
Abstract
Mitochondrial calcium (Ca2+) signals play a central role in cardiac homeostasis and disease. In the healthy heart, mitochondrial Ca2+ levels modulate the rate of oxidative metabolism to match the rate of adenosine triphosphate consumption in the cytosol. During ischemia/reperfusion (I/R) injury, pathologically high levels of Ca2+ in the mitochondrial matrix trigger the opening of the mitochondrial permeability transition pore, which releases solutes and small proteins from the matrix, causing mitochondrial swelling and ultimately leading to cell death. Pharmacological and genetic approaches to tune mitochondrial Ca2+ handling by regulating the activity of the main Ca2+ influx and efflux pathways, i.e., the mitochondrial Ca2+ uniporter and sodium/Ca2+ exchanger, represent promising therapeutic strategies to protect the heart from I/R injury.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cardioprotection refers to the mechanisms and interventions that protect the heart against various forms of damage, minimizing cardiac myocyte loss and thereby preserving cardiac function [84]. This definition applies in principle to any type of cardiac insult, but cardioprotective strategies were predominantly investigated in the context of myocardial ischemia and infarction. While early reperfusion is crucial to prevent infarct expansion, restoration of blood flow to the ischemic myocardium can be accompanied by reperfusion arrhythmias and reversible contractile dysfunction, known as “myocardial stunning” [18, 56]. The concept of myocardial reperfusion as “a double-edged sword” was first formulated in 1985 by Braunwald and Kloner [20]. Henceforth, studies in this field yielded a detailed understanding of the mechanisms underlying ischemia/reperfusion (I/R) injury, and identified a central role for mitochondria in determining cardiac myocyte fate in this context. While being pivotal to energy transduction via oxidative phosphorylation, mitochondria also act as master regulators of different forms of cell death.
Calcium (Ca2+) plays an essential role in cardiac myocyte physiology and disease. In the healthy heart, transient variations in cytosolic Ca2+ levels induce contraction and relaxation of cardiac myocytes, and mitochondrial Ca2+ signals adapt the rate of mitochondrial oxidative metabolism to the rate of adenosine triphosphate (ATP) turnover in the cytosol. During I/R injury, mitochondrial Ca2+ overload is one key mechanism of cardiac myocyte loss by inducing mitochondrial permeability transition. Therefore, therapeutic strategies aimed at modulating intracellular Ca2+ movements represent a viable approach to mitigate I/R injury. In this review, we outline the physiological role of mitochondrial Ca2+ handling, its alterations during cardiac I/R injury, and how mitochondrial Ca2+ signals can be modulated to prevent mitochondrial dysfunction and cardiac myocyte loss upon myocardial reperfusion.
Physiological role of mitochondrial Ca2+ signals
Mechano-energetic coupling
Transient variations in cytosolic Ca2+ levels ([Ca2+]c) trigger contraction and relaxation of cardiac myocytes, consuming large amounts of ATP that are continuously regenerated by oxidative phosphorylation in mitochondria and, to a lesser extent, by glycolysis in the cytosol. When increases in cardiac workload accelerate ATP turnover by myofilaments and ion pumps, the increase in adenosine diphosphate (ADP) delivery to mitochondria partially and transiently dissipates the proton motive force (ΔμH) as complex V (the F1/Fo-ATP synthase) utilizes the proton gradient across the inner mitochondrial membrane (IMM) to catalyze ADP phosphorylation to ATP. Simultaneously, mitochondrial Ca2+ signals adjust the rate of mitochondrial oxidative metabolism by stimulating the activity of three tricarboxylic acid (TCA) cycle enzymes [97], thereby providing more reducing equivalents to the electron transport chain (ETC) to maintain ΔμH [100]. Therefore, ADP and Ca2+ induce a “parallel activation” of mitochondrial oxidative metabolism that enables the heart to sustain abrupt changes in contractility (and, consequently, ATP turnover) (Fig. 1).

Cardiac mechano-energetic coupling. In the healthy heart, oxidative phosphorylation produces 95% of the adenosine triphosphate (ATP) required to fuel excitation–contraction coupling. ATP phosphorylation is catalyzed by the F1/Fo-ATP synthase, which harnesses the electrochemical gradient produced by translocation of protons (H+) across the inner mitochondrial membrane (IMM) by the electron transport chain complexes. In turn, the electron transport chain derives reducing equivalents from the reduced form of nicotinamide adenine dinucleotide (NADH) produced by the tricarboxylic acid (TCA) cycle. Superoxide (.O2−) is a physiological by-product of the respiratory chain activity, and is rapidly dismutated to hydrogen peroxide (H2O2) by the manganese-dependent superoxide dismutase (Mn-SOD). During elevations in cardiac workload, the increased ATP turnover in the cytosol partially dissipates the mitochondrial membrane potential (ΔΨm) and oxidizes the NADH pool. At the same time, calcium (Ca2+) accumulation in the mitochondrial matrix stimulates the TCA cycle dehydrogenases to regenerate NADH and also the reduced form of nicotinamide adenine dinucleotide phosphate (NADPH), which sustain the respiratory chain activity and hydrogen peroxide (H2O2) elimination, respectively. Also under physiological conditions, transient opening of a low-conductance mitochondrial permeability transition pore (mPTP) in the IMM may operate as an alternative Ca2+ efflux pathway in addition to the mitochondrial Na+/Ca2+ exchanger. Figure created with BioRender.com
The reducing equivalents produced from oxidation of nutrients are also used to fuel the activity of antioxidant systems responsible for the elimination of mitochondrial reactive oxygen species (ROS). Namely, the oxidation of malate and isocitrate by the respective dehydrogenases is used to regenerate the reduced form of the cofactor NADPH, which functions as an electron donor for a series of redox reactions that maintain the reduced glutathione (GSH) and thioredoxin (Trx) pools. In turn, GSH and Trx function as reducing agents for the conversion of hydrogen peroxide (H2O2) to water catalyzed by glutathione peroxidase and peroxiredoxin, respectively. An additional source of mitochondrial NADPH is the nicotinamide nucleotide transhydrogenase (NNT), which uses ΔμH to transfer hydride ion equivalents (H−) from NADH to NADPH. Therefore, the TCA cycle is also essential to regenerate the H2O2-eliminating systems of the mitochondrial matrix [78] (Fig. 1).
Mitochondrial Ca2+ signals, therefore, play a central role in cardiac homeostasis, as they allow the heart to respond to sudden changes in workload without incurring in an energetic crisis or inducing oxidative stress. The ability of mitochondrial oxidative metabolism to swiftly adapt to elevations in ATP turnover, coined mechano-energetic coupling, is impaired in acute and chronic cardiovascular disorders, including I/R injury and heart failure (reviewed in [14]).
The mitochondrial Ca2+ uniporter (MCU)
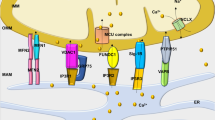
The molecular identity of the channel(s) mediating Ca2+ uptake from the cytosol to the mitochondrial matrix has remained elusive until the early 2010s, when the mitochondrial Ca2+ uniporter (MCU) pore-forming subunit, MCUa, was identified [11, 132]. It became apparent that the MCU has a complex architecture and, besides a tetramer of MCUa subunits forming the channel pore, it comprises at least four other core components: the essential MCU regulatory element, EMRE, and the regulatory proteins MICU1, MICU2, and MICU3 [36]. EMRE is a transmembrane protein that is essential for Ca2+ uptake via the MCU in metazoans [125]. MICU1, 2, and 3 regulate the MCU current depending on Ca2+ levels in the intermembrane space, which they sense via their EF-hand domains. The mechanisms of MICU-mediated regulation of Ca2+ flux through the MCU is still under investigation. There is consensus that MICU1 accounts for the cooperative activation of the MCU at high [Ca2+]c, whereas at low [Ca2+]c, MICU1 physically occludes the pore [122], thus preventing uncontrolled influx of Ca2+ in the mitochondrial matrix at the physiological [Ca2+]c ranges of 100 to 500 nM (so-called gatekeeper function). In line with this model, MICU1 deletion increases mitochondrial Ca2+ levels at low [Ca2+]c, but blunts the rapid MCU-mediated increase in matrix Ca2+ when mitochondria are exposed to high Ca2+ [74, 90, 110].
Because of the low Ca2+ affinity of the uniporter, mitochondrial Ca2+ uptake via the MCU is made possible by the close proximity of mitochondria to the points of Ca2+ release from the sarcoplasmic reticulum (SR). The apposition of the outer mitochondrial membrane and the SR membrane creates specialized microdomains where spatially localized elevations of [Ca2+]c to micromolar levels overcome the threshold required for Ca2+ uptake via the MCU, thus driving Ca2+ influx to the mitochondrial matrix [42]. Several proteins have been implicated in the physical tethering of mitochondria and the SR, including mitofusin (MFN) 1 and 2 and FUN domain containing protein 1 [137]. MFN1 and MFN2 play a primary role in modulating the structural and functional juxtaposition between the SR and mitochondria, as well as the interorganellar exchange of Ca2+ [21, 24].
MFN1 and MFN2 were initially discovered as mediators of mitochondrial fusion [23], the process by which two mitochondria merge into a single, larger mitochondrion. This allows exchange of mitochondrial DNA and other components to compensate for mitochondrial damage. Conversely, mitochondrial fission is the process where one mitochondrion divides into two separate mitochondria. Mitochondrial fission ensures apportioning of mitochondria during cell division and segregation of damaged mitochondrial components for elimination by mitophagy [54]. There is a bidirectional interaction between SR-mitochondria Ca2+ movements and mitochondrial dynamics. Mitochondrial morphology, influenced by the balance between fission and fusion, regulates mitochondrial Ca2+ uptake [82]. Conversely, changes in mitochondrial Ca2+ uptake can also affect fission and fusion dynamics [81]. However, these cross-regulatory mechanisms were mainly investigated in cell systems, and evidence of their role in the intact heart is lacking.
Mitochondrial Ca2+ efflux
The best characterized mitochondrial Ca2+ efflux pathway is the mitochondrial sodium (Na+)/Ca2+ exchanger (NCLX) [22, 113]. The Na+ gradient across the IMM is the primary determinant of NCLX activity, and thereby elevations in cytosolic sodium levels ([Na+]c) accelerate NCLX-mediated Ca2+ extrusion from mitochondria [12, 28]. One Na+-independent mechanism of Ca2+ efflux was recently described [8], but its role in cardiac myocytes has not been investigated thus far. The slower kinetics of mitochondrial Ca2+ efflux compared with MCU-dependent influx account for the progressive increase in matrix Ca2+ that stimulates the TCA cycle dehydrogenases during elevations in heart rate and contractility, which prevents oxidation of mitochondrial pyridine nucleotides [96]. The exact quantities of mitochondrial Ca2+ are not completely resolved; it is unclear whether mitochondria significantly buffer [Ca2+]c and whether matrix Ca2+ oscillates on a beat-to-beat basis, paralleling changes in [Ca2+]c, or integrates cytosolic Ca2+ transients in a frequency-dependent manner [14].
