
Abstract
Neutrophils are not only involved in immune defense against infection but also contribute to the exacerbation of tissue damage after ischemia and reperfusion. We have previously shown that genetic ablation of regulatory Gαi proteins in mice has both protective and deleterious effects on myocardial ischemia reperfusion injury (mIRI), depending on which isoform is deleted. To deepen and analyze these findings in more detail the contribution of Gαi2 proteins in resident cardiac vs circulating blood cells for mIRI was first studied in bone marrow chimeras. In fact, the absence of Gαi2 in all blood cells reduced the extent of mIRI (22,9% infarct size of area at risk (AAR) Gnai2−/− → wt vs 44.0% wt → wt; p < 0.001) whereas the absence of Gαi2 in non-hematopoietic cells increased the infarct damage (66.5% wt → Gnai2−/− vs 44.0% wt → wt; p < 0.001). Previously we have reported the impact of platelet Gαi2 for mIRI. Here, we show that infarct size was substantially reduced when Gαi2 signaling was either genetically ablated in neutrophils/macrophages using LysM-driven Cre recombinase (AAR: 17.9% Gnai2fl/fl LysM-Cre+/tg vs 42.0% Gnai2fl/fl; p < 0.01) or selectively blocked with specific antibodies directed against Gαi2 (AAR: 19.0% (anti-Gαi2) vs 49.0% (IgG); p < 0.001). In addition, the number of platelet-neutrophil complexes (PNCs) in the infarcted area were reduced in both, genetically modified (PNCs: 18 (Gnai2fl/fl; LysM-Cre+/tg) vs 31 (Gnai2fl/fl); p < 0.001) and in anti-Gαi2 antibody-treated (PNCs: 9 (anti-Gαi2) vs 33 (IgG); p < 0.001) mice. Of note, significant infarct-limiting effects were achieved with a single anti-Gαi2 antibody challenge immediately prior to vessel reperfusion without affecting bleeding time, heart rate or cellular distribution of neutrophils. Finally, anti-Gαi2 antibody treatment also inhibited transendothelial migration of human neutrophils (25,885 (IgG) vs 13,225 (anti-Gαi2) neutrophils; p < 0.001), collectively suggesting that a therapeutic concept of functional Gαi2 inhibition during thrombolysis and reperfusion in patients with myocardial infarction should be further considered.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cardiovascular diseases remain still the leading cause of death worldwide [96, 85]. Among them, 8.9 million people worldwide died from ischemic heart disease in 2019, with acute myocardial infarction (MI), for example, responsible for more than 4 million deaths in Europe and northern Asia, and more than 2.4 million deaths in the US each year [79]. The current standard treatment for MI is based on early myocardial reperfusion through thrombolytic therapy or primary percutaneous coronary intervention [79]. Paradoxically, the restoration of perfusion in the ischemic myocardium causes tissue damage leading to myocardial ischemia reperfusion injury (mIRI) [4, 102]. mIRI manifests from a number of pathomechanisms, including formation of reactive oxygen species (ROS), intracellular calcium overload, edema, vasomotion, microembolization, stasis, intravascular cellular aggregates, and capillary destruction and rupture [14, 31, 32, 45]. So far, efforts to translate effective strategies in animal models to the prevention or treatment of mIRI in patients have been less successful [33].
Neutrophils are among the first cells recruited to sites of tissue damage, where they contribute to tissue repair through the removal of cell debris, to the release of proresolving mediators, and to the discharge of microvesicles but they may also damage remaining healthy tissue [52, 64, 92, 95]. In addition, neutrophils are not only essential for removing pathogens or cell debris but also for the resolution of inflammation promoting return to homeostasis [78]. Numerous studies suggest a detrimental role for neutrophils in mIRI. Activated neutrophils exacerbate tissue damage through the release of ROS, proteolytic enzymes, and pro-inflammatory cytokines, resulting in a positive feedback loop of neutrophil recruitment and activation [10, 92]. In addition, neutrophils might contribute to the no-reflow phenomenon, in which the blockage of capillaries by blood cells impedes the reperfusion of ischemic tissue [10, 32]. Massive migration of neutrophils through the endothelium can lead to loss of tight junctions and consequently to dysfunction of the endothelial barrier [91, 98].
Interestingly, platelet-neutrophil interactions play an important role in a wide range of inflammatory conditions, including bacterial infection and sepsis, pulmonary inflammatory and coronary syndromes [62, 89]. Mechanistically, the formation of platelet neutrophil complexes (PNCs) leads to activation of both cell types triggering the release of cytokines, increased production of ROS, as well as expression of adhesion molecules and cell surface receptors [76]. This activation not only facilitates extravasation of neutrophils and their recruitment to the site of inflammation but also to the site of ischemia, also known as sterile imnflammation or alternatively the ischemic inflammatory response (IIR) [29, 75, 83]. Thus, PNCs may contribute to both the thrombotic and inflammatory processes triggered by mIRI [75, 89], which is also reflected by increased numbers of circulating PNCs in the blood of patients with acute coronary syndrome [58, 65, 74]. Furthermore, the number of PNCs in ischemic myocardial tissue correlated with the extent of IIR and mIRI in animal studies [29, 50].
The numerous heterotrimeric G proteins are subdivided according to the degree of homology of the α-subunits. The Gi protein family includes three closely related members, Gαi1–3, which are characterized by their sensitivity towards pertussis toxin [42, 71]. The Gαi1–3 isoforms share 85–95% of amino acid sequence identity and show overlapping expression patterns. While Gαi1 is found primarily in the brain, Gαi2 and Gαi3 are abundantly expressed in the heart, the endothelium, and the immune system [20]. Many biologic functions of leukocytes, including adhesion and chemotaxis, occur through Gαi-mediated signaling [12, 43, 72, 94]. For neutrophilic granulocytes, Gαi2 has been shown to be indispensable for their accumulation at different sites of inflammation and for chemokine-induced intravascular arrest [57, 100, 103]. We have previously shown that inhibition of Gαi2 signaling in platelets has cardio-protective effects [20] and leads to reduced PNC numbers in the ischemic tissue. However, the functional role of Gαi2 specifically in neutrophils remained unclear.
The aim of the present study is to examine the inactivation or inhibition of functional Gαi2 signaling in neutrophils/macrophages in a mouse model of mIRI by applying three independent and complementary experimental approaches: (1) investigating bone marrow (BM) chimeras deficient in Gnai2 only in the hematopoietic compartment, (2) studying mouse mutants deficient in Gnai2 in neutrophils/macrophages using LysM-driven Cre recombinase, and (3) pharmacologic targeting of Gαi2 by i.v.-administered Gαi2-specific antibodies.
Materials and methods
Mice
Global Gnai2-deficient (Gnai2–/–) mice were backcrossed to a C57BL/6N genetic background for more than ten generations and kept in isolated ventilated cages, and were studied in comparison to wild-type (wt) littermate controls [20]. A specific deletion of Gnai2 in granulocytes/macrophages (Gnai2nko) was achieved by crossing Gnai2fl/fl and LysM-Cre+/tg mice [16, 54] and compared to Gnai2fl/fl; LysM-Cre+/+ controls (ctrl). These mice were backcrossed for more than ten generations to a C57BL/6N genetic background excluding genes from the initial 129/Ola background. In accordance with the 3R principle, we did not investigate additional controls, such as Gnai2+/+; LysM-Cre+/tg or Gnai2fl/+; LysM-Cre+/tg mice.
C57BL/6N mice receiving antibody treatment (wtIgG or wtab) were obtained from Charles River (Wilmington, MA, USA). Mice of either sex were used at 8–14 weeks of age. All animal experiments were performed according to the ARRIVE guidelines and have been approved by the Animal Care and Use Committee of the local government (Regierungspräsidium Tübingen) and were in accordance with the German animal laws.
