalyst, were added to the salt (0.91 g, 1.4 mmol) and then this mixture was refluxed for 5 h. After cooling in an ice bath, the red solid was filtered and then crystallized from MeOH/CH3CN to afford (60 %) the desired dye BP, as red solid: 1.08 g, m.p. 196–198 °C; 1H NMR (DMSO-d6, δ ppm) 0.899 (t, 18H, CH3), 1.252 (m, 12H, CH2), 1.470 (m, 12H, CH2), 3.195 (m, 12H, CH2), 5.642 (s, 6H, CH2), 6.674-6.718 (d, 6H, J = 8.8 Hz, ArH), 7.066–7.145 (d, 3H, J = 15.8 Hz, ethylinic H), 7.511-7.553 (d, 6H, J = 8.4 Hz, ArH), 7.647 (s, 3H, ArH), 7.876-7.956 (d, 3H, J = 16 Hz, ethylinic H), 8.030-8.062 (d, 6H, J = 6.4 Hz, ArH), 8.749–8.782 (d, 6H, J = 6.6 Hz, ArH); Anal. calcd. for C72H93N6Br3: C 67.4 %, H 7.3 %, N 6.6 %, Br 18.7 %; found: C 66.94 %, H 7.58 %, N 6.32 %.
The ion exchange reaction was performed using the procedure described in literature [9].
A mixture of 0.15 mmol of the tricationic styrylpyridinium dye and 0.45 mmol of phenyltrialkylborate tetramethylammonium salt in 15 ml of an acetonitrile was boiled for 10 min. Then cold distilled water was added to the resulting hot solution until the turbidity. The mixture was stored in a dark place when the salt crystallized. The precipitate was filtered off and dried.
Results and discussion
Spectroscopic properties of BP dye
The absorption and fluorescence spectra of the tri-cationic hemicyanine dye (BP) were measured in three organic solvents. Its spectral data are compiled in Table 1.
The UV–vis spectroscopic measurements of tri-cationic hemicyanine dye BP (Fig. 1) show high-intensity broad absorption band with maximum at about 500 nm corresponding to the S0 → S1 state due to intramolecular charge transfer (ICT) from electron-donating dibutylamino group to electron accepting pyridinium moiety. Independing on the solvents, the extinction coefficient value of this transition equals about 50,000 M−1 cm−1. Figure 1 shows the normalized absorption spectra of the dye in methanol, DMF and AcCN. The 4-(N,N-dimethylaminostyryl)pyridinium perchlorate (SP1) absorption spectrum is also shown for comparison. Relative to the monomeric hemicyanine dye, a red shift in the absorption maxima was observed for the BP. The mechanism causing the red shift observed for trichromophoric dyes in comparison to its monomeric equivalent is not clear. It may be speculated that the additional electron acceptors, being in proximity to one belonging to light absorbing chromophore, causes an additional increase of electron affinity of the electron-accepting moiety of light absorbing chromophore. The results supporting this hypothesis may come from the paper published by Huang et al. who described the properties of similar types of dichromophoric hemicyanine dyes [15]. These authors have documented that the red shift of the absorption maximum for the dichromophoric dyes in comparison to monochromophoric ones is decreasing as the number of methylene units separating chromophores is increasing. The red shift (in DMF) for the dye with 3 methylene units is about 700 cm−1, for 5 units about 340 cm−1, and for 12 units only 90 cm−1. The specific influence of the neighboring electron accepting group on light absorbing molecule may have crucial importance on possible two-photon absorption properties of the dye described in this paper.

Normalized electronic absorption spectra of mono- and tri-cationic hemicyanine dyes in different solvents at 293 K.59 × 45mm (600 × 600 DPI)
Like absorption spectra, the dye also shows broad and structureless emission spectra with maximum of about 640 nm and Stokes shifts of about 4,000 cm−1. A large Stokes shift in the BP generally, is ascribed to a different charge distribution in the excited state and the ground state of the molecule. In the molecule, the positive charge is transferred from pyridinium nitrogen (benzenoid form) to dibutylamino group (quinoid form), thereby increasing the dipole moment.