In conclusion, mitochondrial Ca2+ handling plays a relevant physiological role in matching ATP supply with demand in cardiac myocytes. Ca2+ levels in the mitochondrial matrix are finely tuned by the balance between Ca2+ uptake via the MCU and Ca2+ extrusion via the NCLX, driven by the Na+ gradient across the IMM. The structural and functional interaction between SR and mitochondria is pivotal to the transmission of Ca2+ signals to the mitochondrial matrix.
Mitochondrial Ca2+ overload and permeability transition in I/R injury
Consequences of ischemia on cardiac myocyte ion handling
Alterations in mitochondrial Ca2+ handling have important consequences on cardiac myocyte function and viability. In chronic heart failure, insufficient mitochondrial Ca2+ accumulation causes bioenergetic mismatch and oxidative stress during transitions of cardiac workload [78, 79]. The opposite alteration, i.e., mitochondrial Ca2+ overload, is a major driver of cardiac myocyte loss in the context of I/R injury and therefore, represents a potential target for cardioprotection.
During myocardial ischemia, the reduction in oxygen (O2) and nutrient supply to the myocardium halts mitochondrial oxidative metabolism. The arrest of oxidative phosphorylation leads to a rapid decline in cellular ATP and phosphocreatine below the levels required to sustain cardiac myocyte contraction and relaxation. Subsequently, the activity of plasma membrane ATP-dependent Na+ pumps decreases, resulting in an increase in [Na+]c and potassium (K+) efflux from the cell. A compensatory increase in anaerobic glycolysis leads to H+ accumulation in the cytosol [130], which decreases myofilament Ca2+ affinity [35] and leads to further Na+ influx via the sarcolemmal Na+/H+ exchanger (NHE) [49]. The increase in [Na+]c shifts the driving force of the sarcolemmal Na+/Ca2+ exchanger (NCX) toward reverse mode, thus favoring Ca2+ influx from the extracellular space [66]. This, together with Ca2+ entry via L-type Ca2+ channels and impaired SR Ca2+ reuptake due to reduced SR Ca2+ ATPase (SERCA) activity, contributes to [Ca2+]c elevation, which is transmitted to the mitochondrial matrix (Fig. 2). Ultimately, prolonged depletion of ATP generates a hyperosmolar environment that promotes cell swelling and leads to disruption of the protein synthetic apparatus, causing cell death.

Mechanisms of cardiac ischemia/reperfusion injury. Panel A. Ischemia. During ischemia, the lack of oxygen (O2) abruptly halts oxidative phosphorylation and induces a metabolic switch to anaerobic glycolysis, which increases lactate production and induces intracellular acidosis. The reduced availability of ATP stops cardiac myocyte contraction and relaxation and alters intracellular ion concentrations. Reduced activity of the sodium (Na+)/potassium (K+) ATPase, together with the increased activity of the Na+/H+ exchanger due to intracellular acidosis, induce Na+ and Ca2+ accumulation in the cytosol. The increase in calcium (Ca2+) levels is transmitted to the mitochondrial matrix via the mitochondrial Ca2+ uniporter. Mitochondrial Ca2+ accumulation progressively dissipates the mitochondrial membrane potential (ΔΨm), which can ultimately trigger mitochondrial permeability transition and necrotic cell death. Panel B. Reperfusion. When blood flow is restored, the activity of the respiratory chain resumes, but succinate accumulated during ischemia fuels superoxide (O2−) production at complex I via reverse electron transport. The massive production of mitochondrial reactive oxygen species (ROS), together with mitochondrial Ca2+ accumulation and partial restoration of intracellular pH, triggers irreversible opening of the large-conductance mitochondrial permeability transition pore (mPTP), which allows release of ions, solutes and ROS from the mitochondrial matrix. ROS release from the mPTP and other redox-sensitive channels, such as the inner mitochondrial anion channel (IMAC), can induce ROS production from neighboring mitochondria, a process coined ROS-induced ROS release. Figure created with BioRender.com
Consequences of reperfusion on cardiac myocyte ion handling
If blood flow is restored before the ischemic myocardium is irreversibly damaged, the recovery of sarcolemmal Na+/K+ ATPase and NHE activity rapidly restores intracellular Na+ and K+ levels and pH, respectively. However, reperfusion can result, paradoxically, in the death of cardiac myocytes that are not otherwise irreversibly injured by ischemia, a process coined I/R injury. The underlying mechanism is in part related to the increased influx of leukocytes and aberrant immune responses in the ischemic tissue upon reperfusion [34]. In addition, cell death during reperfusion can be triggered by cardiac myocyte-specific processes that result from the abrupt restoration of O2 availability.
Mitochondrial Ca2+ overload is considered a major contributor to irreversible mitochondrial damage and subsequent cardiac myocyte death during I/R injury [38, 48]. Uncontrolled Ca2+ release from the SR leads to abnormal [Ca2+]c oscillation and mitochondrial Ca2+ overload in the early stages of reperfusion [1, 124]. There are two potential pathways of Ca2+ influx in the matrix from the cytosol during I/R injury, i.e., the MCU and/or the NCLX operating in reverse mode [7], but their relative contribution is still unresolved. The mechanisms linking mitochondrial Ca2+ overload to cell death have been debated, but there is now substantial evidence indicating that excess matrix Ca2+, together with oxidative stress occurring during reperfusion, triggers the opening of a large pore in the IMM, coined the mitochondrial permeability transition pore (mPTP), which leads to the release of proteins and solutes up to ~ 1.5 kDa from the matrix.
The mitochondrial permeability transition pore
It has long been known that a non-selective increase in permeability occurs in mitochondrial membrane after exposure to Ca2+, but this was considered a consequence of membrane damage until the seminal studies by Hunter and Haworth in the 1970s first proposed the concept of mitochondrial permeability transition [52, 62]. The existence of the mPTP was supported by the subsequent identification of a large-conductance channel in the IMM [77] and the observation that permeability transition can be inhibited by cyclosporine A (CsA), which argues against a non-specific disruption of the IMM accounting for permeability transition [29, 40]. The inhibitory effect exerted by CsA was shown to be dependent on a matrix peptidyl prolyl cis–trans isomerase, cyclophilin D (CypD) [45, 134], whose genetic ablation desensitizes the mPTP to Ca2+ [9, 104].
The molecular composition of the mPTP is still under investigation. The adenine nucleotide translocator (ANT) was first proposed as a mPTP component based on the modulatory effects of its inhibitors on permeability transition [61]. However, genetic ablation of ANT does not abolish mPTP opening, although it increases by tenfold the Ca2+ threshold for permeability transition [80]. Another candidate mPTP component is the F1/Fo-ATP synthase, which can generate Ca2+-induced high-conductance currents when reconstituted in lipid bilayers [44]. A unifying and now widely accepted model posits that permeability transition is mediated by two types of pores, one with low and one with high conductance. The former opens transiently and operates to re-equilibrate ion concentrations across the IMM under physiological conditions, whereas the latter, by allowing the leakage of larger solutes, induces important alterations on mitochondrial structure that can ultimately lead to cell death (for a recent review see [19]). ANT is a candidate component of the low-conductance mPTP [106], whereas F1/Fo-ATP synthase dimers might form the high-conductance channel within their Fo subunits in response to structural rearrangements induced by Ca2+ binding to the F1 subunit [43, 118]. CypD, the target of CsA, functions as a chaperone protein that promotes the conformational modification of the F1/Fo-ATP synthase by binding to its regulatory oligomycin sensitivity-conferring protein (OSCP) subunit [43].
The threshold for mitochondrial Ca2+ required to trigger permeability transition is modulated by multiple factors. In particular, mPTP opening is promoted by low ADP levels, dissipation of ΔμH, and production of ROS, and antagonized by magnesium (Mg2+) and pH values < 7 [19]. Despite the increase in mitochondrial Ca2+, permeability transition is inhibited by high ADP levels and the acidic cellular milieu induced by ischemia [3]. Therefore, the majority of mPTP-mediated cardiac myocyte loss occurs during the early stage of reperfusion, when ATP levels increase and ion pumps reprise their activity to restore cellular pH [32, 46]. In addition, mPTP opening is promoted by the increase in mitochondrial ROS production occurring during reperfusion, discussed in the next section.
One additional mechanism of mPTP regulation involves dynamin-related protein 1 (Drp1), an essential mediator of mitochondrial fission. During mitochondrial fission, Drp1 is recruited to the outer mitochondrial membrane, where it forms a ring-like structure that constricts the dividing mitochondrion [87]. Although mitochondrial fission and Drp1 are essential for normal cardiac development and homeostasis [65], enhanced mitochondrial fission driven by Drp1 recruitment to mitochondria exacerbates cardiac myocyte loss during I/R injury. Indeed, acute pharmacological or genetic inhibition of Drp1 protects the heart from I/R injury by preventing mPTP opening [108]. The mechanism of cardioprotection afforded by acute Drp1 ablation might be explained by increased Ca2+ retention capacity and decreased ROS production of larger, undivided mitochondria compared with fragmented mitochondria generated by excessive fission [117]. For a more comprehensive discussion on the role of mitochondrial dynamics in cardiac homeostasis and cardioprotection, we refer the reader to another review of this series [54].
The prolonged and widespread opening of the high-conductance mPTP during reperfusion injury aggravates the redistribution of ions and solutes and causes osmotic swelling of the mitochondrial matrix. Altogether, the arrest of oxidative metabolism, collapse of membrane potential, and irreversible mitochondrial and cellular alterations lead to cell necrosis. Of note, mitochondrial permeability transition can also induce apoptosis mediated by the release of proapoptotic factors normally sequestered within mitochondria, such as cytochrome c. Since apoptosis is an ATP-dependent process, ATP availability is the key factor determining the mode of cell death after mitochondrial permeability transition [75].
Mitochondrial reactive oxygen species in I/R injury
Mitochondrial ROS are a physiological by-product of aerobic metabolism. The incomplete reduction of O2 gives rise to superoxide (O2.−), an extremely reactive and short-lived form of ROS that can cause cellular damage by subtracting electrons to lipids, nucleic acids, or proteins. The respiratory chain, and namely complex I and complex III, are considered major cellular sources of superoxide [102]. To dispose of superoxide, mitochondria are equipped with micromolar concentrations of manganese-dependent superoxide dismutase (Mn-SOD), which dismutates superoxide into H2O2. As described above, H2O2 is converted to water by matrix peroxidases, i.e., peroxiredoxin and glutathione peroxidases, which derive their reducing equivalents from NADPH and thereby, from the TCA cycle.