Generation of bone marrow chimeric mice
BM chimeric mice were generated as previously described [50]. Briefly, donor mice (8 to 10 weeks old, 20 to 25 g body weight (b.w.)) were sacrificed, and BM was harvested by flushing tibia and femur. Then cells were centrifuged at 400 × g for 5 min, resuspended and counted. Recipient mice (8 to 10 weeks of age, 20 to 25 g b.w.) were irradiated with a total dose of 10 Gy. After irradiation, the recipients were reconstituted with 1 × 107 BM cells in 150 μl of 0.9% sodium chloride via tail vein injection. In the first 2 weeks after BM transplantation chimera were fed with water containing tetracycline (100 mg/L) and housed in microisolators for 6–8 weeks before experimentation. In total, four groups of BM chimeric mice were generated: (1) BM cells from wt mice were transplanted into irradiated Gnai2-deficient mice [wt → Gnai2−/−] and (2) vice versa [Gnai2−/− → wt]. In addition, BM cells were transplanted (3) from wt mice into irradiated wt mice [wt → wt] and (4) from Gnai2-deficient mice into irradiated Gnai2-deficient mice [Gnai2−/− → Gnai2−/−] as controls (Supplemental Fig. 1A). The efficiency of BM transplantation was verified by immunoblotting (Supplemental Fig. 1B).
Murine model of myocardial ischemia
The previously described hanging weight system was applied as a well-established and routinely used murine model of mIRI [20, 48, 49]. Briefly, anesthetized mice (pentobarbital, 80 mg/kg b.w.) (Sigma-Aldrich, St. Louis, MO, USA) were placed on a temperature-controlled heating table. Animals were intubated, ventilated and left parasternal thoracotomy was maintained to display the left coronary artery. Coronary artery occlusion was performed for 1 hour and subsequently, the hearts were exposed to a 2-hour reperfusion period. Mice receiving Gαi2-specific antibodies (for details of the antibody see Sect. “Immunoblot analysis of Gαi2 expression”) were treated with the antibodies 5 min before onset of reperfusion. Afterward blood was collected for detection of troponin I levels and hearts were removed for subsequent analyses. The extent of infarct size was determined according to the state of the art as defined by current guidelines by calculating the percentage of infarction compared with the area at risk (AAR) [13, 59,60,61]. For this purpose, a double staining technique using triphenyl tetrazolium chloride (TTC) to mark vital and necrotic tissue and Evans Blue staining to negatively mark the AAR was used [27]. Images were taken with an Olympus SZX12 microscope. Planimetric determination of infarct size and AAR was performed using ImageJ Software.
Electrocardiogram (ECG) records
Mice were anesthetized with pentobarbital-Na+ (70 mg/kg i.p.) and were positioned on a small animal physiological monitoring system (HPMS, Harvard Apparatus, Holliston, MA, USA) equipped with a tablet (No. 75-1501). Temperature was kept at 37.5 °C throughout the experiment using a rectal probe connected to an integrated heating platform. Paws were placed on gel coated ECG surface electrodes and recordings were performed according to the manufacturer’s instruction manual. Baseline ECG was measured for 5 min, then either anti-Gαi1/i2 (1 µg/mouse) or IgG control (1 µg/mouse) antibodies were administered by i.v. injection. After 5 min, postinjection ECG responses were monitored continuously for a period of 2 min. The experimenter who evaluated the recordings had no information about whether anti-Gαi1/i2 or IgG control antibodies were used. Recorded data were converted to CSV format and signals were displayed in Excel.
Troponin I measurement
To measure troponin I in the serum of ischemic mice, blood was collected after reperfusion. Serum from chimeric mice was analyzed with the ADVIA Centaur Immunoassay system using the TnI Ultra chemoliuminescent assay (Siemens Healthcare Diagnostics, Fehrenwald, Germany). Serum from mice subjected to other experiments was analyzed using the ELISA kit SEA478Mu (for murine cardiac troponin I Type 3 (TNNI3) as recommended by the manufacturer’s instructions (Cloude-Clone Corp., Houston, TX, USA).
Detection of PNCs in heart tissue
To detect PNCs within the AAR, hearts were removed after one hour ischemia followed by one hour of reperfusion and processed for further analyses as described previously [20]. Heart sections of 5 µm thickness were embedded in Tissue-Tek (Fisher Scientific, Schwerte, Germany). Immunohistochemical staining was performed with Vectastain ABC Kit (Linaris, Wertheim, Germany). After inhibiting the non-specific binding sites with Avidin blocking solution (Vector, Burlingame, CA, USA), sections were incubated with primary antibodies (rabbit anti-mouse CD41; AbD Serotec, Düsseldorf, Germany) overnight at 4 °C. Tissue sections were then incubated with biotinylated anti-rabbit immunoglobulin for 1 hour followed by Vectastain ABC reagent for 30 min, and then developed using 3,3′-diaminodbenzidine (DAB) substrate (Fisher Scientific, Schwerte, Germany). For neutrophil staining, the procedure was repeated using rat anti-mouse neutrophil antibodies (rat anti-mouse Ly-6-B2/clone 4; AbD Serotec, Düsseldorf, Germany) and histogreen as substrate (Linaris, Wertheim, Germany). Counterstaining was performed using nuclear fast red (Linaris, Wertheim, Germany). Images were taken with the Leitz DM IRB inverted microscope with the Zeiss AxioCamMRC. Of note, the pathologist was blinded to genotype and BM status.
Measurement of PNC formation in vitro
Mice were anaesthetized with pentobarbital (80 mg/kg b.w.). Blood was drawn from the inferior vena cava and mixed with 3.2% sodium citrate solution (Merck, Darmstadt, Germany) in a 1:1 ratio. Blood samples were stimulated with 20 µM ADP (Sigma-Aldrich, St. Louis, MO, USA) or PBS (Sigma-Aldrich, St. Louis, MO, USA) and incubated at 37 °C for 15 min. Subsequently, the samples were stained with anti-CD45, anti-Ly-6G/C and anti-CD41 antibodies (Biolegend, San Diego, CA, USA) at room temperature for 15 min. Thereafter, erythrocytes were lysed in 2 ml BD FACS lysing solution (BD Bioscience, San Jose, CA, USA). Samples were measured on a BD FACS Canto II and analyzed using FlowJo software (FlowJo, Ashland, OR, USA). PNCs were identified as CD45+ Ly-6G/C+ CD41+ events and expressed as percentage of CD45+ Ly-6G/C+ neutrophils. In addition, neutrophil numbers from PBS-treated samples were expressed as percentage of CD45+ leukocytes and compared between animals to rule out variations in blood composition [29].
Isolation of human neutrophils
Following approval by the Institutional Review Board of Tübingen University Hospital (ethics committee; 483/2021BO2), human blood samples were taken from healthy donors after written informed consent, and neutrophils were isolated as follows. Whole blood was mixed with dextran solution (2% Dextran, 0.9% NaCl) in a 1:1 ratio and incubated for 30 min until two phases had formed. The upper phase was loaded on Biocoll (1.077 g/ml, Bio&Sell, Feucht, Germany) in a 3:2 ratio and density gradient centrifugation was performed for 30 min at 2000 × g without brake. After removing the supernatant, the pellet containing erythrocytes and granulocytes was resuspended in erythrocyte lysis buffer (C-C-Pro, Vogtei, Germany), incubated for 10 min and subsequently centrifuged for 10 min at 2.000 × g without brake. The remaining pellet containing neutrophils was washed once in PBS and then resuspended in keratinocyte base medium (CELLnTECH, Bern, Switzerland) containing 1.7 mM CaCl2. Cell concentration was brought to 3 × 106 cells/ml.
Isolation of neutrophils from Gnai2 nko mice
For chemotaxis analysis cells from BM were prepared by flushing tibia and femur of euthanized mice with PBS (Sigma-Aldrich, St. Louis, MO, USA).
For immunoblot analysis, mice were injected intraperitoneally with 300 µl of 4% thioglycollate solution (BD, Franklin Lakes, NJ, USA). Four hours later, the mice were sacrificed, and the peritoneum was flushed with cold PBS (Sigma-Aldrich, St. Louis, MO, USA).