The fluorescence quantum yields of the dye are very low in organic solvents. This fact may be the result of ultrafast internal twisting processes in the excited state of the molecule. It has been confirmed by several reports [7, 16] that fluorescence quantum yields of monomeric hemicyanine dyes depend on the solvent polarity and decrease in high polar solvents. This is attributed to the formation of a nonradiative TICT state which is the most efficient deactivation process of the excited state of the dyes. A polar solvent prefers the formation of the TICT state and this, in turn, increases the nonradiative transition of the excited state [17, 18]. In the case of studied dye, the rotation about its separate bonds occurs in the excited state. The deactivation of the states formed by a twist of the double bond or by a twist of the dibutylamino group should be radiationless in character. This causes lower fluorescence intensity.
To understand more about the excited-state dynamics, fluorescence decay lifetimes of the dye were measured in the three solvents. In order to extract the quantitative values of the decay lifetimes from the experimental fluorescence curves, the tri-exponential decay functions were used for nonlinear curve fitting. From the tri-exponential decay functions, the fast decay lifetime for BP in DMF is fitted to the value of about 33 ps (τ 1), the middle to 168 ps (τ 2) and the slow one to 1,000 ps (τ 3). Based on the results listed in Table 2, according to Eq. 4, the averaged time (τ av) was calculated:
The measured and the evaluated photophysical properties for the BP dye, e.g., fluorescence quantum yields (ϕ f), lifetime (τ av), radiative (k r) and non-radiative (k nr) rate constants are presented in Table 2. The rate constants of the radiative and nonradiative processes of the excited singlet state were calculated according to the relations 5 and 6:
The data compiled in Table 2 show that for tested dye, the non-radiative transition rates are 2 orders of magnitude faster than the radiative rates. The non-radiative rate includes contribution of the excited singlet state to the photoisomerization reaction, the singlet-triplet intersystem crossing and the internal conversion processes.
The spectroscopic and lifetime results indicate the existence of emitting multiple species of the styrylpyridinium dye. The multiple fluorescence can in principle be due to:
-
1.
a possible transition of π → π * locally excited state (LE) to the charge-transfer (CT) state
-
2.
the intramolecular relaxation of the π → π * locally excited state involving rotations of the single bonds of the olefinic styryl group.
Kinetic study of multifunctional acrylate polymerization
The steps determining the reaction of the radical initiated polymerization via inter- or intramolecular electron transfer process (PET) are dependent on the nature of the dye and the electron donor (or acceptor). That is why photopolymerization experiments were carried out to illustrate the relationship between the rate of polymerization and the composition of the photoinitiating system (concentration and structure of the photoinitiator).
The efficiency of radical polymerization photoinitiated by 1,1′,1″-(benzene-1,3,5-triyltrimethanetriyl)-tris(N,N-dibutylaminostyrylpyridinium borate) was evaluated by monitoring the rate of heat evolution during the radical polymerization of multiacrylate (TMPTA). The polymerization was initiated with the visible light. It was found that irradiation of monomer mixture (TMPTA–MP) containing the sensitizer (BP) or co-initiator alone did not result in polymer formation under the experimental conditions (irradiation at λ = 514 nm).
Influence of photoinitiator concentration
It is well-known that the photoinitiator concentration plays a key role in photopolymerization. In conventional UV/Vis photopolymerization, rate of polymerization increases when more initiator is used; however, it decreases rapidly if too much initiator is added. This effect is rendered to the “inter filter effect” and becomes more significant for photoinitiators with a high molar extinction coefficient. Therefore, the polymerization will proceed efficiently if the initiator concentration guarantees that the whole incident light will be absorbed by the sample throughout its volume.
As shown in Fig. 2, it was found that as the photoinitiator concentration increases, the rate of polymerization increases and reaches a maximum followed by continuous decrease. The highest rate of polymerization was achieved at initiator concentration of 0.001 M.

Rate of polymerization vs. photoinitiators concentration. 115 × 92mm (600 × 600 DPI)
This indicated that the photoinitiator concentration of 0.001 M is optimal for the experimental condition and that, at this concentration, the whole incident light is absorbed across the entire polymerizing formulation layer. Thus, all kinetic measurements for hemicyanine borate salts were carried out at this concentration.
Influence of co-initiator structure
The mechanism of photoinitiation, consistent with Schuster and co-workers’ [19] observations for cyanine–alkyltriarylborate pair has been previously suggested [2]. Electron transfer from the borate anion to the singlet excited state of cyanine molecule leads to the formation of cyanine radical anion and boranyl radical which rapidly decomposes producing an alkyl radical. The latter is the most likely initiator of acrylates polymerization. What is more, the studies of Necker et al. [20] revealed that in the case of phenyltrialkylborates, the second product formed after an electron transfer process, phenyldialkylborane, is also capable of initiating a chain radical reaction. It can react either with the oxygen centered radical product of addition of the alkyl radical to the vinyl bond of monomers or with the excited state of the sensitizer [21] forming additional radicals.