Mitochondrial production of ROS plays a central role in the pathophysiology of I/R injury [57]. The burst of mitochondrial superoxide occurring at the onset of reperfusion was initially considered a non-specific consequence of the restored availability of O2, but there is now robust evidence indicating that reverse electron transport (RET) at complex I of the respiratory chain is the primary source of superoxide during reperfusion [25]. The mechanistic underpinnings of this process and its role in I/R injury are discussed in detail in another review of this series [120]. Here, we outline the concept of redox-optimized ROS balance, a unifying framework that explains how the overflow of ROS is determined by the redox environment of the cell [5] and ROS-induced ROS release, a self-amplifying process that can exacerbate cardiac myocyte loss and induce arrhythmias during I/R injury [143].
Redox-optimized ROS balance
Initial observations in isolated mitochondria demonstrated that ROS production is maximized under conditions of little electron flow, high mitochondrial membrane potential (ΔΨm), and a fully reduced NADH pool. In this context, ROS production can be alleviated by “mild” uncoupling [101]. These observations stand at odds with experiments in intact cardiac myocytes, in which mitochondrial uncoupling increases ROS emission. The concept of redox-optimized ROS balance solves this apparent contradiction by proposing that the extent of ROS overflow is determined by the overall intracellular redox environment, which includes the redox couples involved in electron transport, i.e., NADH/NAD+ and the ubiquinone pool, and those involved in ROS elimination, i.e., NADPH/NADP and the glutathione pool. Oxidative stress occurs at either extreme of redox potential, that is, when the intramitochondrial environment is either highly reduced or highly oxidized [5] (Fig. 3). Mitochondria normally operate at intermediate redox states, characterized by low ROS production at the respiratory chain and sufficient levels of reduced NADPH to prevent H2O2 emission.

Copyright Elsevier
Redox-optimized ROS balance. The graph illustrates the relationship between the redox environment of cardiac myocytes and the emission of reactive oxygen species (ROS). ROS emission occurs when the intracellular and/or intramitochondrial environments are either highly reduced (right side of the figure) or highly oxidized (left). Under physiological conditions, the cell and mitochondria operate at intermediate redox state (gray-shaded area), at which ROS production is controlled by the antioxidant systems. Reproduced with permission from [5].
Redox-optimized ROS balance also explains the increase in ROS production observed during hypoxia, a seeming paradox when considering the dependence of superoxide production on O2 concentration. According to this concept, hypoxic cells and mitochondria operate in the right arm of the redox extreme curve (Fig. 3), implying that they exhibit highly reduced redox potentials and augmented superoxide production due to the low electron flow [5]. However, since ROS formation at the ETC also depends linearly on the availability of O2 per se [10], superoxide formation during hypoxia is still limited despite a highly reduced redox state of the ETC. At reperfusion (and thereby, reoxygenation), the increase in O2 meets a highly reduced respiratory chain, provoking a “burst” of ROS production and release in the very first seconds and minutes of reperfusion, which levels off once the redox state of the respiratory chain becomes more oxidized due to the reinstallment of electron flux for ATP production. These processes add (and are part of) the concept of the burst-like release of ROS due to reverse electron transfer provoked by succinate accumulation during ischemia. Furthermore, a depletion of glutathione via multidrug resistance protein 1 (MRP-1) during ischemia depletes the antioxidative defense and therefore, adds to the burst of ROS during reperfusion [64].
Mitochondria, however, can also be pushed toward the more oxidized extreme of the redox spectrum under pathological conditions. In systolic heart failure, insufficient Ca2+-dependent stimulation of the TCA cycle chronically depletes mitochondrial antioxidant systems, leading to oxidative stress that drives maladaptive remodeling and arrhythmias [78, 91]. Furthermore, an acute oxidation of the mitochondrial NAD(P)H pool can be induced by the opening of redox-sensitive ion channels in the IMM, including the mPTP, which is the basis for ROS-induced ROS release.
ROS-induced ROS release
Mitochondria permeability transition can exacerbate mitochondrial ROS production via a process coined ROS-induced ROS release [144]. This process involves the ROS-dependent induction of redox-sensitive ion channels within the IMM, such as the mPTP [143], inner mitochondrial anion channel (IMAC) [2, 4], and mitochondrial ATP-dependent K+ channels (mKATP) [31, 76] of nearby or neighboring mitochondria. Opening of these channels dissipates ΔΨm and oxidizes the NADH and NADPH pools, consequently diminishing antioxidant defenses and causing ROS generation. This ROS-induced ROS release, coupled with IMAC activation, fosters synchronized oscillations of ΔΨm, establishing “metabolic sinks” that render specific myocardial regions electrically inactive, thereby contributing to re-entrant arrhythmias during cardiac I/R [2]. It is unclear whether the IMAC coincides with the low-conductance, reversible mPTP described above, whose opening occurs at lower levels of oxidative stress and can be reversed, or whether it represents a distinct entity. In addition, ROS-induced ROS release has been observed through interactions between distinct ROS sources. ROS derived from NADPH oxidase 2 can elevate mitochondrial ROS [30, 76, 95] and vice versa, mitochondrial ROS can exacerbate NADPH oxidase-induced ROS production [83].
In conclusion, although mitochondrial ROS production is increased also under hypoxic conditions, the largest quantity of ROS is produced during reperfusion, primarily via reverse electron transport at complex I. Besides the ETC, there are other important sources of ROS in mitochondria that have been implicated in I/R injury, such as monoamine oxidases (discussed in another review of this series [73]), NADPH oxidases [141], and p66shc [119]. Oxidative stress, in concert with mitochondrial Ca2+ overload, can trigger mitochondrial permeability transition and ROS-induced ROS release, exacerbating cellular damage and creating a substrate for ventricular arrhythmias during I/R injury.
Role of mitochondrial Ca2+ in ischemic conditioning
Ischemic conditioning refers to the activation of a cardioprotective program induced by repeated brief episodes of coronary occlusion and reperfusion preceding or following the prolonged insult. Ischemic conditioning in the heart was first described in a series of seminal studies by the group of Keith Reimer, who showed that brief, intermittent cycles of I/R delay the rate of ATP depletion and markedly reduce infarct size in hearts subsequently subjected to sustained ischemia [103]. Subsequent studies demonstrated that a protective effect against prolonged ischemia is achieved also when intermittent ischemia is applied during early reperfusion, a process coined ischemic post-conditioning [142], and when they involve extracardiac tissues, known as remote conditioning [41]. The mechanisms underlying ischemic pre-, post-, and remote conditioning (IPC, POC, and RIC, respectively) are manifold [55], and involve the release of soluble mediators including adenosine, cytokines, and neurohormones. In turn, these mediators bind to specific receptors expressed by cardiac myocytes and activate intracellular signaling cascades, including the reperfusion injury salvage kinase (RISK) pathway, survival activating factor enhancement (SAFE) pathway, and a pathway involving endothelial nitric oxide synthase, protein kinase G and protein kinase C. These pathways primarily converge on mitochondria as the end effectors of cardioprotection [55]. Namely, modulation of mPTP opening is considered the key mediator of cardioprotection induced by ischemic conditioning. Other mitochondrial ion channels, i.e., mKATP and connexin 43 (Cx43), are also known to modulate susceptibility to myocardial I/R injury [51, 55] (Fig. 4).

Role of mitochondria in ischemic conditioning. The main signaling pathways activated during ischemic conditioning include the reperfusion injury salvage kinase (RISK) pathway, the survival activating factor enhancement (SAFE) pathway, and a pathway involving activation of endothelial nitric oxide synthase (eNOS), protein kinase G (PKG) and protein kinase C (PKC), which mainly converge on mitochondria as the end effector of cardioprotection. Opening of the mitochondrial permeability transition pore (mPTP), which is triggered by mitochondrial Ca2+ overload and reactive oxygen species (ROS) production, is a major cause of cardiac myocyte loss during ischemia/reperfusion injury. Modulation of mitochondrial permeability transition is considered a central mediator of cardioprotection induced by ischemic conditioning. Intracellular signaling pathways activated by intermittent cycles of ischemia and reperfusion reduce susceptibility to mPTP opening via multiple mechanisms, including direct mPTP inhibition, mitochondrial translocation of connexin 43 (Cx43), activation of mitochondrial potassium (K+) uptake via the mitochondrial ATP-dependent K+ channel (mKATP), and reduced formation of reactive oxygen species (ROS). In addition, modulation of Ca2+ exchange between the sarcoplasmic reticulum (SR) and mitochondria contributes to the cardioprotection afforded by ischemic conditioning. Namely, decreased Ca2+ release via ryanodine receptor type 2 (RyR2) and delayed phospholamban (PLN) phosphorylation might prevent exaggerate SR Ca2+ release and consequent mitochondrial Ca2+ overload during reperfusion. Cyt c cytochrome c, GPCR G protein-coupled receptor, GSK-3β glycogen synthase kinase-3β, JAK Janus kinase, MCU mitochondrial calcium uniporter, MFN1/2 mitofusin ½, PKA protein kinase A, SERCA sarcoplasmic reticulum Ca2+ ATPase, SR, sarcoplasmic reticulum, STAT3 signal transducer and activator of transcription 3; TNFR tumor necrosis factor receptor. Figure created with BioRender.com
Mitochondrial ATP-dependent K+ channels
ATP-dependent K+ channels (KATP) are present on both the sarcolemma and IMM and their activation is regulated by the ATP/ADP ratio [67]. When ATP levels drop, opening of sarcolemmal KATP functions as an “energy-sparing” mechanism by shortening action potential duration, consequently reducing cytosolic Ca2+ entry and depressing contractility [39]. Initially, ischemic conditioning was attributed to sarcolemmal KATP based on the observation that KATP openers mimicked IPC, while KATP inhibitors abolished it [139]. However, later studies showed limited correlation between action potential shortening and cardioprotection conferred by KATP inhibitors [47], KATP opener protection in unstimulated cells [89], and negligible protection from agents targeting only sarcolemmal KATP [126]. These findings shifted the focus to mitochondrial KATP (mKATP) as mediators of IPC. Since K+ influx osmotically drives water in the mitochondrial matrix, mKATP activity is a key regulator of mitochondrial volume. Multiple mechanisms were proposed to explain the cardioprotective effect of mKATP opening. First, K+ influx partially dissipates ΔμH, thereby reducing the driving force for mitochondrial Ca2+ uptake [26, 92]. In addition, mKATP opening might modulate mitochondrial permeability transition indirectly, by activating cardioprotective signaling pathways. For instance, the mKATP opener diazoxide and the K+ ionophore valinomycin elicit PKCε-dependent phosphorylation of the mPTP by inducing mild ROS release from the matrix [27].