Neutrophils from BM or peritoneal lavage were purified by immunomagnetic separation according to the manufacturer instructions (MACS neutrophil isolation kit, Miltenyi, Bergisch Gladbach, Germany). Purity was confirmed to be above 90% by FACS analysis using CD11b, CD45 and Ly-6G antibodies (BioLegend, San Diego, CA, USA).
Isolation of macrophages from Gnai2 nko mice
BM cells were prepared by flushing tibia and femur of euthanized mice with PBS (Sigma-Aldrich, St. Louis, MO, USA). For macrophage maturation, 1 × 106 cells were seeded in 10 cm dishes (bacterial ones) and cultured for one week in the presence of L929 supernatant.
Immunoblot analysis of Gαi2 expression
Isolated neutrophils or macrophages from Gnai2nko mice were counted in a hemocytometer, resuspended in SDS lysis buffer (21 mM Tris–HCl, 0.67% SDS, 19 mg/ml 2-mercaptoethanol, 0.4 mM PMSF) at a concentration of 2 × 107 cells/ml and stored at −20 °C. Finally, 2 × 105 neutrophils or macrophages were loaded on 9% SDS polyacrylamide gels containing 6 M urea and blotted onto PVDF membranes (Merck Millipore, Burlington, MA, USA). As controls, lysates from 2 × 105 splenocytes isolated from Gnai2−/− and wt mice were loaded.
To verify BM transplantations, heart tissue and 100 µl blood were homogenized in RIPA buffer and protein concentrations were measured by standard BCA method following the manufacturers’ instructions (Thermo Fisher Scientific, Rockford, IL, USA). Protein lysates (30 µg heart and 15 µg blood) were loaded on 12% SDS polyacrylamide gels and blotted onto nitrocellulose membranes as described [47].
Gαi2 protein expression was visualized by immunodetection using rabbit polyclonal anti-Gαi1/i2 antibodies (1:2000). The anti-Gαi2-specific antibodies are directed against the extreme C-terminus of Gαi2 which is known to be essential for functional interaction with its coupling GPCR [70]. Similar to the PTX-mediated ADP-ribosylation of a cysteine in the C-terminus of the Gα subunit, this also leads to a functional uncoupling between receptor and G protein. The antibodies were raised against a 21 amino acid comprising C-terminal peptide derived from Gαi2, which is identical to Gαi1 [11, 55]; Gαi2 can be easily distinguished from Gαi1 by a different mobility characteristic in the urea-supplemented polyacrylamide gels described above [54, 55, 73, 100], the use of horseradish peroxidase (HRP)-conjugated anti-rabbit-IgG secondary antibodies (1:2000) (Cell Signaling Technology, Danvers, MA, USA) and the Westar Supernova ECL detection system (Cyanagen, Bologna, Italy). Equal loading was verified with rabbit anti-β-actin (1:10,000) (abcam, Cambridge, United Kingdom) or anti-GAPDH (Santa Cruz Biotechnology, Dallas, USA) antibodies. For recombination analysis in Gnai2nko mice, Gαi2-protein levels were quantified by densitometry using the ImageJ software (NIH, Bethesda, MD, USA) and were normalized to β-actin levels of the same samples.
Immunoblot analysis of antibody uptake by neutrophils
Isolated human neutrophils were incubated with either anti-Gαi2-specific antibodies [54,55,56, 100] or anti-IgG antibodies (rabbit-IgG isotype control; Invitrogen, Carlsbad, CA, USA) at 37 °C for one hour. To specifically assess cytosolic uptake of the antibodies, cells were washed twice with PBS and then treated with digitonin (50 µg/ml in PBS w/o Ca2+ and Mg2+) for 5 min at room temperature followed by another 25 min on ice. Afterward, samples were spun down to separate cytosol from membrane and nuclei (membranous fraction). Supernatant (cytosolic fraction) and membranous fraction were mixed with 4 × Laemmli buffer (Carl Roth, Karlsruhe, Germany). Lysates of 4 × 106 neutrophils were loaded on 7% SDS polyacrylamide gels and blotted onto PVDF membranes (Merck Millipore, Burlington, MA, USA). The uptaken antibodies were visualized by immunodetection using HRP-conjugated anti-rabbit IgG (1:100) (Cell Signaling Technology, Danvers, MA, USA) and the Westar Supernova ECL detection system (Cyanagen, Bologna, Italy). To check for purity of cytosol and membranous fraction anti-Hsp90 (Cell Signaling Technology, Danvers, MA, USA, 1:1000) and anti-EEA antibodies (Cell Signaling Technology, Danvers, MA, USA, 1:1000) were used, respectively.
Immunofluorescent analysis of antibody uptake by neutrophils
Isolated human neutrophils were incubated with either anti-Gαi2-specific antibodies [54,55,56, 100] or IgG antibodies (rabbit-IgG isotype control; Invitrogen, Carlsbad, CA, USA) at 37 °C for one hour. To specifically detect localisation of the antibodies in different cellular compartments, cells were washed twice with PBS and then stained with lysotracker for one hour. Finally, cells were immobilized on glas slides and fixed with 4% formaldehyde.
A Leica Stellaris 8 confocal microscope was used to capture and accurately visualize the antibody-labeled fluorescence-stained neutrophil granulocytes with either anti-IgG or anti-Gαi2-antibodies. The images were taken using a 63 × objective. The standard Leica LAS X software (Leica Application Suite X; version 3.7.3.23245) was used for extended zooming, executed Z stacks, Z-stack video animations and 3D representations with Z-stack cross sections.
Endothelial transmigration assay of neutrophils in vitro
Transendothelial migration assays were performed as described previously [50]. Briefly, the upper chamber of transwell inserts (Fisher Scientific, Schwerte, Germany) covered with Human Microvascular Endothelial Cells-1 (HMEC-1) cell monolayers was filled with 1 × 106 neutrophils. To establish a chemotactic gradient each lower chamber was filled with 1 μM N-formyl-methionyl-leucyl-phenylalanine (fMLP; Merck, Darmstadt, Germany). Neutrophil transmigration studies were performed at 37 °C for 1 hour. Transmigration was assessed by quantification of the enzymatic activity of the azurophilic neutrophil granule protein myeloid peroxidase in the lower chamber. Neutrophils were preincubated with either Gαi2- [54,55,56, 100] or IgG antibodies (rabbit-IgG isotype control; Invitrogen, Carlsbad, CA, USA).
Chemotactic migration assay
For chemotaxis analysis MACS-isolated neutrophils were resuspended in RPMI and diluted (6 × 106 cells/ml). To maintain their viability, they were kept at 37 °C.
To control and analyze neutrophil migration, µ-Slide IbiTreat® chemotaxis chambers (Ibidi GmbH, Gräfelfing, Germany) were used. For murine neutrophil experiments a collagen gel (rat tail collagen 1.6 mg/ml) was prepared with a final cell concentration of 3 × 106 cells/ml. The gel was inserted in the middle chamber and incubated for 30 min at 37 °C to polymerize. Afterward, the lateral chambers were filled with RPMI and fMLP was added to one side with a final concentration of 10 µM right before starting the imaging process. The imaging was performed with the Leica THUNDER Imager Live Cell & 3D Cell Culture System (Leica Microsystems GmbH, Wetzlar, Germany) using a 20× (0.8 NA) objective and brightfield illumination. Multiple image tiles were recorded per condition which covered a total area of 900 × 1800 µm. Time-lapse sequences were obtained for 30 min in total with a frame rate of one image per minute. Neutrophils of Gnai2nko and control animals were imaged simultaneously.
To analyze chemotaxis after Gαi2 antibody treatment, human neutrophils were preincubated with anti-Gαi2-specific antibodies (1:100) or with IgG (1:100) for 30 min, loaded in µ-Slide Collagen IV chemotaxis chambers (Ibidi GmbH, Gräfelfing, Germany) and stimulated with 5 µM fMLP.