Thus, the overall efficiency of the photoinitiation of radical polymerization can be affected by: (1) the rate of the primary electron transfer process, (2) the rate of carbon-boron bond cleavage, (3) the rate of secondary processes, and finally (4) the reactivity of free radicals formed.
Figures 3, 4, 5, and 6 present the family of kinetic curves recorded during an argon-ion laser initiated polymerization of a solution composed of 1 mL of MP, 9 mL of TMPTA, and different photoinitiating systems with a photoinitiator concentration of 0.001 M. The rates of photoinitiated polymerization measured for all photoredox pairs under the study are compiled in Table 3.

Family of kinetic curves recorded during the measurements of the heat flow emitted during the photoinitiated polymerization of the TMPTA-MP (9:1) mixture initiated by hemicyanine borates marked in the figure. The photoinitiator concentration was 0.001 M, I0 = 100 mW/cm2. The applied systems possess identical chromophore and different borates. Inset: Double-bond conversion vs. time for the photoinitiators. 59 × 45 mm (600 × 600 DPI)

Double-bond conversion vs. time for the photoinitiators marked in the figure. The photoinitiator concentration was 0.001 M, I0 = 100 mW/cm2. The curves illustrate the influence of the substituent in borate anion on photoinitiating ability of the systems.60 × 45 mm (600 × 600 DPI)

Comparison of the TMPTA/MP (9/1) mixture polymerization initiated by BPB7 and BPB2. Inset: Double-bond conversion vs. time for the photoinitiators. 60 × 45 mm (600 × 600 DPI)

Comparison of the TMPTA/MP (9/1) mixture polymerization initiated by BPB7, titanocene and DIBFB7 systems as measured by photo-DSC with the incident light intensity of 100 mW/cm2. DIBF—5,7-diiodo-3- butoxy-6-fluorone (triplet-state photoinitiator), its concentration was 0.001 M. [22]. 60 × 46 mm (600 × 600 DPI)
It was found that there is a marked difference between the rates of photoinitiation of polymerization under the conditions described above when the trialkylphenylborates of different structure are used as electron donors. Figure 3 shows that the rates of the polymerization using BPB7, BPB8, and BPB9 are almost the same. The conversion of carbon–carbon double bonds reaches about 60 % after 30 s of irradiation. The curing process is slow when BPB5 is used under the same conditions. Photopolymerization proceeds gradually, and conversion was 30–35 % at 6–7 min. The lower cure efficiency with the BPB5 may be related to the lower reactivity of the free radical formed after photoinduced electron transfer process. The order of reactivity of borate anions as co-initiators in forming polymers increases as follows: phenyltrimethyl-, phenyltriethyl-, phenyltri-sec-butyl-, phenyltri-n-butyl-, and phenyltriisopentylborate. The overall reactivity of the borate salts in TMPTA photopolymerization depends on the ease of their oxidation, radical bond order and their stability. The reactivity of alkyl radicals decreases with increasing bond order of carbon atoms with a single electron. Borates producing the more stable radicals are more reactive then others. From borates examined, different radical species may be formed having the following stability order: isopentyl > n-butyl > sec-butyl > ethyl > methyl. Similar observations were described by Neckers and co-workers [22], who tested the series of tetraorganoborates in the presence of 5,7-diiodo-3-butoxy-6-fluorone, the triplet-state sensitizer.
Additionally, it was found that the photoinitiation efficiency of the tested hemicyanine borates depends on a character of a substituent in phenyl ring of the electron donor moiety (Fig. 4). In the group of phenyltriethylborates, only one type of radical (ethyl) can be produced. In that case, the oxidation potential of borate salts can affect the initiation efficiency. 4-Phenoxyphenyltriethylborate (B63), which bears an electron releasing substituent, has an oxidation potential of about 0.68 V, while the 4-cyanophenyltriethylborate (B65), which bears a strong electron withdrawing group, has an oxidation potential of about 0.95 V. The best photoinitiating abilities exhibit the photoredox pairs possessing electron-donating substituents in the borate molecule, the worst borates with cyano and chlorine substituents. Generally, the substitution on the benzene ring strongly influences the reduction properties of phenyltriethylborate salts and plays crucial role in the initiating radical polymerization.