A limitation of these studies is that they are based on pharmacological inducers or inhibitors of mKATP opening, while investigations employing in vivo genetic interventions on the channel are hindered by the elusive molecular identity of mKATP. CCDC51, a protein with previously unknown function, was identified to interact with the known mKATP regulator ATP Binding Cassette protein 8 (ABCB8) to form a channel with mKATP-like properties [109]. Accordingly, genetic ablation of CCDC51 protects the heart against I/R injury [109]. An alternative and paradigm-shifting model posits that the F1/Fo-ATP synthase (complex V of the ETC) can use both H+ and K+ to synthesize ATP and represents the primary way for K+ to enter mitochondria [70]. By mediating this previously unrecognized K+ flux, the F1/Fo-ATP synthase might also function as a recruitable mKATP channel, whereby mitochondrial K+ influx and consequent increase in matrix volume result in desensitization of the mPTP [71]. Further expanding this model, the same authors discovered that two members of the survival proteins family, Bcl-xL and Mcl-1, modulate chemo-mechanical efficiency of the F1/Fo-ATP synthase to function as mKATP [71]. Altogether, further studies in vivo are warranted to confirm this suggested role of the ATP synthase as mKATP.
Mitochondrial connexin 43
Connexin 43 (Cx43) is a major component of gap junctions, i.e., intercellular channels that allow diffusion of ions and small molecules between adjacent cardiac myocytes. The cardioprotective effect of IPC, but not POC, is abolished in mouse hearts with a heterozygous deletion of Cx43 gene [58, 127]. The effect of IPC is also lost when isolated Cx43-deficient cardiac myocytes are exposed to simulated ischemia (i.e., hypoxia and acidosis) in vitro, suggesting that cell–cell communication mediated by Cx43 is not the sole mechanism for its protective effect [88]. Subsequent studies linked Cx43-dependent cardioprotection to its localization to subsarcolemmal mitochondria, where its expression is increased by IPC [15, 17]. Mechanistically, IPC induces Cx43 translocation to the IMM via the translocase of the outer membrane (TOM) complex [123]. However, the precise mechanisms by which mitochondrial translocation of Cx43 protects the heart remain unclear. Cx43 interacts with several mitochondrial proteins [33] and modulates critical mitochondrial functions, including complex I respiration [16], ROS formation [53], and K+ and Ca2+ uptake [99, 131], all of which potentially contribute to its cardioprotective activity. An additional layer of complexity arises from Cx43 phosphorylation by at least three kinases: mitogen-activated protein kinase (MAPK), protein kinase C (PKC), and casein kinase 1 (CK1), the latter of which is determinant for the cardioprotection by IPC, in the absence of any effect on ROS formation and mPTP opening in isolated mitochondria [60]. Altogether, these findings suggest a potentially multifaceted role of Cx43 in cardioprotection, which partly depends on its effects on mitochondrial ROS and Ca2+ and is modulated by CK1 phosphorylation via yet unknown mechanisms.
Role of mitochondrial Ca2+ and permeability transition in ischemic conditioning
The mPTP is considered a crucial effector of ischemic conditioning. The prevailing view posits that ischemic conditioning is mediated by transient (low-conductance) opening of the mPTP inducing a controlled release of Ca2+ from mitochondria, which alleviates Ca2+ overload and prevents sustained mPTP opening during prolonged ischemia [50]. Indeed, inhibitors of mPTP opening abolish the protection associated with both IPC and POC [6, 50]. Elucidating the precise mechanisms by which intracellular signaling cascades elicited by intermittent ischemia modulate mPTP opening remains an active area of research. One potential link between cardioprotective signaling and mPTP modulation is glycogen synthase kinase-3β (GSK-3β), which acts as a downstream target for multiple kinases and whose phosphorylation limits mitochondrial permeability transition [72]. Furthermore, activation of the Janus kinase 2 (JAK2)/signal transducer and activator of transcription 3 (STAT3) axis, which can be induced by a moderate increase in ROS production, mediates the cardioprotective effect of POC by preserving mitochondrial respiration and increasing mitochondrial Ca2+ retention capacity [59, 135, 136] (Fig. 4).
Modulation of Ca2+ exchange between the SR and mitochondria contributes to the cardioprotection afforded by ischemic conditioning. One mechanism implicated in IPC-mediated cardioprotection is a decrease in Ca2+ release from the SR via the type 2 ryanodine receptor (RyR2) upon reperfusion [133], which lowers mitochondrial Ca2+ levels and potentially also explains the reduction in reperfusion-induced arrhythmias observed with IPC [128]. Moreover, at the onset of reperfusion, rapid phosphorylation of phospholamban relieves its inhibition of SERCA contributing to SR and, consequently, mitochondrial Ca2+ overload. By delaying phospholamban phosphorylation, POC might promote cellular Ca2+ extrusion via the NCX and prevent exaggerate SR Ca2+ release [68] (Fig. 4).
Altogether, modulation of mPTP opening emerges as the end effector of cardioprotection induced by ischemic conditioning. As detailed in the following section, strategies that attenuate mitochondrial Ca2+ overload effectively limit mPTP opening and subsequent cardiomyocyte death during I/R injury. Conversely, mitochondrial ROS production has a Janus-faced role. While excessive ROS generation during reperfusion is a well-established mediator of cellular damage, a moderate ROS emission through mKATP and mPTP appears to be required for the cardioprotective effects of ischemic conditioning [50, 129]. Several key questions remain to be definitively addressed, e.g., the precise threshold of mitochondrial Ca2+ uptake that triggers irreversible overload and the specific molecular links between cardioprotective signaling cascades involved in ischemic conditioning and the modulation of mPTP opening.
Modulation of mitochondrial Ca2+ handling for cardioprotection
Pharmacological and genetic inhibition of the MCU
The central role played by mitochondrial Ca2+ overload in triggering permeability transition and consequent cardiac myocyte loss implies that preventing mitochondrial Ca2+ uptake represents a viable therapeutic approach to mitigate myocardial damage during reperfusion. Early studies using ruthenium red to block mitochondrial Ca2+ uptake during reperfusion showed reduced myocardial O2 consumption, preserved contractile function, and decreased myocardial damage in the isolated rat heart ex vivo [13]. Ruthenium red, however, inhibits the enzymatic activity of several other proteins involved in excitation–contraction coupling and Ca2+ handling, including SERCA and RyR2 [145], which could partly account for the observed benefit. Subsequent studies using the more specific MCU inhibitor ruthenium 360 (Ru360) confirmed the protective effect of blocking mitochondrial Ca2+ uptake on myocardial performance in isolated rat hearts [69]. Conversely, the MCU activator spermine blunts the protective effect of ischemic conditioning [135, 140].
The discovery of the molecular identity of the MCU complex paved the way to a series of studies that investigated the effects of genetic silencing of the MCU pore and regulatory components on I/R injury. Surprisingly, global constitutive knockout of the MCUa gene (Mcua-/-) in mice did not affect viability and cardiac function, but impaired exercise capacity [115]. Cardiac mitochondria isolated from Mcua-/- mice did not exhibit rapid, high-capacity mitochondrial Ca2+ uptake nor mPTP opening after prolonged exposure to high Ca2+ [115]. Nevertheless, Mcua-/- mice were not protected against cardiac I/R injury, and could not be protected by treatment with CsA [115]. In stark contrast, mice with conditional and cardiac myocyte-specific deletion of Mcua during adult life exhibited a 50% reduction in infarct size after I/R injury [85, 94]. Taken together, Mcua deletion before birth does not protect against I/R injury, whereas inducible Mcua silencing has a strong cardioprotective effect. Another potential approach to decrease MCUa levels is to induce degradation of Mcua mRNA with miR-25, which is protective in vitro but was never tested in vivo [114].
MCUb overexpression
MCUb is a paralog of MCUa and functions as a negative regulator of MCU flux by forming hetero-oligomers with MCUa, thus altering the stoichiometry and function of the uniporter [121]. Both constitutive and inducible cardiac myocyte-specific MCUb overexpression confer protection against ischemic injury [63, 86]. Under physiological conditions, the MCU complex of cardiac myocytes does not comprise MCUb subunits. Cardiac stressors such as ischemia induce MCUb expression and thereby decrease Ca2+ uptake via incorporation of MCUb in the MCU complex. However, MCUb induction occurs too late after the onset of ischemia to protect the heart from acute I/R injury [86]. Accordingly, mice lacking MCUb do not show differences in acute cardiac injury following I/R, but exhibit substantially worse left ventricular dilatation and contractile dysfunction 2 to 4 weeks after the ischemic event [63]. Altogether, the results of these studies indicate that, although MCUb upregulation occurs too late to mitigate acute cardiac myocyte loss during I/R, it represents an adaptive mechanism that prevents further cardiac myocyte death and scar expansion by reducing the sensitivity of mitochondria to elevated [Ca2+]c [86].
MICU1 modulation
MICU1 exerts an important regulatory role on MCU flux. Physiologically, the MICU1/MCU ratio determines tissue-specific differences in the [Ca2+]c threshold for Ca2+ uptake [111]. In myocardial samples from patients with heart failure, MICU1 levels and the MICU1/MCU ratio are increased [112], which might explain the differences in MCU current measured in mitoplasts (i.e., mitochondria stripped of the outer membrane) isolated from myocardial samples of heart failure patients [98]. On the other hand, mitochondrial localization of MICU1 mediated by the translocase of the outer membrane 70 (TOM70) is impaired after myocardial I/R, which decreases the MICU1/MCU ratio [138]. MICU1 silencing aggravates mitochondrial Ca2+ overload during I/R injury, thereby worsening apoptosis, cardiac remodeling, and contractile dysfunction [138]. On these grounds, it has been proposed that the gatekeeping function exerted by MICU1 on MCU flux can be exploited to protect the heart from I/R injury.
NCLX
One additional strategy to prevent Ca2+-induced cell death is to accelerate Ca2+ extrusion from the mitochondrial matrix by increasing NCLX activity. Cardiac-specific overexpression of the NCLX reduces superoxide production, prevents cardiac myocyte loss and preserves contractile function in mice subjected to I/R [93]. Vice versa, cardiac myocyte-specific deletion of NCLX gene in the adult mouse heart leads to mitochondrial Ca2+ overload and mPTP opening that result in severe systolic dysfunction and death within days after ablation of mitochondrial Ca2+ efflux [93]. Cardiac dysfunction and lethality induced by NCLX silencing are rescued by genetic deletion of CypD, which underscores the importance of mPTP opening as a mediator of cell death induced by mitochondrial Ca2+ overload [93].