Chemotaxis analysis
To analyze neutrophil migration image tiles were stitched using FIJI/ImageJ (NIH, Bethesda, MD, USA) [90] and cells were segmented using WEKA Trainable Segmentation [6]. The segmented cells were filtered based on cell size (< 10 µm2, > 50 µm2) to exclude cell debris and segmentation artifacts from uneven background. Binary images of segmented neutrophils were then imported to Bitplane Imaris (Version 9.8) for cell tracking. To exclude dead and immobile cells, we filtered the tracks for track length (> 100 µm for murine and > 220 µm for human cells) before plotting migration speed, track length and persistence. The Chemotaxis and Migration Tool 2.0 (Ibidi GmbH) was used to determine the forward migration index towards fMLP (FMI(x)).
Statistical analysis
To analyze differences between more than two normally distributed groups, a one-way ANOVA with Newman–Keuls or Tukey’s multiple comparison test was performed as indicated in the Figure legends. For differences between more than two not normally distributed groups one-way ANOVA with Kruskal–Wallis test for multiple comparison was performed. To analyze differences between two normally distributed groups, an unpaired Student’s t test was used. P < 0.05 was considered statistically significant.
Results
Hematopoietic Gnai2-deficiency reduces cardiac ischemia reperfusion injury
Our previous studies indicated that a global Gnai2-deficiency aggravates mIRI [47]. To provide a first insight whether this effect depends on hematopoietic or tissue resident cells, BM transplantations were performed in mice in which the BM was destroyed by lethal irradiation prior to mIRI (for time line of this myocardial ischemia-reperfusion injury model see Suppl. Fig. 1a). Four experimental groups were analyzed in the study as depicted in Suppl. Fig. 1b. Successful reconstitution was confirmed by immunoblot analysis (Suppl. Fig. 1c). Results from control transplantation models (Suppl. Fig. 2, wt → wt and Gnai2−/− → Gnai2−/−) were consistent with those from non-transplanted wt- and Gnai2−/−-animals [47], confirming that the global absence of Gαi2 exacerbates mIRI. Gnai2-deficiency in resident tissue cells (wt → Gnai2−/−) but not in BM cells (Gnai2−/− → wt), increased infarct sizes (n = 6–7), which are considered the most robust endpoints of cardioprotection studies [34], to an extent observed in mice with global Gnai2-deficiency (Gnai2−/− → Gnai2−/−) (Suppl. Fig. 2a–c). In parallel, serum levels of troponin I (n = 3–4) were also elevated. In contrast, selective Gnai2-deficiency in the hematopoietic compartment (Gnai2−/− → wt) protected against mIRI (Suppl. Fig. 2a–c). Although serum levels of troponin I were similar to wt → wt BM chimeras (Suppl. Fig. 2b), infarct sizes were significantly reduced compared to all groups studied (Suppl. Fig. 2a).
Neutrophil-specific Gnai2-deficiency reduces mIRI
The interplay between platelets and neutrophils profoundly determines the outcome of mIRI [49, 50, 62]. The specific absence of Gαi2 in platelets extensively reduces mIRI, which is accompanied by decreased PNC counts [20]. To address the role of neutrophil Gαi2 in mIRI, LysM-Cre+/tg mice [16] were crossed with mice carrying floxed Gnai2 alleles [41] to generate a neutrophil/macrophage-specific conditional Gnai2 knockout (Gnai2nko) and ctrl, which were subsequently exposed to the mIRI model (Fig. 1 and Suppl. Fig. 3). All genotypes were obtained in the expected Mendelian ratios and were viable and fertile. Although our focus is on neutrophils, as it is well described, that in acute models of mIRI neutrophils are the main drivers of injury, whereas the impact of macrophages is still under investigation at this early time point [5, 8, 51], we confirmed recombination efficiency in both neutrophils and macrophages (Supppl. Fig. 3). Importantly, recombination was detectable only in genomic DNA derived from Gnai2nko samples, as indicated by the additional 390 bp band representing the knockout locus (Suppl. Fig. 3a). To additionally verify recombination on the protein level, immunoblot analysis of cell-suspensions from neutrophils and macrophages from these mice showed 66% and 98% reduction in Gαi2 protein content, respectively (Suppl. Fig. 3b–e), which is comparable to other studies using neutrophils and macrophages from LysM-Cre mice [1, 40].

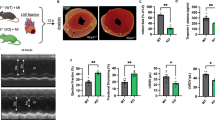
Neutrophil-specific Gnai2-deficiency protects from mIRI. a Mouse model to achieve neutrophil-specific Gnai2-deficiency (Gnai2nko). b Infarct size determined after 1 h of myocardial ischemia followed by 2 h reperfusion in Gnai2nko (blue; Gnai2nko; genotype: Gnai2fl/fl; LysM-Cre+/tg) mice compared to ctrl (black; ctrl; genotype: Gnai2fl/fl; LysM-Cre+/+). Percentage of necrotic tissue within the AAR is significantly reduced in Gnai2nko mice (n = 6–7 mice). Representative images are shown in Suppl. Fig. 4a. c Corresponding serum troponin I levels of Gnai2nko and ctrl animals (n = 6–7 mice). d Representative platelet-neutrophil complex (PNC) staining of ctrl and Gnai2nko heart sections. PNCs were stained for neutrophils (anti-Ly-6B2; blue) and platelets (anti-CD41; black). Scale bar 100 µm e Number of PNCs present in histologic sections of Gnai2nko hearts (blue) is significantly reduced (n = 3 animals; with 3 slices counted per animal). Statistical differences were calculated using Students’ t test. Data are shown as mean ± SEM; **p < 0.01; ***p < 0.001 as indicated
Interestingly, Gnai2nko mice subjected to mIRI, had on average almost 60% smaller infarct sizes and also greatly reduced serum levels of troponin I compared to their ctrl (Fig. 1b,c and Suppl. Fig. 4a). PNCs are important drivers of inflammation [29, 62] and their accumulation in the ischemic tissue is associated with the extent of IIR [50, 66]. In Gnai2nko mice the number of PNCs in the infarcted area was reduced by 43% compared to ctrl (Fig. 1d, e).
To address, whether the lack of Gαi2 in neutrophils results in decreased formation of PNCs, whole-blood samples were stimulated with ADP in vitro and then the formation of PNCs was quantified by flow cytometry (Suppl. Fig. 5). ADP stimulation of the blood resulted in similar PNC formation in Gnai2nko and control animals (Suppl. Fig. 5a), while the percentage of neutrophils in the blood was similar between both genotypes (Suppl. Fig. 5b).
Taken together, these results suggest that the protection of Gnai2nko mice from mIRI is driven by impaired PNC migration, reduced IIR and/or chemotaxis but not due to hampered PNC formation as we have previously found for platelet-derived Gαi2 [20].
Impaired chemotaxis of Gnai2-deficient neutrophils
To study neutrophil chemotaxis in vitro, the directional migration of neutrophils to fMLP, a Gαi-dependent chemoattractant, was monitored by live-cell imaging [53, 67, 101]. In control neutrophils, fMLP induced an increase in track lengths (Fig. 2a and Suppl. Fig. 6a) and a stronger orientation of cell tracks towards the chemokine gradient (forward migration index, FMI; Fig. 2b) compared with cells not exposed to a chemotactic stimulus.
In contrast, fMLP-induced track length was significantly shorter in Gnai2-deficient neutrophils, which was also reflected in much lower FMI compared with control cells. Thus, the lack of Gαi2 in neutrophils impairs movement efficiency and directional migration toward fMLP, suggesting that this results in an impaired PNC migration protecting from mIRI.