What is more, in the photoinitiating systems shown in Fig. 5, the chromophore structure and the radicals formed from borate anions after PET are the same. This causes that the reduction potential of hemicyanine dye and the reactivity of butyl radical cannot affect the initiation efficiency. Since the borate salts are efficient electron donor in the photoredox pairs, the difference in their reactivity in the system investigated is also related to substantial differences in the oxidation potentials of the borates. The tetramethylammonium n-butyltriphenylborate has an oxidation potential of about 1.2 V. This is higher than that of tetramethylammonium phenyltrialkylborates, meaning that phenyltrialkylborate salts are much more reductive than n-butyltriphenylborate salt. Moreover, the photoinduced one electron oxidation of phenyltrialkylborate anion by the excited state of styrylpyridinium cation produces substantially more than one equivalent of the alkyl radicals. This was confirmed by measuring the quantity of an adduct formed during irradiation of a deaerated solution containing phenyltrialkylborate and methyl methacrylate in benzene/acetonitrile [23]. The obtained result was in good agreement with the Polykarpov and Neckers [20] studies.
Data presented in Fig. 6 show that the polymerization photoinitiation ability of tri-cationic photoinitiating system is better than the photoinitiating ability observed for DIBF, common triplet-state photoinitiator with the same borate salt used as an electron donor [22]. It is also noteworthy that the BPB7 photoredox pair exhibits only slightly worse photoinitiating ability as typical commercial photoinitiators—titanocene for which the quantum yield of polymerization is about 340, a conversion of monomer double bonds—about 60 % and the polymerization rate equals 29 μmol/s.
Influence of thermodynamics parameter
As mentioned earlier, initiation of polymerization via a photoinduced intermolecular electron transfer process involves many steps, including the photoinduced electron transfer from an electron donor to the excited state of the dye, or from an excited electron donor to the ground state of an electron acceptor, followed by secondary reactions, which yield a neutral radical that initiates polymerization. For the case when the rate of electron transfer reaction is much lower than the rate of diffusion controlled processes that occur after electron transfer, the rate of polymerization should increase when the thermodynamic driving force of electron transfer increases. On the other hand, for a process controlled by diffusion, the reactivity of free radicals formed as a result of the electron transfer process can limit the rate of polymerization photoinitiation.
The free energy activation for the electron transfer process from a donor D to an excited acceptor A is expressed by the Rehm–Weller equation [24]:
where: E 00 is the excited-state energy of either electron acceptor or electron donor (dye molecule in many cases), E(D/D •+) is the oxidation potential of the donor, and E(A •−/A) is the reduction potential of the acceptor. The last term in Eq. 7 accounts the Coulomb energy associated with the separation of charges in the electron transfer products. This factor very often can be ignored taking into account the overall value of ΔG el .
For the photoinitiating systems possessing different borate salts as co-initiators the values of free energy of electron transfer process were estimated using the oxidation potentials of the co-initiators (ranging from 0.618 to 1.14 V versus SCE), the excitation energies of the sensitizers (BP) equals 2.16 eV and the sensitizer reduction potentials (−0.442 eV versus SCE) (see Tables 2 and 3). The reduction potential of the dye and the oxidation potentials of the borate salts were measured by cyclic voltammetry. The data was acquired at scan rates of 500 mV s−1 in acetonitrile containing 0.1 M tetrabutylammonium perchlorate as supporting electrolyte.
The values of ΔG el for tested photoinitiating systems oscillate in the range from −0.57 to −1.1 eV. The calculations clearly show that for the tested photoredox pairs the electron transfer process is thermodynamically allowed. This, in turn, allows to predict that the tested dye in combination with borate anion effectively generate free radical that can start polymerization of the acrylic monomer.
Translating this to the practical application of the Marcus equation [2, 4] in the polymerization photoinitiation process, one can anticipate that the rules regulating the primary processes might also be applied for the secondary reactions. The results of such treatment presented in Fig. 7 seem to confirm such an expectation.

Relationship between the free energy of activation of electron transfer process and the rate of polymerization initiated by the series of phenyltrialkylborate salts. 62 × 49 mm (600 × 600 DPI)
From the inspection of the data presented in Fig. 7, it is clear that the rate of free radical polymerization initiated by the photoredox pairs composed of BP hemicyanine dye cation and phenyltrialkylborate anions increases as the driving force of the electron transfer reaction increases. It is noteworthy that this behavior is predicted by the classical theory of the photoinduced electron transfer process [2, 3].