Mitofusin 2
In principle, modulation of mitochondrial Ca2+ signals can also be achieved by regulating the structural and functional proximity of mitochondria and the SR. However, the role of proteins mediating SR-mitochondria tethering in cardiac physiology and pathology remains incompletely resolved. In particular, it is debated whether MFN2 functions as a tether or a spacer in the SR-mitochondria contact sites [37, 105]. In cell lines and mice in which MFN2 is genetically deleted, mitochondrial Ca2+ uptake and the Ca2+-mediated bioenergetic adaptation of cardiac myocytes are blunted, and mice are protected against Ca2+ overload-induced mPTP opening, indicating that the SR-mitochondria juxtaposition is decreased by MFN2 ablation [24, 105, 116]. This concept was subsequently challenged by Filadi and colleagues, who proposed that MFN2 ablation in fact potentiates interorganellar communication, thereby sensitizing MFN2-deficient cells to Ca2+ overload-mediated cell death [37]. These conflicting results might be explained by differences in genetic manipulation (inducible vs constitutive knockout) and cellular/animal models. An additional factor of complexity is the different effect of acute vs chronic modulation of SR/mitochondria tethering. The results of a recent study suggest that while acute changes in tethering may cause dysfunction, chronic enhancement of SR/mitochondria contact sites from early life induces an adaptive remodeling of the organelles that improves the resilience to stressors associated with mitochondrial Ca2+ overload, including adrenergic stimulation and I/R injury [107].
Conclusions
Taken together, mitochondrial Ca2+ handling is pivotal to the regulation of mitochondrial redox state, which impacts ATP production and mitochondrial antioxidative capacity. While in systolic heart failure, decreased mitochondrial Ca2+ uptake and steady-state Ca2+ accumulation hampers TCA cycle activation, which leads to oxidation of pyridine nucleotides and elevated ROS emission, excessive mitochondrial uptake during I/R injury provokes mPTP opening and cell death. Since most of the evidence for the role of the MCU and NCLX on cardiac physiology and pathology derives from genetic mouse models, and the physiological role and regulation of mitochondrial steady-state Ca2+ is very different between mice and human (due to a tenfold difference in resting heart rate), a note of caution is raised when interpreting results from genetic mouse studies. Nevertheless, considering the opposing defects in terms of mitochondrial Ca2+ uptake in systolic heart failure versus I/R injury, targeting mitochondrial Ca2+ handling therapeutically can be a double-edged sword, but may be successful when tailored to the respective pathological conditions. Additional research is warranted to better understand the fine-tuning of mitochondrial Ca2+ handling, its alterations in different pathological conditions, but also species (and potentially, sex-) dependent differences to further establish such therapies.
References
Abdallah Y, Kasseckert SA, Iraqi W, Said M, Shahzad T, Erdogan A, Neuhof C, Gündüz D, Schlüter K-D, Tillmanns H, Piper HM, Reusch HP, Ladilov Y (2011) Interplay between Ca2+ cycling and mitochondrial permeability transition pores promotes reperfusion-induced injury of cardiac myocytes. J Cell Mol Med 15:2478–2485. https://doi.org/10.1111/j.1582-4934.2010.01249.x
Akar FG, Aon MA, Tomaselli GF, O’Rourke B (2005) The mitochondrial origin of postischemic arrhythmias. J Clin Invest 115:3527–3535. https://doi.org/10.1172/JCI25371
Antoniel M, Jones K, Antonucci S, Spolaore B, Fogolari F, Petronilli V, Giorgio V, Carraro M, Di Lisa F, Forte M, Szabó I, Lippe G, Bernardi P (2018) The unique histidine in OSCP subunit of F-ATP synthase mediates inhibition of the permeability transition pore by acidic pH. EMBO Rep 19:257–268. https://doi.org/10.15252/embr.201744705
Aon MA, Cortassa S, Marbán E, O’Rourke B (2003) Synchronized whole cell oscillations in mitochondrial metabolism triggered by a local release of reactive oxygen species in cardiac myocytes. J Biol Chem 278:44735–44744. https://doi.org/10.1074/jbc.M302673200
Aon MA, Cortassa S, O’rourke B (2010) Redox-optimized ROS balance: a unifying hypothesis. Biochim Biophys Acta Bioenergetics 1797:865–877
Argaud L, Gateau-Roesch O, Raisky O, Loufouat J, Robert D, Ovize M (2005) Postconditioning inhibits mitochondrial permeability transition. Circulation 111:194–197. https://doi.org/10.1161/01.CIR.0000151290.04952.3B
Ashok D, Papanicolaou K, Sidor A, Wang M, Solhjoo S, Liu T, O’Rourke B (2023) Mitochondrial membrane potential instability on reperfusion after ischemia does not depend on mitochondrial Ca2+ uptake. J Biol Chem 299:104708
Austin S, Mekis R, Mohammed SEM, Scalise M, Wang W-A, Galluccio M, Pfeiffer C, Borovec T, Parapatics K, Vitko D et al (2022) TMBIM5 is the Ca2+/H+ antiporter of mammalian mitochondria. EMBO Rep 23:e54978
Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW et al (2005) Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 434:658–662
Balaban RS, Nemoto S, Finkel T (2005) Mitochondria, oxidants, and aging. Cell 120:483–495
Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL, Koteliansky V, Mootha VK (2011) Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476:341–345. https://doi.org/10.1038/nature10234
Bay J, Kohlhaas M, Maack C (2013) Intracellular Na+ and cardiac metabolism. J Mol Cell Cardiol 61:20–27
Benzi RH, Lerch R (1992) Dissociation between contractile function and oxidative metabolism in postischemic myocardium. attenuation by ruthenium red administered during reperfusion. Circ Res 71:567–576. https://doi.org/10.1161/01.res.71.3.567
Bertero E, Maack C (2018) Calcium signaling and reactive oxygen species in mitochondria. Circ Res 122:1460–1478
Boengler K, Dodoni G, Rodriguez-Sinovas A, Cabestrero A, Ruiz-Meana M, Gres P, Konietzka I, Lopez-Iglesias C, Garcia-Dorado D, Di Lisa F, Heusch G, Schulz R (2005) Connexin 43 in cardiomyocyte mitochondria and its increase by ischemic preconditioning. Cardiovasc Res 67:234–244. https://doi.org/10.1016/j.cardiores.2005.04.014
Boengler K, Ruiz-Meana M, Gent S, Ungefug E, Soetkamp D, Miro-Casas E, Cabestrero A, Fernandez-Sanz C, Semenzato M, Di Lisa F, Rohrbach S, Garcia-Dorado D, Heusch G, Schulz R (2012) Mitochondrial connexin 43 impacts on respiratory complex I activity and mitochondrial oxygen consumption. J Cell Mol Med 16:1649–1655. https://doi.org/10.1111/j.1582-4934.2011.01516.x
Boengler K, Stahlhofen S, van de Sand A, Gres P, Ruiz-Meana M, Garcia-Dorado D, Heusch G, Schulz R (2009) Presence of connexin 43 in subsarcolemmal, but not in interfibrillar cardiomyocyte mitochondria. Basic Res Cardiol 104:141–147. https://doi.org/10.1007/s00395-009-0007-5
Bolli R, Marbán E (1999) Molecular and cellular mechanisms of myocardial stunning. Physiol Rev 79:609–634
Bonora M, Giorgi C, Pinton P (2022) Molecular mechanisms and consequences of mitochondrial permeability transition. Nat Rev Mol Cell Biol 23:266–285. https://doi.org/10.1038/s41580-021-00433-y
Braunwald E, Kloner RA et al (1985) Myocardial reperfusion: a double-edged sword? J Clin Invest 76:1713–1719
De Brito OM, Scorrano L (2008) Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456:605–610
Carafoli E, Tiozzo R, Lugli G, Crovetti F, Kratzing C (1974) The release of calcium from heart mitochondria by sodium. J Mol Cell Cardiol 6:361–371
Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC (2003) Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol 160:189–200
Chen Y, Csordás G, Jowdy C, Schneider TG, Csordás N, Wang W, Liu Y, Kohlhaas M, Meiser M, Bergem S et al (2012) Mitofusin 2-containing mitochondrial-reticular microdomains direct rapid cardiomyocyte bioenergetic responses via interorganelle Ca2+ crosstalk. Circ Res 111:863–875
Chouchani ET, Pell VR, Gaude E, Aksentijević D, Sundier SY, Robb EL, Logan A, Nadtochiy SM, Ord ENJ, Smith AC, Eyassu F, Shirley R, Hu C-H, Dare AJ, James AM, Rogatti S, Hartley RC, Eaton S, Costa ASH, Brookes PS, Davidson SM, Duchen MR, Saeb-Parsy K, Shattock MJ, Robinson AJ, Work LM, Frezza C, Krieg T, Murphy MP (2014) Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 515:431–435. https://doi.org/10.1038/nature13909
Costa ADT, Garlid KD (2008) Intramitochondrial signaling: interactions among mitoKATP, PKC$\varepsilon$, ROS, and MPT. Am J Physiol Circ Physiol 295:H874–H882
Costa ADT, Jakob R, Costa CL, Andrukhiv K, West IC, Garlid KD (2006) The mechanism by which the mitochondrial ATP-sensitive K+ channel opening and H2O2 inhibit the mitochondrial permeability transition. J Biol Chem 281:20801–20808. https://doi.org/10.1074/jbc.M600959200
Cox DA, Matlib MA (1993) A role for the mitochondrial Na (+)-Ca2+ exchanger in the regulation of oxidative phosphorylation in isolated heart mitochondria. J Biol Chem 268:938–947
Crompton M, Ellinger H, Costi A (1988) Inhibition by cyclosporin A of a Ca2+-dependent pore in heart mitochondria activated by inorganic phosphate and oxidative stress. Biochem J 255:357