Deletion and inhibition of Gαi2 reduce neutrophil migration. a Migration of murine neutrophils from control (gray; genotype: Gnai2fl/fl; LysM-Cre+/+) and Gnai2nko (blue; genotype: Gnai2fl/fl; LysM-Cre+/tg) mice towards fMLP gradients was monitored by live-cell imaging. The length of fMLP-induced migration tracks (µm) was significantly reduced in Gnai2nko (light blue) neutrophils (***p < 0.001, n = 282–723 cells per group of four independent experiments). b fMLP-induced x-forward migration index (FMI, efficiency of the forward migration of the cells in direction of the x-axis towards the chemotactic gradient) was significantly reduced in Gnai2nko neutrophils (***p < 0.001, n = 448–723 cells per group of four independent experiments). c Directed migration of human neutrophils towards a fMLP-gradient was monitored by live-cell imaging. Human neutrophils were treated with either IgG (gray) or Gαi2-specific (orange) antibodies 30 min prior to the assay (***p < 0.001, n = 924–1098 cells per group of four independent experiments). d The fMLP-induced FMI was significantly reduced in Gαi2-specific antibody-treated (orange) neutrophils (***p < 0.001, n = 924–1098 cells per group of four independent experiments). e fMLP-triggered transmigration of human neutrophils through HMEC-1 monolayer was significantly reduced when treated with Gαi2-specific (orange) antibodies. As controls IgG-antibody-treated cells were used (gray). The results are expressed as mean ± SEM (n = 3 donors; ***p < 0.001). Statistical differences were calculated using Kruskal–Wallis (a–d) or Students’ t (e) test
Gαi2-specific antibody treatment reduces neutrophil migration and endothelial transmigration
To test whether blocking Gαi2 signaling also leads to decreased migration, isolated human neutrophils were treated with Gαi2-specific antibodies [54,55,56] and their migration pattern was monitored (Suppl. Fig. 6b). Although, fMLP-stimulated track length was not affected (Fig. 2c), antibody treatment actually impaired directed migration FMI (Fig. 2d). To rapidly reach the affected tissue after ischemia, neutrophils must first cross the endothelial barrier. Therefore, we also analyzed fMLP-induced transendothelial migration of human neutrophils (Fig. 2e). Interestingly, Gαi2-specific antibody treatment also reduced the number of transmigrated neutrophils by 50% compared with control IgG-antibody treatment (Fig. 2e), which could indicate a functionally relevant significance of neutrophil Gαi2 for mIRI.
Gαi2-specific antibody treatment protects from mIRI
To evaluate the effects of antibody-mediated blockage of neutrophil Gi signal transduction on mIRI in our mouse model, wt animals were treated with either control IgG or Gαi2-specific antibodies immediately before reperfusion (Fig. 3a, b). Treatment with Gαi2-specific antibodies resulted in more than 50% reduction in infarct size compared with IgG-treated animals (Fig. 3c and Suppl. Fig. 4b). Consistent with the protective effect on infarct size, troponin I levels (Fig. 4d) and the numbers of PNCs present in infarct tissue (Fig. 4e, f) were also reduced.

Gαi2-specific antibodies protect from mIRI. a Timeline for mIRI and antibody treatment. Mice were subjected to 1 h ischemia and then treated with antibodies 5 min before onset of reperfusion. b Wild type mice received either IgG (wtIgG; black) or Gαi2-specific (wtab; green) antibodies (2 µg/mouse) via the tail vein. c Infarct size in antibody-treated mice. Percentage of necrotic tissue within the AAR is significantly reduced in wtab mice (n = 6 mice). Representative images are shown in Suppl. Fig. 4b. d Correlating serum troponin I levels are significantly reduced in wtab mice (n = 6). e Representative platelet-neutrophil complex (PNC) staining of wtIgG and wtab heart sections. PNCs were stained for neutrophils (anti-Ly-6B2; blue) and platelets (anti-CD41; black). Scale bar 100 µm f Number of PNCs present in histologic sections of wtab hearts (green) is significantly reduced (n = 3 animals; with three slices counted per animal). Data are shown as mean ± SEM; *p < 0.05; ***p < 0.001 as indicated. Statistical differences were calculated using Students’ t test
Notably, antibody treatment did not affect heart rate, bleeding time, organ weight or cellular composition in BM, blood and spleen (Suppl. Figs. 7, 8). In conclusion, a single injection of Gαi2-specific antibodies prior to reperfusion protects from mIRI.
Gαi2-specific antibodies are detectable intracellularly in neutrophils
As Gi proteins are located at the inner leaflet of the plasma membrane Gαi2-specific antibodies have to enter the cell to target Gαi2. In the cytosol of human neutrophils, we detected IgG in both Gαi2-specific and isotype IgG-treated cells (Fig. 4a). The absence of Early Endosome Antigen 1 (EEA1), but the detection of Heat Shock Protein 90 (HSP90) confirmed the purity of the cytosolic fraction. Of note, both antibodies were also detected in the membranous fraction, with higher levels of Gαi2-specific antibodies (Fig. 4b). In addition, confocal microscopy revealed different subcellular localization of phagolysosomes and the uptaken antibodies in human neutrophils (Fig. 4c).

Antibodies are taken up by neutrophils. Antibody visualization in a cytosolic and b membranous fractions of 1 × 106 human neutrophils by immunoblot analysis. Details of the experimental setup are described in Material and Methods. IgG and Gαi2-specific antibodies were detected by using HRP-coupled anti-rabbit-IgG antibodies. As positive control IgG antibodies were loaded and are depicted on the right. EEA1 and HSP90 served as controls for purity of cytosol and membrane fractions, respectively. One representative immunoblot out of three independent experiments is shown. c Confocal microscopy of human neutrophils treated with either IgG or Gαi2-specific antibodies, detected by using anti-rabbit-IgG antibodies (green). Subsequently phagolysosomes were stained with lysotracker (red). One representative Z-stack image is shown. The reconstructed 3D z-stacks (Suppl. Fig. 10) and movies can be found in the supplemental material
Overall, we demonstrated that the antibodies are taken up by neutrophils and that functional inhibition of the intracellular Gαi2 protein is thus possible.
Discussion
Numerous studies document that mIRI is driven in part by inflammation through multiple interacting pathways, with a notable detrimental role of neutrophils, particularly in the initial phase [91, 98]. Neutrophils are activated during acute MI [21] and rapidly infiltrate the ischemic cardiac tissue together with platelets as PNCs [24]. In addition, in vitro findings show that neutrophils directly damage cardiomyocytes [25]. For instance, they are the primary sources of ROS during myocardial ischemia [22] and their depletion resulted in a striking reduction in infarct sizes in mice, dogs and pigs [30, 50, 82]. We have previously shown, that inhibition of Gαi2 signaling in platelets has cardio-protective effects [20]. However, the functional role of Gαi2 specifically in neutrophils remained unclear.
In the present study three different, independent and complementary experimental approaches were applied to examine its role in an acute mouse model of mIRI by inactivating or inhibiting neutrophil Gαi2 signaling. First, we performed mIRI experiments with murine Gnai2-deficient BM chimeras, followed by studies in mice displaying a neutrophil/macrophage-specific knockout of Gnai2. Moreover, in an experimental interventional therapeutic approach, we treated wt mice with Gαi2-specific antibodies. All these approaches showed the same, a massive reduction of cardiac tissue damage. The reduction in infarct size was associated with a reduced chemotaxis and endothelial transmigration of neutrophils in vitro and a reduced presence of PNCs in the infarcted tissue in vivo. These findings strongly suggest a key role of neutrophil Gαi2 in the early phase of mIRI, whereas the functional importance of concomitant genetically targeted macrophages during this early stages of mIRI remains to be clarified. Accordingly, we observed that a blockade of Gαi2 signaling resulted in an inhibition of human neutrophil functions.
This supports our hypothesis that the loss of function of human neutrophils as a result of inhibition of Gαi2 signaling should lead to a reduction in reperfusion injury in the myocardium. Therefore, we envision that neutrophil Gαi2 can be a potential target for early pharmacologic treatment of mIRI.