Influence of light intensity
Photoinduced electron transfer process in donor–acceptor systems besides the initiation of radical polymerization causes the color loss of the primary absorber. The effect is crucial during the thick monomer layer, due to the bleaching of the initiator, the polymerization reaction may occur at depth as well as at the surface. It is obvious that polymerization at a certain depth is possible when the dye bleaches because the change of the depth of polymerization is controlled by the rate of the bleaching process [2, 3]. Figure 8 illustrates changes of the electronic absorption spectra of BPB7 during an argon-ion laser irradiation.

Changes of the electronic absorption spectra of BPB7 during an argon-ion laser irradiation (I0 = 30 mW) in acetonitrile. Time of irradiation marked in the figure. 62 × 49 mm (600 × 600 DPI)
It is easy to imagine that for a thick layer, the front of polymerization is moving as the dye bleaches at the depth, thus the bleaching volume of the irradiated system should be directly proportional to the light intensity. Also analysis of the kinetics of radical polymerization indicates that the linear relationship is observed when one radical obtained after PET is an initiating radical while the second type of radical terminates the chain [2, 3].
Figure 9 presents relationship between the rate of polymerization of TMPTA-MP (9:1) mixture as a function of power of laser beam, which initiated polymerization in the presence of BPB2 photoredox pair.

Photopolymerization kinetic traces for the TMPTA polymerization initiated by BPB65 at different light intensity. Inset: Rate of polymerization vs. the light intensity for selected hemicyanine borate systems. The photoinitiator concentration was 0.001 M. 116 × 92 mm (600 × 600 DPI)
Measurements of the rate of polymerization with different power densities, as shown in Fig. 9, indicate that the rate of polymerization varies proportionally to the square root of the light intensity. Based on the measurements, one can conclude that polymerization photoinitiated by hemicyanine borate photoredox pairs proceeds by the conventional mechanism, in which bimolecular termination occurs by a reaction between two macroradicals. This suggests that the free radicals formed from hemicyanine ion after a photoinduced electron-transfer process does not act as terminator of the polymer chains [2].
Conclusion
1,1′,1″(Benzene-1,3,5-triyltrimethanetriyl)-tris(N,N-dibutylaminostyrylpyridinium borate) can be used as effective visible-light photoinitiators for multiacrylate polymerization. The highest initiation efficiency with quantum yield of polymerization above 200 were observed for the BPB61, BPB7, BPB8, and BPB9 photoredox pairs. Generally, the photoinitiating ability of tested systems depends on both the driving force of the electron transfer process between an electron donor and an electron acceptor and the reactivity of the free radical resulting from the secondary processes occurring after photoinduced electron transfer process. The study revealed that the tricationic hemicyanine borate salts seem to be very good candidates as visible-light photoinitiators of acrylates and can find practical application in modern technologies based on photocuring.
References
Matyjaszewski K, Davis TP (2002) Handbook of radical polymerization. Wiley, Hoboken
Pączkowski J, Kabatc J, Jędrzejewska B (2008) Polymethine dyes as fluorescent probes and visible-light photoinitiators for free radical polymerization. In: Strekowski L (ed) Heterocyclic polymethine dyes. In: Gupta RR (ed) Topics in heterocyclic chemistry (series title), vol. 14. Springer, Heidelberg, pp 200–220
Pączkowski J (2006) Electron-transfer photoinitiators of free radical polymerization. The effect of the co-iniciator structure on photoinitiation ability. In: Fouassier JP (ed) Photochemistry and UV curing: New trends. Research Signpost, Kerala, India, pp 101–116
Marcus RA (1965) On the theory of electron-transfer reactions. VI. Unified treatment for homogeneous and electrode reactions. J Chem Phys 43:679–701
Pączkowski J, Pietrzak M, Kucybała Z (1996) Generalization of the kinetic scheme for photoinduced polymerization via an intermolecular electron transfer process. 2. Application of the Marcus theory. Macromolecules 29:5057–5063