Dai D-F, Chen T, Szeto H, Nieves-Cintrón M, Kutyavin V, Santana LF, Rabinovitch PS (2011) Mitochondrial targeted antioxidant Peptide ameliorates hypertensive cardiomyopathy. J Am Coll Cardiol 58:73–82. https://doi.org/10.1016/j.jacc.2010.12.044
Dai D-F, Johnson SC, Villarin JJ, Chin MT, Nieves-Cintrón M, Chen T, Marcinek DJ, Dorn GW 2nd, Kang YJ, Prolla TA, Santana LF, Rabinovitch PS (2011) Mitochondrial oxidative stress mediates angiotensin II-induced cardiac hypertrophy and Galphaq overexpression-induced heart failure. Circ Res 108:837–846. https://doi.org/10.1161/CIRCRESAHA.110.232306
Davidson SM, Yellon DM, Murphy MP, Duchen MR (2012) Slow calcium waves and redox changes precede mitochondrial permeability transition pore opening in the intact heart during hypoxia and reoxygenation. Cardiovasc Res 93:445–453
Denuc A, Núñez E, Calvo E, Loureiro M, Miro-Casas E, Guarás A, Vázquez J, Garcia-Dorado D (2016) New protein-protein interactions of mitochondrial connexin 43 in mouse heart. J Cell Mol Med 20:794–803. https://doi.org/10.1111/jcmm.12792
Eltzschig HK, Eckle T (2011) Ischemia and reperfusion—from mechanism to translation. Nat Med 17:1391–1401
Fabiato A, Fabiato F (1978) Effects of pH on the myofilaments and the sarcoplasmic reticulum of skinned cells from cardiace and skeletal muscles. J Physiol 276:233–255
Fan M, Zhang J, Tsai C-W, Orlando BJ, Rodriguez M, Xu Y, Liao M, Tsai M-F, Feng L (2020) Structure and mechanism of the mitochondrial Ca2+ uniporter holocomplex. Nature 582:129–133
Filadi R, Greotti E, Turacchio G, Luini A, Pozzan T, Pizzo P (2015) Mitofusin 2 ablation increases endoplasmic reticulum-mitochondria coupling. Proc Natl Acad Sci U S A 112:E2174–E2181. https://doi.org/10.1073/pnas.1504880112
Finkel T, Menazza S, Holmström KM, Parks RJ, Liu J, Sun J, Liu J, Pan X, Murphy E (2015) The ins and outs of mitochondrial calcium. Circ Res 116:1810–1819
Flagg TP, Enkvetchakul D, Koster JC, Nichols CG (2010) Muscle KATP channels: recent insights to energy sensing and myoprotection. Physiol Rev 90:799–829. https://doi.org/10.1152/physrev.00027.2009
Fournier N, Ducet G, Crevat A (1987) Action of cyclosporine on mitochondrial calcium fluxes. J Bioenerg Biomembr 19:297–303
Gho BC, Schoemaker RG, van den Doel MA, Duncker DJ, Verdouw PD (1996) Myocardial protection by brief ischemia in noncardiac tissue. Circulation 94:2193–2200. https://doi.org/10.1161/01.cir.94.9.2193
Giacomello M, Drago I, Bortolozzi M, Scorzeto M, Gianelle A, Pizzo P, Pozzan T (2010) Ca2+ hot spots on the mitochondrial surface are generated by Ca2+ mobilization from stores, but not by activation of store-operated Ca2+ channels. Mol Cell 38:280–290
Giorgio V, Burchell V, Schiavone M, Bassot C, Minervini G, Petronilli V, Argenton F, Forte M, Tosatto S, Lippe G, Bernardi P (2017) Ca(2+) binding to F-ATP synthase β subunit triggers the mitochondrial permeability transition. EMBO Rep 18:1065–1076. https://doi.org/10.15252/embr.201643354
Giorgio V, von Stockum S, Antoniel M, Fabbro A, Fogolari F, Forte M, Glick GD, Petronilli V, Zoratti M, Szabó I, Lippe G, Bernardi P (2013) Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc Natl Acad Sci USA 110:5887–5892. https://doi.org/10.1073/pnas.1217823110
Griffiths EJ, Halestrap AP (1991) Further evidence that cyclosporin a protects mitochondria from calcium overload by inhibiting a matrix peptidyl-prolyl cis-trans isomerase Implications for the immunosuppressive and toxic effects of cyclosporine. Biochem. https://doi.org/10.1042/bj2740611
Griffiths EJ, Halestrap AP (1995) Mitochondrial non-specific pores remain closed during cardiac ischaemia, but open upon reperfusion. Biochem J 307:93–98
Grover GJ, D’Alonzo AJ, Parham CS, Darbenzio RB (1995) Cardioprotection with the KATP opener cromakalim is not correlated with ischemic myocardial action potential duration. J Cardiovasc Pharmacol 26:145–152. https://doi.org/10.1097/00005344-199507000-00023
Halestrap AP (2006) Calcium, mitochondria and reperfusion injury: a pore way to die. Biochem Soc Trans 34:232–237
Hartmann M, Decking UKM (1999) Blocking Na+–H+ exchange by cariporide reduces Na+-overload in ischemia and is cardioprotective. J Mol Cell Cardiol 31:1985–1995
Hausenloy D, Wynne A, Duchen M, Yellon D (2004) Transient mitochondrial permeability transition pore opening mediates preconditioning-induced protection. Circulation 109:1714–1717. https://doi.org/10.1161/01.CIR.0000126294.81407.7D
Hausenloy DJ, Schulz R, Girao H, Kwak BR, De Stefani D, Rizzuto R, Bernardi P, Di Lisa F (2020) Mitochondrial ion channels as targets for cardioprotection. J Cell Mol Med 24:7102–7114. https://doi.org/10.1111/jcmm.15341
Haworth RA, Hunter DR (1979) The Ca2+-induced membrane transition in mitochondria: II. Nature of the Ca2+ trigger site. Arch Biochem Biophys 195:460–467
Heinzel FR, Luo Y, Li X, Boengler K, Buechert A, García-Dorado D, Di Lisa F, Schulz R, Heusch G (2005) Impairment of diazoxide-induced formation of reactive oxygen species and loss of cardioprotection in connexin 43 deficient mice. Circ Res 97:583–586. https://doi.org/10.1161/01.RES.0000181171.65293.65
Hernandez-Resendiz S, Prakash A, Loo SJ, Semenzato M, Chinda K, Crespo-Avilan GE, Dam LC, Lu S, Scorrano L, Hausenloy DJ (2023) Targeting mitochondrial shape: at the heart of cardioprotection. Basic Res Cardiol 118:49. https://doi.org/10.1007/s00395-023-01019-9
Heusch G (2015) Molecular basis of cardioprotection: signal transduction in ischemic pre-, post-, and remote conditioning. Circ Res 116:674–699
Heusch G (2021) Myocardial stunning and hibernation revisited. Nat Rev Cardiol 18:522–536. https://doi.org/10.1038/s41569-021-00506-7
Heusch G, Andreadou I, Bell R, Bertero E, Botker H-E, Davidson SM, Downey J, Eaton P, Ferdinandy P, Gersh BJ, Giacca M, Hausenloy DJ, Ibanez B, Krieg T, Maack C, Schulz R, Sellke F, Shah AM, Thiele H, Yellon DM, Di Lisa F (2023) Health position paper and redox perspectives on reactive oxygen species as signals and targets of cardioprotection. Redox Biol 67:102894. https://doi.org/10.1016/j.redox.2023.102894
Heusch G, Büchert A, Feldhaus S, Schulz R (2006) No loss of cardioprotection by postconditioning in connexin 43-deficient mice. Basic Res Cardiol 101:354–356. https://doi.org/10.1007/s00395-006-0589-0
Heusch G, Musiolik J, Gedik N, Skyschally A (2011) Mitochondrial STAT3 activation and cardioprotection by ischemic postconditioning in pigs with regional myocardial ischemia/reperfusion. Circ Res 109:1302–1308. https://doi.org/10.1161/CIRCRESAHA.111.255604
Hirschhäuser C, Lissoni A, Görge PM, Lampe PD, Heger J, Schlüter K-D, Leybaert L, Schulz R, Boengler K (2021) Connexin 43 phosphorylation by casein kinase 1 is essential for the cardioprotection by ischemic preconditioning. Basic Res Cardiol 116:21. https://doi.org/10.1007/s00395-021-00861-z
Hunter DR, Haworth RA (1979) The Ca2+-induced membrane transition in mitochondria: I. The protective mechanisms Arch Biochem Biophys 195:453–459. https://doi.org/10.1016/0003-9861(79)90371-0
Hunter DR, Haworth RA, Southard JH (1976) Relationship between configuration, function, and permeability in calcium-treated mitochondria. J Biol Chem 251:5069–5077
Huo J, Lu S, Kwong JQ, Bround MJ, Grimes KM, Sargent MA, Brown ME, Davis ME, Bers DM, Molkentin JD (2020) MCUb induction protects the heart from postischemic remodeling. Circ Res 127:379–390. https://doi.org/10.1161/CIRCRESAHA.119.316369
Ichihara G, Katsumata Y, Sugiura Y, Matsuoka Y, Maeda R, Endo J, Anzai A, Shirakawa K, Moriyama H, Kitakata H, Hiraide T, Goto S, Ko S, Iwasawa Y, Sugai K, Daigo K, Goto S, Sato K, Yamada K-I, Suematsu M, Ieda M, Sano M (2023) MRP1-dependent extracellular release of glutathione induces cardiomyocyte ferroptosis after ischemia-reperfusion. Circ Res 133:861–876. https://doi.org/10.1161/CIRCRESAHA.123.323517
Ikeda Y, Shirakabe A, Maejima Y, Zhai P, Sciarretta S, Toli J, Nomura M, Mihara K, Egashira K, Ohishi M, Abdellatif M, Sadoshima J (2015) Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ Res 116:264–278. https://doi.org/10.1161/CIRCRESAHA.116.303356
Imahashi K, Pott C, Goldhaber JI, Steenbergen C, Philipson KD, Murphy E (2005) Cardiac-specific ablation of the Na+-Ca2+ exchanger confers protection against ischemia/reperfusion injury. Circ Res 97:916–921
Inoue I, Nagase H, Kishi K, Higuti T (1991) ATP-sensitive K+ channel in the mitochondrial inner membrane. Nature 352:244–247. https://doi.org/10.1038/352244a0
Inserte J, Hernando V, Ruiz-Meana M, Poncelas-Nozal M, Fernández C, Agulló L, Sartorio C, Vilardosa U, Garcia-Dorado D (2014) Delayed phospholamban phosphorylation in post-conditioned heart favours Ca2+ normalization and contributes to protection. Cardiovasc Res 103:542–553. https://doi.org/10.1093/cvr/cvu163
de Jesús G-R, Guerrero-Hernández A, Guerrero-Serna G, Rodríguez-Zavala JS, Zazueta C (2005) Inhibition of the mitochondrial calcium uniporter by the oxo-bridged dinuclear ruthenium amine complex (Ru360) prevents from irreversible injury in postischemic rat heart. FEBS J 272:3477–3488. https://doi.org/10.1111/j.1742-4658.2005.04771.x