We showed that platelet Gαi2 signaling is essential for the activation of neutrophils to form PNCs [20]. Accordingly, ADP-stimulated PNC formation in global Gnai2-deficient mice was reduced (Suppl. Fig. 9a) but intact in Gnai2nko mice and neutrophils treated with Gαi2-specific antibodies (Suppl. Figs. 5 and 9) [20, 29, 50, 62]. Thus, it is unlikely that the reduced infarct sizes after deletion of Gnai2 in neutrophils can be explained by impaired PNC formation. Rather, our findings suggest a different function of Gαi2 in neutrophils for their activation upon binding platelets and/or in neutrophil recruitment that is independent of their interaction with platelets.
Reperfusion injury contributes significantly to loss of myocardial tissue upon infarction. As inflammation is a driver of mIRI, its underlying pathways have been targeted in many experimental studies in search for therapy. Trials have not only included broad anti-inflammatory interventions with glucocorticoids, non-steroid anti-inflammatory drugs (NSAIDs) or immunomodulatory agents but also targeted anti-inflammatory interventions directed against the complement system, cytokines, the TGF-β system, integrins, and selectins [38, 88]. Some of them specifically aimed to prevent neutrophil recruitment to the infarct site by blocking cell adhesion molecules. In particular, blockade of CD18 significantly reduced mIRI in animal studies [88], but failed in clinical trials using antibodies directed against CD18 or CD11/CD18 [9, 26, 87]. Likewise, antibodies directed against components of the complement system were able to reduce mIRI in animal models [77, 97], but were unsuccessful in clinical trials [7]. At present, only the use of low-dose colchicine in long-term management may be considered in the current ESC Guidelines for the management of acute coronary syndromes of the European Society of Cardiology [14]. The advantage of our approach is that, for the first time, it is possible to directly interfere with neutrophil signaling and to block neutrophil migration in acute mIRI instead of interfering with platelet activation. Of course, there are still a number of gaps that need to be addressed to successfully translate our basic animal findings to human patients. These include producing a humanized monoclonal antibody, ensuring the uptake of the antibody restricted to neutrophils and its specific interference with Gαi2 signaling. Finally, the antibody has to be tested in humans and must successfully complete clinical trials.
An immediate question regarding the therapeutic use of an anti-Gαi2 antibody is whether after systemic administration it can reach its target, as G proteins are physiologically localized inside the cell. Accordingly, the antibodies used must actually enter an intact cell to exert their blocking effect by binding to the G proteins. We used a functional active polyclonal peptide antibody that binds to the extreme C-terminus of Gαi2 in a selective and specific manner [55]. The antibody is not only directed to a region that is important for receptor-G protein interaction but also for effector regulation by steric hindrance [2, 37, 72]. Indeed, our data show that it is a functionally active antibody that is effective in vivo. Previously it has been shown that global deficiency of Gnai2 greatly reduces neutrophil recruitment to sites of inflammation, e.g., in LPS-induced lung-inflammation, LPS-induced peritonitis, skin Arthus reaction and lung Arthus reaction [100, 103]. In this study, targeting intracellularly localized Gαi2 by Gαi2-specific antibodies disrupted transendothelial migration of neutrophils. In line with these findings, we demonstrated a strong reduction of damage in an acute murine model of mIRI. As a prerequisite for the effect of intravenously injected antibodies on blood-cell Gαi2 proteins, the antibodies had to enter the cells without ending up in phagolysosomes. Indeed, the uptake of antibodies into living cells has been described in the pathogenesis of several autoimmune diseases, such as systemic lupus erythematodes (SLE) [3, 69]. However, the mechanisms whereby an antibody may penetrate into the cell are still debated [86, 93]. Possible mechanisms for the entry of antibodies include binding to Fc-receptors and subsequent endocytosis, as well as transport via nucleotide transporters, clathrin-associated vesicles or even free transit [69, 86]. Even if our study does not make a contribution to these details, we can still exclude that Gαi2-specific antibodies are found in phagolysosomes causing their rapid degradation. Nevertheless, the exact mechanism of how this antibody is acting on neutrophils or macrophages remains to be clarified in further studies.
Targeting neutrophils for cardioprotection is challenging. Different strategies have been pursued either targeting neutrophil-triggered inflammation, inhibiting neutrophil activity, or reducing the neutrophil accumulation in the infarct area [5, 17,18,19]. These already included approaches to intervene in the G protein signaling pathway, in these cases in the Gs protein-mediated signaling axis, to achieve a cardio-protective effect or reduce the infarct size. Metoprolol, a so-called β1-selective adrenoreceptor antagonist, is probably best known [17, 28, 44, 63, 99]. According to the ESC Guidelines for the management of acute coronary syndromes [14], i.v. administration of metoprolol should be applied before reperfusion therapy. Metoprolol was found to reduce heart rate, infarct size, and inflammation. However, the results following metoporolol in various preclinical animal studies are controversial [36, 39]. Whereas, on the one hand various neutrophil functions, including neutrophil-platelet interaction, neutrophil migration and reduction of coronary microvascular obstruction were reported to be hampered, others could not confirm the reduction of infarct size reduction and area of no-reflow [17, 46]. Not only a number of possible reasons for these apparent contradictions are discussed, which may be related to the animal model used, genetic variations, the experimental conditions, but also the need for further detailed studies on clinically relevant animal models is emphasized [36, 39, 45, 84]. But the molecular mechanism of action also needs to be clarified in more detail: thus, it is currently assumed that metoprolol exerts its infarct-reducing effects through anti-inflammatory inhibition of neutrophil migration and stunning of coronary microvascular obstruction via non-canonical mechanisms, since classical inhibition of the cAMP signaling pathway is unlikely to affect neutrophil migration [17, 28, 36]. Further support for this assumption is coming from studies targeting phosphodiesterase-4 subtype B (PDE4B) for cardioprotection in acute MI via neutrophils and microcirculation [99]. Despite uncertainty about the mode of action of metoprolol or PDE4B inhibition on neutrophils and other target cells that protect them from mIRI, alternative pathways involving biased signaling are conceivable [72].
Limitations
Although, we provide strong evidence for a protective effect of Gαi2 inhibition on acute mIRI, the underlying molecular mechanisms preventing a worsening or prolonged inflammatory repsonse remain to be further elucidated. In addition, future studies should focus on the impact of neutrophil Gαi2-signaling on coronary microvascular obstruction by analyzing the no-reflow phenomenon in more detail. For instance, it is still controversial whether intravascular neutrophil plugging is pivotal or whether neutrophil-mediated damage by ROS, proteolytic enzymes, and lipoxygenase products as well as interactions with the endothelium, platelets, and perhaps with myocytes contribute secondarily to the no-reflow phenomenon [15, 23, 68, 80, 81]. Since we conducted our studies using an acute model of mIRI, the role of neutrophil Gαi2 in tissue repair also needs to be clarified, but is beyond the scope of this study. In addition, in this study we did not investigate whether Gαi2-signaling in macrophages has an effect on cardiac damage, no-reflow phenomenon and tissue repair [35], but this will be clarified in future studies.
Conclusion
The present study suggests that cell-specific Gαi2 could be a potential new target for pharmacologic (co-)treatment of mIRI. The absence of Gαi2 in blood cells as well as the neutrophil/macrophage-specific deletion of Gnai2 reduced cardiac tissue damage, troponin I levels and PNCs in the infarcted tissue. Furthermore, the specific anti-Gαi2 antibody directed against the functional important C-terminus (1) inhibited the effect on (a) the development of mIRI damage, (b) the invasion of PNCs into the affected area and (c) the typical increase in serum troponin I levels in our murine IRI model but also (2) reduced directed chemotaxis of human neutrophils, and (3) massively inhibited transendothelial migration of human neutrophils. We therefore conclude that our antibody is actually capable of exerting a specific effect in both neutrophils and the mIRI model. The identification of its exact mode of action is beyond of the scope of the present study but will be analyzed in future. Importantly, in our mIRI mouse model, a single treatment with Gαi2-specific antibodies shortly before reperfusion was highly effective and would perfectly meet the therapeutic demands considering the clinical circumstances of MI patients. Further efforts are clearly needed to test the translational potential of our findings.