Jędrzejewska B, Pietrzak M, Rafiński Z (2011) Phenyltrialkylborates as co-initiators with cyanine dyes in visible light polymerization of acrylates. Polymer 52:2110–2119
Jędrzejewska B, Kabatc J, Pączkowski J (2007) Dichromophoric hemicyanine dyes. Synthesis and spectroscopic investigation. Dyes Pigm 74:262–268
Brandrup J, Immergut EH (1989) Polymer handbook, 3rd edn. Wiley, New York, p II/298
Jędrzejewska B, Jeziórska J, Paczkowski J (2008) Studies on an argon laser-induced photopolymerization employing both mono- and bischromophoric hemicyanine dye-borate complex as a photoinitiator. J Photochem Photobiol A: Chem 195:105–115
Jędrzejewska B, Pietrzak M, Pączkowski J (2010) Tetramethylammonium phenyltrialkylborates as co-initiators with novel two-cationic styrylbenzimidazolium dyes in highly efficient, visible light polymerization of acrylate. J Photochem Photobiol A: Chem 214:276–283
Hassoon S, Neckers DC (1995) Electron transfer photoreduction of 5,7-diiodo-3-butoxy-6-fluorone with tetrabutylammonium triphenylbutylborate and N,N-dimethyl-2,6-diisopropylaniline. J Phys Chem 99:9416–9424
Sarker AM, Polykarpov AY, de Raaff AM, Marino TI, Neckers DC (1996) Visible light photopolymerization employing 2,4-diiodo-6-butoxy-3-fluorone and tetraorganyl borate salts as photoinitiators. J Polym Sci: Part A-Polym Chem 34:2817–2824
Pietrzak M, Jędrzejewska B (2011) Synthesis of tetramethylammonium phenyltrialkylborate salts by the addition of alkyl lithium reagents to a triorganylborane or organoboranylhalides. J Organomet Chem 696:2135–2141
Mishra A, Haram NS (2004) New push–pull type dendritic stilbazolium dyes: synthesis, photophysical and electrochemical investigation. Dyes Pigm 63:191–202
Huang Y, Cheng T, Li F, Luo C, Huang C-H, Cai Z, Zeng X, Zhou J (2002) Photophysical studies on the mono- and dichromophoric hemicyanine dyes II. Solvent effects and dynamic fluorescence spectra study in chloroform and in LB films. J Phys Chem B 106:10031–10040
Fromherz P (1995) Monopole-dipole model for symmetrical solvatochromism of hemicyanine dyes. J Phys Chem 99:7188–7192
Strehmel B, Seifert H, Rettig W (1997) Photophysical properties of fluorescence probes. 2. A model of multiple fluorescence for stilbazolium dyes studied by global analysis and quantum chemical calculations. J Phys Chem B 101:2232–2243
Szczepan M, Rettig W, Bricks YL, Słomiński YL, Tolmachev AI (1999) Unsymmetric cyanines: chemical rigidization and photophysical properties. J Photochem Photobiol A: Chem 124:75–84
Chatterjee S, Davis PD, Gottschalk P, Kurz ME, Sauerwein B, Yang X, Schuster GB (1990) Photochemistry of carbocyanine alkyltriphenylborate salts intra-ion-pair electron transfer and the chemistry of boranyl radicals. J Am Chem Soc 112:6329–6338
Polykarpov AY, Neckers DC (1995) Tetramethylammonium phenyltrialkylborates in the photoinduced electron transfer reaction with benzophenone. Generation of alkyl radicals and their addition to activated alkenes. Tetrahedron Lett 36:5483–5486
Wilkinson G, Stone FGA, Abel EW (1982) The synthesis, reactions and structures of organometallic compounds. In: Wilkinson, G., Stone, F. G. A., Abel, E. W (eds) Comprehensive organometallic chemistry, vol. 7. Pergamon Press, Oxford, pp. 291–293, 335
Polykarpov AY, Hassoon S, Neckers DC (1996) Tetramethylammonium tetraorganylborates as co-initiators with 5,7-diiodo-3-butoxy-6-fluorone in visible light polymerization of acrylates. Macromolecules 29:8274–8276
Pietrzak M, Jędrzejewska B, Pączkowski J (2009) Unusually highly efficient, singlet state, visible light photoinitiators based on styrylbenzimidazolium phenyltributylborate photoredox pairs for vinyl monomers free radical polymerization. J Polym Sci Part A-Polym Chem 47:4119–4129
Rehm D, Weller A (1970) Kinetics of fluorescence quenching by electron and hydrogen-atom transfer. Isr J Chem 8:259–271
Acknowledgments
This work was supported by the Ministry of Science and Higher Education (MNiSW) (BS 12/2010)
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Jędrzejewska, B. Factors affecting the TMPTA radical polymerization photoinitiated by phenyltrialkylborates paired with tri-cationic hemicyanine dye. Kinetic studies. Colloid Polym Sci 291, 2225–2236 (2013). https://doi.org/10.1007/s00396-013-2964-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00396-013-2964-3