Juhaszova M, Kobrinsky E, Zorov DB, Nuss HB, Yaniv Y, Fishbein KW, De Cabo R, Montoliu L, Gabelli SB, Aon MA et al (2022) ATP synthase K+-and H+-fluxes drive ATP synthesis and enable mitochondrial K+-“uniporter” function: II Ion and ATP synthase flux regulation. Function 3:zqac001
Juhaszova M, Kobrinsky E, Zorov DB, Nuss HB, Yaniv Y, Fishbein KW, De Cabo R, Montoliu L, Gabelli SB, Aon MA et al (2022) ATP synthase K+-and H+-fluxes drive ATP synthesis and enable mitochondrial K+-“uniporter” function I characterization of ion fluxes. Function 3:zqab065
Juhaszova M, Zorov DB, Kim S-H, Pepe S, Fu Q, Fishbein KW, Ziman BD, Wang S, Ytrehus K, Antos CL, Olson EN, Sollott SJ (2004) Glycogen synthase kinase-3beta mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J Clin Invest 113:1535–1549. https://doi.org/10.1172/JCI19906
Kaludercic N, Arusei RJ, Di Lisa F (2023) Recent advances on the role of monoamine oxidases in cardiac pathophysiology. Basic Res Cardiol 118:41. https://doi.org/10.1007/s00395-023-01012-2
Kamer KJ, Grabarek Z, Mootha VK (2017) High-affinity cooperative Ca2+ binding by MICU 1–MICU 2 serves as an on–off switch for the uniporter. EMBO Rep 18:1397–1411
Kim J-S, He L, Lemasters JJ (2003) Mitochondrial permeability transition: a common pathway to necrosis and apoptosis. Biochem Biophys Res Commun 304:463–470. https://doi.org/10.1016/s0006-291x(03)00618-1
Kimura S, Zhang G-X, Nishiyama A, Shokoji T, Yao L, Fan Y-Y, Rahman M, Suzuki T, Maeta H, Abe Y (2005) Role of NAD(P)H oxidase- and mitochondria-derived reactive oxygen species in cardioprotection of ischemic reperfusion injury by angiotensin II. Hypertens. https://doi.org/10.1161/01.HYP.0000163462.98381.7f
Kinnally KW, Campo ML, Tedeschi H (1989) Mitochondrial channel activity studied by patch-clamping mitoplasts. J Bioenerg Biomembr 21:497–506
Kohlhaas M, Liu T, Knopp A, Zeller T, Ong MF, Böhm M, O’Rourke B, Maack C (2010) Elevated cytosolic Na+ increases mitochondrial formation of reactive oxygen species in failing cardiac myocytes. Circulation 121:1606–1613
Kohlhaas M, Maack C (2010) Adverse bioenergetic consequences of Na+-Ca2+ exchanger–mediated Ca2+ influx in cardiac myocytes. Circulation 122:2273–2280
Kokoszka JE, Waymire KG, Levy SE, Sligh JE, Cai J, Jones DP, MacGregor GR, Wallace DC (2004) The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature 427:461–465. https://doi.org/10.1038/nature02229
Koval OM, Nguyen EK, Santhana V, Fidler TP, Sebag SC, Rasmussen TP, Mittauer DJ, Strack S, Goswami PC, Abel ED, Grumbach IM (2019) Loss of MCU prevents mitochondrial fusion in G(1)-S phase and blocks cell cycle progression and proliferation. Sci Signal. https://doi.org/10.1126/scisignal.aav1439
Kowaltowski AJ, Menezes-Filho SL, Assali E, Gonçalves IG, Abreu P, Miller N, Nolasco P, Laurindo FRM, Bruni-Cardoso A, Shirihai O (2019) Mitochondrial morphology regulates organellar Ca2+ uptake and changes cellular Ca2+ homeostasis. bioRxiv 12:624981
Kröller-Schön S, Steven S, Kossmann S, Scholz A, Daub S, Oelze M, Xia N, Hausding M, Mikhed Y, Zinssius E, Mader M, Stamm P, Treiber N, Scharffetter-Kochanek K, Li H, Schulz E, Wenzel P, Münzel T, Daiber A (2014) Molecular mechanisms of the crosstalk between mitochondria and NADPH oxidase through reactive oxygen species-studies in white blood cells and in animal models. Antioxid Redox Signal 20:247–266. https://doi.org/10.1089/ars.2012.4953
Kübler W, Haass M (1996) Cardioprotection: definition, classification, and fundamental principles. Heart 75:330
Kwong JQ, Lu X, Correll RN, Schwanekamp JA, Vagnozzi RJ, Sargent MA, York AJ, Zhang J, Bers DM, Molkentin JD (2015) The mitochondrial calcium uniporter selectively matches metabolic output to acute contractile stress in the heart. Cell Rep 12:15–22
Lambert JP, Luongo TS, Tomar D, Jadiya P, Gao E, Zhang X, Lucchese AM, Kolmetzky DW, Shah NS, Elrod JW (2019) MCUB regulates the molecular composition of the mitochondrial calcium uniporter channel to limit mitochondrial calcium overload during stress. Circulation 140:1720–1733. https://doi.org/10.1161/CIRCULATIONAHA.118.037968
Legesse-Miller A, Massol RH, Kirchhausen T (2003) Constriction and Dnm1p recruitment are distinct processes in mitochondrial fission. Mol Biol Cell 14:1953–1963. https://doi.org/10.1091/mbc.e02-10-0657
Li X, Heinzel FR, Boengler K, Schulz R, Heusch G (2004) Role of connexin 43 in ischemic preconditioning does not involve intercellular communication through gap junctions. J Mol Cell Cardiol 36:161–163. https://doi.org/10.1016/j.yjmcc.2003.10.019
Liang BT (1996) Direct preconditioning of cardiac ventricular myocytes via adenosine A1 receptor and KATP channel. Am J Physiol 271:H1769–H1777. https://doi.org/10.1152/ajpheart.1996.271.5.H1769
Liu JC, Liu J, Holmström KM, Menazza S, Parks RJ, Fergusson MM, Yu Z-X, Springer DA, Halsey C, Liu C et al (2016) MICU1 serves as a molecular gatekeeper to prevent in vivo mitochondrial calcium overload. Cell Rep 16:1561–1573
Liu T, Takimoto E, Dimaano VL, DeMazumder D, Kettlewell S, Smith G, Sidor A, Abraham TP, O’Rourke B (2014) Inhibiting mitochondrial Na+/Ca2+ exchange prevents sudden death in a Guinea pig model of heart failure. Circ Res 115:44–54
Liu Y, Sato T, O’Rourke B, Marban E (1998) Mitochondrial ATP-dependent potassium channels: novel effectors of cardioprotection? Circulation 97:2463–2469. https://doi.org/10.1161/01.cir.97.24.2463
Luongo TS, Lambert JP, Gross P, Nwokedi M, Lombardi AA, Shanmughapriya S, Carpenter AC, Kolmetzky D, Gao E, Van Berlo JH et al (2017) The mitochondrial Na+/Ca2+ exchanger is essential for Ca2+ homeostasis and viability. Nature 545:93–97
Luongo TS, Lambert JP, Yuan A, Zhang X, Gross P, Song J, Shanmughapriya S, Gao E, Jain M, Houser SR et al (2015) The mitochondrial calcium uniporter matches energetic supply with cardiac workload during stress and modulates permeability transition. Cell Rep 12:23–34
Maack C, Böhm M (2011) Targeting mitochondrial oxidative stress in heart failure throttling the afterburner. J Am Coll Cardiol 58:83–86
Maack C, Cortassa S, Aon MA, Ganesan AN, Liu T, O’Rourke B (2006) Elevated cytosolic Na+ decreases mitochondrial Ca2+ uptake during excitation-contraction coupling and impairs energetic adaptation in cardiac myocytes. Circ Res 99:172–182. https://doi.org/10.1161/01.RES.0000232546.92777.05
McCormack JG, Denton RM (1993) The role of intramitochondrial Ca2+ in the regulation of oxidative phosphorylation in mammalian tissues. Biochem Soc Trans 21:793–799
Michels G, Khan IF, Endres-Becker J, Rottlaender D, Herzig S, Ruhparwar A, Wahlers T, Hoppe UC (2009) Regulation of the human cardiac mitochondrial Ca 2+ uptake by 2 different voltage-gated Ca 2+ channels. Circulation 119:2435–2443. https://doi.org/10.1161/CIRCULATIONAHA.108.835389
Miro-Casas E, Ruiz-Meana M, Agullo E, Stahlhofen S, Rodríguez-Sinovas A, Cabestrero A, Jorge I, Torre I, Vazquez J, Boengler K, Schulz R, Heusch G, Garcia-Dorado D (2009) Connexin43 in cardiomyocyte mitochondria contributes to mitochondrial potassium uptake. Cardiovasc Res 83:747–756. https://doi.org/10.1093/cvr/cvp157
Mitchell P (2011) Chemiosmotic coupling in oxidative and photosynthetic phosphorylation. Biochim Biophys Acta Bioenerget 1807:1507–1538
Miwa S, Brand MD (2003) Mitochondrial matrix reactive oxygen species production is very sensitive to mild uncoupling. Biochem Soc Trans 31:1300–1301. https://doi.org/10.1042/bst0311300
Murphy MP (2009) How mitochondria produce reactive oxygen species. Biochem J 417:1–13
Murry CE, Jennings RB, Reimer KA (1986) Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation 74:1124–1136. https://doi.org/10.1161/01.CIR.74.5.1124
Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, Inohara H, Kubo T, Tsujimoto Y (2005) Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature 434:652–658. https://doi.org/10.1038/nature03317
Naon D, Zaninello M, Giacomello M, Varanita T, Grespi F, Lakshminaranayan S, Serafini A, Semenzato M, Herkenne S, Hernández-Alvarez MI, Zorzano A, De Stefani D, Dorn GW 2nd, Scorrano L (2016) Critical reappraisal confirms that Mitofusin 2 is an endoplasmic reticulum-mitochondria tether. Proc Natl Acad Sci U S A 113:11249–11254. https://doi.org/10.1073/pnas.1606786113
Neginskaya MA, Solesio ME, Berezhnaya EV, Amodeo GF, Mnatsakanyan N, Jonas EA, Pavlov EV (2019) ATP synthase C-subunit-deficient mitochondria have a small cyclosporine a-sensitive channel, but lack the permeability transition pore. Cell Rep 26:11-17.e2. https://doi.org/10.1016/j.celrep.2018.12.033
Nichtová Z, Fernandez-Sanz C, De La Fuente S, Yuan Y, Hurst S, Lanvermann S, Tsai H-Y, Weaver D, Baggett A, Thompson C et al (2023) Enhanced mitochondria-SR tethering triggers adaptive cardiac muscle remodeling. Circ Res 132:e171–e187
Ong S-B, Subrayan S, Lim SY, Yellon DM, Davidson SM, Hausenloy DJ (2010) Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation 121:2012–2022. https://doi.org/10.1161/CIRCULATIONAHA.109.906610
Paggio A, Checchetto V, Campo A, Menabò R, Di Marco G, Di Lisa F, Szabo I, Rizzuto R, De Stefani D (2019) Identification of an ATP-sensitive potassium channel in mitochondria. Nature 572:609–613