Data availability
All data will be available on request.
Abbreviations
- AAR:
-
Area at risk
- BM:
-
Bone marrow
- FMI:
-
Forward migration index
- fMLP:
-
N-formylmethionyl-leucyl-phenylalanine
- GPCR:
-
G protein-coupled receptor
- HMEC-1:
-
Human microvascular endothelial cells
- IIR:
-
Ischemic inflammatory response
- MI:
-
Myocardial infarction
- (m)IRI:
-
Myocardial ischemia reperfusion injury
- PBS:
-
Phosphate-buffered saline
- PNC:
-
Platelet-neutrophil complex
- ROS:
-
Reactive oxygen species
- TTC:
-
Triphenyl tetrazolium chloride
- wt:
-
wild-type
References
Abram CL, Roberge GL, Hu Y, Lowell CA (2014) Comparative analysis of the efficiency and specificity of myeloid-Cre deleting strains using ROSA-EYFP reporter mice. J Immunol Methods 408:89–100. https://doi.org/10.1016/j.jim.2014.05.009
Ahnert-Hilger G, Nurnberg B, Exner T, Schafer T, Jahn R (1998) The heterotrimeric G protein Go2 regulates catecholamine uptake by secretory vesicles. EMBO J 17:406–413. https://doi.org/10.1093/emboj/17.2.406
Alarcon-Segovia D, Ruiz-Arguelles A (1978) Decreased circulating thymus-derived cells with receptors for the Fc portion of immunoglobulin G in systemic lupus erythematosus. J Clin Invest 62:1390–1394. https://doi.org/10.1172/JCI109260
Algoet M, Janssens S, Himmelreich U, Gsell W, Pusovnik M, Van den Eynde J, Oosterlinck W (2023) Myocardial ischemia-reperfusion injury and the influence of inflammation. Trends Cardiovasc Med 33:357–366. https://doi.org/10.1016/j.tcm.2022.02.005
Andreadou I, Cabrera-Fuentes HA, Devaux Y, Frangogiannis NG, Frantz S, Guzik T, Liehn EA, Gomes CPC, Schulz R, Hausenloy DJ (2019) Immune cells as targets for cardioprotection: new players and novel therapeutic opportunities. Cardiovasc Res 115:1117–1130. https://doi.org/10.1093/cvr/cvz050
Arganda-Carreras I, Andrey P (2017) Designing image analysis pipelines in light microscopy: a rational approach. Methods Mol Biol 1563:185–207. https://doi.org/10.1007/978-1-4939-6810-7_13
Armstrong PW, Granger CB (2007) Pexelizumab and the APEX AMI trial. JAMA 297:1881; author reply 1881–1882. https://doi.org/10.1001/jama.297.17.1881-b
Bajpai G, Bredemeyer A, Li W, Zaitsev K, Koenig AL, Lokshina I, Mohan J, Ivey B, Hsiao HM, Weinheimer C, Kovacs A, Epelman S, Artyomov M, Kreisel D, Lavine KJ (2019) Tissue resident CCR2- and CCR2+ cardiac macrophages differentially orchestrate monocyte recruitment and fate specification following myocardial injury. Circ Res 124:263–278. https://doi.org/10.1161/CIRCRESAHA.118.314028
Baran KW, Nguyen M, McKendall GR, Lambrew CT, Dykstra G, Palmeri ST, Gibbons RJ, Borzak S, Sobel BE, Gourlay SG, Rundle AC, Gibson CM, Barron HV, Limitation of Myocardial Infarction Following Thrombolysis in Acute Myocardial Infarction Study Group (2001) Double-blind, randomized trial of an anti-CD18 antibody in conjunction with recombinant tissue plasminogen activator for acute myocardial infarction: limitation of myocardial infarction following thrombolysis in acute myocardial infarction (LIMIT AMI) study. Circulation 104:2778–2783. https://doi.org/10.1161/hc4801.100236
Baxter GF (2002) The neutrophil as a mediator of myocardial ischemia-reperfusion injury: time to move on. Basic Res Cardiol 97:268–275. https://doi.org/10.1007/s00395-002-0366-7
Beer-Hammer S, Lee SC, Mauriac SA, Leiss V, Groh IAM, Novakovic A, Piekorz RP, Bucher K, Chen C, Ni K, Singer W, Harasztosi C, Schimmang T, Zimmermann U, Pfeffer K, Birnbaumer L, Forge A, Montcouquiol M, Knipper M, Nurnberg B, Ruttiger L (2018) Galphai proteins are indispensable for hearing. Cell Physiol Biochem 47:1509–1532. https://doi.org/10.1159/000490867
Bengtsson T, Stendahl O, Andersson T (1986) The role of the cytosolic free Ca2+ transient for fMet-Leu-Phe induced actin polymerization in human neutrophils. Eur J Cell Biol 42:338–343
Botker HE, Hausenloy D, Andreadou I, Antonucci S, Boengler K, Davidson SM, Deshwal S, Devaux Y, Di Lisa F, Di Sante M, Efentakis P, Femmino S, Garcia-Dorado D, Giricz Z, Ibanez B, Iliodromitis E, Kaludercic N, Kleinbongard P, Neuhauser M, Ovize M, Pagliaro P, Rahbek-Schmidt M, Ruiz-Meana M, Schluter KD, Schulz R, Skyschally A, Wilder C, Yellon DM, Ferdinandy P, Heusch G (2018) Practical guidelines for rigor and reproducibility in preclinical and clinical studies on cardioprotection. Basic Res Cardiol 113:39. https://doi.org/10.1007/s00395-018-0696-8
Byrne RA, Rossello X, Coughlan JJ, Barbato E, Berry C, Chieffo A, Claeys MJ, Dan GA, Dweck MR, Galbraith M, Gilard M, Hinterbuchner L, Jankowska EA, Juni P, Kimura T, Kunadian V, Leosdottir M, Lorusso R, Pedretti RFE, Rigopoulos AG, Rubini Gimenez M, Thiele H, Vranckx P, Wassmann S, Wenger NK, Ibanez B, Group ESCSD (2023) 2023 ESC Guidelines for the management of acute coronary syndromes. Eur Heart J 44:3720–3826. https://doi.org/10.1093/eurheartj/ehad191
Carlson RE, Schott RJ, Buda AJ (1989) Neutrophil depletion fails to modify myocardial no reflow and functional recovery after coronary reperfusion. J Am Coll Cardiol 14:1803–1813. https://doi.org/10.1016/0735-1097(89)90036-3
Clausen BE, Burkhardt C, Reith W, Renkawitz R, Forster I (1999) Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res 8:265–277
Clemente-Moragon A, Gomez M, Villena-Gutierrez R, Lalama DV, Garcia-Prieto J, Martinez F, Sanchez-Cabo F, Fuster V, Oliver E, Ibanez B (2020) Metoprolol exerts a non-class effect against ischaemia-reperfusion injury by abrogating exacerbated inflammation. Eur Heart J 41:4425–4440. https://doi.org/10.1093/eurheartj/ehaa733
Clemente-Moragon A, Martinez-Milla J, Oliver E, Santos A, Flandes J, Fernandez I, Rodriguez-Gonzalez L, Serrano Del Castillo C, Ioan AM, Lopez-Alvarez M, Gomez-Talavera S, Galan-Arriola C, Fuster V, Perez-Calvo C, Ibanez B (2021) Metoprolol in critically Ill patients with COVID-19. J Am Coll Cardiol 78:1001–1011. https://doi.org/10.1016/j.jacc.2021.07.003
Clemente-Moragon A, Oliver E, Calle D, Cusso L, Gomez M, Pradillo JM, Castejon R, Rallon N, Benito JM, Fernandez-Ferro JC, Carneado-Ruiz J, Moro MA, Sanchez-Gonzalez J, Fuster V, Cortes-Canteli M, Desco M, Ibanez B (2023) Neutrophil beta(1) adrenoceptor blockade blunts stroke-associated neuroinflammation. Br J Pharmacol 180:459–478. https://doi.org/10.1111/bph.15963