Paillard M, Csordás G, Huang K-T, Várnai P, Joseph SK, Hajnóczky G (2018) MICU1 interacts with the D-ring of the MCU pore to control its Ca2+ flux and sensitivity to Ru360. Mol Cell 72:778–785
Paillard M, Csordás G, Szanda G, Golenár T, Debattisti V, Bartok A, Wang N, Moffat C, Seifert EL, Spät A, Hajnóczky G (2017) Tissue-specific mitochondrial decoding of cytoplasmic Ca2+ signals is controlled by the stoichiometry of MICU1/2 and MCU. Cell Rep 18:2291–2300. https://doi.org/10.1016/j.celrep.2017.02.032
Paillard M, Huang K-T, Weaver D, Lambert JP, Elrod JW, Hajnóczky G (2022) Altered composition of the mitochondrial Ca2+ uniporter in the failing human heart. Cell Calcium 105:102618
Palty R, Silverman WF, Hershfinkel M, Caporale T, Sensi SL, Parnis J, Nolte C, Fishman D, Shoshan-Barmatz V, Herrmann S et al (2010) NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc Natl Acad Sci 107:436–441
Pan L, Huang B-J, Ma X-E, Wang S-Y, Feng J, Lv F, Liu Y, Liu Y, Li C-M, Liang D-D, Li J, Xu L, Chen Y-H (2015) MiR-25 protects cardiomyocytes against oxidative damage by targeting the mitochondrial calcium uniporter. Int J Mol Sci 16:5420–5433. https://doi.org/10.3390/ijms16035420
Pan X, Liu J, Nguyen T, Liu C, Sun J, Teng Y, Fergusson MM, Rovira II, Allen M, Springer DA et al (2013) The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat Cell Biol 15:1464–1472
Papanicolaou KN, Khairallah RJ, Ngoh GA, Chikando A, Luptak I, O’Shea KM, Riley DD, Lugus JJ, Colucci WS, Lederer WJ et al (2011) Mitofusin-2 maintains mitochondrial structure and contributes to stress-induced permeability transition in cardiac myocytes. Mol Cell Biol 31:1309–1328
Piao L, Fang Y-H, Fisher M, Hamanaka RB, Ousta A, Wu R, Mutlu GM, Garcia AJ 3rd, Archer SL, Sharp WW (2024) Dynamin-related protein 1 is a critical regulator of mitochondrial calcium homeostasis during myocardial ischemia/reperfusion injury. FASEB J Off Publ Fed Am Soc Exp Biol 38:e23379. https://doi.org/10.1096/fj.202301040RR
Pinke G, Zhou L, Sazanov LA (2020) Cryo-EM structure of the entire mammalian F-type ATP synthase. Nat Struct Mol Biol 27:1077–1085. https://doi.org/10.1038/s41594-020-0503-8
Pinton P, Rimessi A, Marchi S, Orsini F, Migliaccio E, Giorgio M, Contursi C, Minucci S, Mantovani F, Wieckowski MR, Del Sal G, Pelicci PG, Rizzuto R (2007) Protein kinase C beta and prolyl isomerase 1 regulate mitochondrial effects of the life-span determinant p66Shc. Science 315:659–663. https://doi.org/10.1126/science.1135380
Prag HA, Murphy MP, Krieg T (2023) Preventing mitochondrial reverse electron transport as a strategy for cardioprotection. Basic Res Cardiol 118:34. https://doi.org/10.1007/s00395-023-01002-4
Raffaello A, De Stefani D, Sabbadin D, Teardo E, Merli G, Picard A, Checchetto V, Moro S, Szabò I, Rizzuto R (2013) The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J 32:2362–2376. https://doi.org/10.1038/emboj.2013.157
Rodr\’\iguez-Prados M, Berezhnaya E, Castromonte MT, Menezes-Filho SL, Paillard M, Hajnóczky G, (2023) MICU1 occludes the mitochondrial calcium uniporter in divalent-free conditions. Proc Natl Acad Sci 120:e2218999120
Rodriguez-Sinovas A, Boengler K, Cabestrero A, Gres P, Morente M, Ruiz-Meana M, Konietzka I, Miró E, Totzeck A, Heusch G, Schulz R, Garcia-Dorado D (2006) Translocation of connexin 43 to the inner mitochondrial membrane of cardiomyocytes through the heat shock protein 90-dependent TOM pathway and its importance for cardioprotection. Circ Res 99:93–101. https://doi.org/10.1161/01.RES.0000230315.56904.de
Ruiz-Meana M, Abellán A, Miró-Casas E, Agulló E, Garcia-Dorado D (2009) Role of sarcoplasmic reticulum in mitochondrial permeability transition and cardiomyocyte death during reperfusion. Am J Physiol Heart Circ Physiol 297:H1281–H1289. https://doi.org/10.1152/ajpheart.00435.2009
Sancak Y, Markhard AL, Kitami T, Kovács-Bogdán E, Kamer KJ, Udeshi ND, Carr SA, Chaudhuri D, Clapham DE, Li AA et al (2013) EMRE is an essential component of the mitochondrial calcium uniporter complex. Science 342:1379–1382
Sato T, Sasaki N, Seharaseyon J, O’Rourke B, Marbán E (2000) Selective pharmacological agents implicate mitochondrial but not sarcolemmal K(ATP) channels in ischemic cardioprotection. Circulation 101:2418–2423. https://doi.org/10.1161/01.cir.101.20.2418
Schwanke U, Konietzka I, Duschin A, Li X, Schulz R, Heusch G (2002) No ischemic preconditioning in heterozygous connexin43-deficient mice. Am J Physiol Heart Circ Physiol 283:H1740–H1742. https://doi.org/10.1152/ajpheart.00442.2002
Shiki K, Hearse DJ (1987) Preconditioning of ischemic myocardium: reperfusion-induced arrhythmias. Am J Physiol 253:H1470–H1476. https://doi.org/10.1152/ajpheart.1987.253.6.H1470
Skyschally A, Schulz R, Gres P, Korth H-G, Heusch G (2003) Attenuation of ischemic preconditioning in pigs by scavenging of free oxyradicals with ascorbic acid. Am J Physiol Heart Circ Physiol 284:H698-703. https://doi.org/10.1152/ajpheart.00693.2002
Smith GL, Donoso P, Bauer CJ, Eisner DA (1993) Relationship between intracellular pH and metabolite concentrations during metabolic inhibition in isolated ferret heart. J Physiol 472:11–22
Srisakuldee W, Makazan Z, Nickel BE, Zhang F, Thliveris JA, Pasumarthi KBS, Kardami E (2014) The FGF-2-triggered protection of cardiac subsarcolemmal mitochondria from calcium overload is mitochondrial connexin 43-dependent. Cardiovasc Res 103:72–80. https://doi.org/10.1093/cvr/cvu066
De Stefani D, Raffaello A, Teardo E, Szabò I, Rizzuto R (2011) A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476:336–340
Tani M, Asakura Y, Hasegawa H, Shinmura K, Ebihara Y, Nakamura Y (1996) Effect of preconditioning on ryanodine-sensitive Ca2+ release from sarcoplasmic reticulum of rat heart. Am J Physiol 271:H876–H881. https://doi.org/10.1152/ajpheart.1996.271.3.H876
Tanveer A, Virji S, Andreeva L, Totty NF, Hsuan JJ, Ward JM, Crompton M (1996) Involvement of cyclophilin D in the activation of a mitochondrial pore by Ca2+ and oxidant stress. Eur J Biochem 238:166–172
Wu L, Tan J-L, Chen Z-Y, Huang G (2019) Cardioprotection of post-ischemic moderate ROS against ischemia/reperfusion via STAT3-induced the inhibition of MCU opening. Basic Res Cardiol 114:39. https://doi.org/10.1007/s00395-019-0747-9
Wu L, Tan J-L, Wang Z-H, Chen Y-X, Gao L, Liu J-L, Shi Y-H, Endoh M, Yang H-T (2015) ROS generated during early reperfusion contribute to intermittent hypobaric hypoxia-afforded cardioprotection against postischemia-induced Ca(2+) overload and contractile dysfunction via the JAK2/STAT3 pathway. J Mol Cell Cardiol 81:150–161. https://doi.org/10.1016/j.yjmcc.2015.02.015
Wu S, Lu Q, Wang Q, Ding Y, Ma Z, Mao X, Huang K, Xie Z, Zou M-H (2017) Binding of FUN14 domain containing 1 with inositol 1, 4, 5-trisphosphate receptor in mitochondria-associated endoplasmic reticulum membranes maintains mitochondrial dynamics and function in hearts in vivo. Circulation 136:2248–2266
Xue Q, Pei H, Liu Q, Zhao M, Sun J, Gao E, Ma X, Tao L (2017) MICU1 protects against myocardial ischemia/reperfusion injury and its control by the importer receptor Tom70. Cell Death Dis 8:e2923. https://doi.org/10.1038/cddis.2017.280
Yao Z, Gross GJ (1994) Activation of ATP-sensitive potassium channels lowers threshold for ischemic preconditioning in dogs. Am J Physiol 267:H1888–H1894. https://doi.org/10.1152/ajpheart.1994.267.5.H1888
Zhang S-Z, Gao Q, Cao C-M, Bruce IC, Xia Q (2006) Involvement of the mitochondrial calcium uniporter in cardioprotection by ischemic preconditioning. Life Sci 78:738–745. https://doi.org/10.1016/j.lfs.2005.05.076
Zhang Y, Murugesan P, Huang K, Cai H (2020) NADPH oxidases and oxidase crosstalk in cardiovascular diseases: novel therapeutic targets. Nat Rev Cardiol 17:170–194. https://doi.org/10.1038/s41569-019-0260-8
Zhao Z-Q, Corvera JS, Halkos ME, Kerendi F, Wang N-P, Guyton RA, Vinten-Johansen J (2003) Inhibition of myocardial injury by ischemic postconditioning during reperfusion: comparison with ischemic preconditioning. Am J Physiol Heart Circ Physiol 285:H579–H588. https://doi.org/10.1152/ajpheart.01064.2002
Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ (2000) Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med 192:1001–1014. https://doi.org/10.1084/jem.192.7.1001
Zorov DB, Juhaszova M, Sollott SJ (2014) Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev 94:909–950. https://doi.org/10.1152/physrev.00026.2013
Zucchi R, Ronca-Testoni S (1997) The sarcoplasmic reticulum Ca2+ channel/ryanodine receptor: modulation by endogenous effectors, drugs and disease states. Pharmacol Rev 49:1–51
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest related to this work.
Additional information
This article is part of the topical collection “Mitochondria at the heart of cardioprotection”.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bertero, E., Popoiu, TA. & Maack, C. Mitochondrial calcium in cardiac ischemia/reperfusion injury and cardioprotection. Basic Res Cardiol 119, 569–585 (2024). https://doi.org/10.1007/s00395-024-01060-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00395-024-01060-2