Devanathan V, Hagedorn I, Köhler D, Pexa K, Cherpokova D, Kraft P, Singh M, Rosenberger P, Stoll G, Birnbaumer L, Piekorz RP, Beer-Hammer S, Nieswandt B, Nürnberg B (2015) Platelet Gi protein Galphai2 is an essential mediator of thrombo-inflammatory organ damage in mice. Proc Natl Acad Sci U S A 112:6491–6496. https://doi.org/10.1073/pnas.1505887112
Dinerman JL, Mehta JL, Saldeen TG, Emerson S, Wallin R, Davda R, Davidson A (1990) Increased neutrophil elastase release in unstable angina pectoris and acute myocardial infarction. J Am Coll Cardiol 15:1559–1563. https://doi.org/10.1016/0735-1097(90)92826-n
Duilio C, Ambrosio G, Kuppusamy P, DiPaula A, Becker LC, Zweier JL (2001) Neutrophils are primary source of O2 radicals during reperfusion after prolonged myocardial ischemia. Am J Physiol Heart Circ Physiol 280:H2649–H2657. https://doi.org/10.1152/ajpheart.2001.280.6.H2649
Eeckhout E, Kern MJ (2001) The coronary no-reflow phenomenon: a review of mechanisms and therapies. Eur Heart J 22:729–739. https://doi.org/10.1053/euhj.2000.2172
Engler RL, Dahlgren MD, Peterson MA, Dobbs A, Schmid-Schonbein GW (1986) Accumulation of polymorphonuclear leukocytes during 3-h experimental myocardial ischemia. Am J Physiol 251:H93–H100. https://doi.org/10.1152/ajpheart.1986.251.1.H93
Entman ML, Youker K, Shoji T, Kukielka G, Shappell SB, Taylor AA, Smith CW (1992) Neutrophil induced oxidative injury of cardiac myocytes. A compartmented system requiring CD11b/CD18-ICAM-1 adherence. J Clin Invest 90:1335–1345. https://doi.org/10.1172/JCI115999
Faxon DP, Gibbons RJ, Chronos NA, Gurbel PA, Sheehan F, Investigators H-M (2002) The effect of blockade of the CD11/CD18 integrin receptor on infarct size in patients with acute myocardial infarction treated with direct angioplasty: the results of the HALT-MI study. J Am Coll Cardiol 40:1199–1204. https://doi.org/10.1016/s0735-1097(02)02136-8
Fishbein MC, Meerbaum S, Rit J, Lando U, Kanmatsuse K, Mercier JC, Corday E, Ganz W (1981) Early phase acute myocardial infarct size quantification: validation of the triphenyl tetrazolium chloride tissue enzyme staining technique. Am Heart J 101:593–600. https://doi.org/10.1016/0002-8703(81)90226-x
Garcia-Prieto J, Villena-Gutierrez R, Gomez M, Bernardo E, Pun-Garcia A, Garcia-Lunar I, Crainiciuc G, Fernandez-Jimenez R, Sreeramkumar V, Bourio-Martinez R, Garcia-Ruiz JM, Del Valle AS, Sanz-Rosa D, Pizarro G, Fernandez-Ortiz A, Hidalgo A, Fuster V, Ibanez B (2017) Neutrophil stunning by metoprolol reduces infarct size. Nat Commun 8:14780. https://doi.org/10.1038/ncomms14780
Granja TF, Köhler D, Leiss V, Eggstein C, Nürnberg B, Rosenberger P, Beer-Hammer S (2022) Platelets and the cybernetic regulation of ischemic inflammatory responses through PNC formation regulated by extracellular nucleotide metabolism and signaling. Cells 11:3009. https://doi.org/10.3390/cells11193009
Hatori N, Roberts RL, Tadokoro H, Ryden L, Satomura K, Fishbein MC, Stiehm ER, Corday E, Drury JK (1991) Differences in infarct size with lidocaine as compared with bretylium tosylate in acute myocardial ischemia and reperfusion in pigs. J Cardiovasc Pharmacol 18:581–588. https://doi.org/10.1097/00005344-199110000-00015
He J, Liu D, Zhao L, Zhou D, Rong J, Zhang L, Xia Z (2022) Myocardial ischemia/reperfusion injury: mechanisms of injury and implications for management (Review). Exp Ther Med 23:430. https://doi.org/10.3892/etm.2022.11357
Heusch G (2016) The coronary circulation as a target of cardioprotection. Circ Res 118:1643–1658. https://doi.org/10.1161/CIRCRESAHA.116.308640
Heusch G (2019) Coronary microvascular obstruction: the new frontier in cardioprotection. Basic Res Cardiol 114:45. https://doi.org/10.1007/s00395-019-0756-8
Heusch G (2020) Myocardial ischaemia-reperfusion injury and cardioprotection in perspective. Nat Rev Cardiol 17:773–789. https://doi.org/10.1038/s41569-020-0403-y
Heusch G (2024) Myocardial ischemia/reperfusion: translational pathophysiology of ischemic heart disease. Med 5:10–31. https://doi.org/10.1016/j.medj.2023.12.007
Heusch G, Kleinbongard P (2020) Is metoprolol more cardioprotective than other beta-blockers? Eur Heart J 41:4441–4443. https://doi.org/10.1093/eurheartj/ehaa764
Holtje M, von Jagow B, Pahner I, Lautenschlager M, Hortnagl H, Nurnberg B, Jahn R, Ahnert-Hilger G (2000) The neuronal monoamine transporter VMAT2 is regulated by the trimeric GTPase Go(2). J Neurosci 20:2131–2141. https://doi.org/10.1523/JNEUROSCI.20-06-02131.2000
Huang S, Frangogiannis NG (2018) Anti-inflammatory therapies in myocardial infarction: failures, hopes and challenges. Br J Pharmacol 175:1377–1400. https://doi.org/10.1111/bph.14155
Ibanez B (2023) A tale of pigs, beta-blockers and genetic variants. Basic Res Cardiol 118:27. https://doi.org/10.1007/s00395-023-00998-z
Jakubzick C, Bogunovic M, Bonito AJ, Kuan EL, Merad M, Randolph GJ (2008) Lymph-migrating, tissue-derived dendritic cells are minor constituents within steady-state lymph nodes. J Exp Med 205:2839–2850. https://doi.org/10.1084/jem.20081430
Jiang M, Boulay G, Spicher K, Peyton MJ, Brabet P, Birnbaumer L, Rudolph U (1997) Inactivation of the Gɑi2 and G ɑo genes by homologous recombination. Recept Channels 5:187–192
Kaziro Y, Itoh H, Kozasa T, Nakafuku M, Satoh T (1991) Structure and function of signal-transducing GTP-binding proteins. Annu Rev Biochem 60:349–400. https://doi.org/10.1146/annurev.bi.60.070191.002025
Kehrl JH (2016) The impact of RGS and other G-protein regulatory proteins on Galphai-mediated signaling in immunity. Biochem Pharmacol 114:40–52. https://doi.org/10.1016/j.bcp.2016.04.005
Kleinbongard P (2020) Cardioprotection by early metoprolol-attenuation of ischemic vs. reperfusion injury? Basic Res Cardiol 115:54. https://doi.org/10.1007/s00395-020-0814-2
Kleinbongard P, Heusch G (2022) A fresh look at coronary microembolization. Nat Rev Cardiol 19:265–280. https://doi.org/10.1038/s41569-021-00632-2
Kleinbongard P, Lieder HR, Skyschally A, Heusch G (2023) No robust reduction of infarct size and no-reflow by metoprolol pretreatment in adult Gottingen minipigs. Basic Res Cardiol 118:23. https://doi.org/10.1007/s00395-023-00993-4
Köhler D, Devanathan V, de Oliveira B, Franz C, Eldh T, Novakovic A, Roth JM, Granja T, Birnbaumer L, Rosenberger P, Beer-Hammer S, Nürnberg B (2014) Gαi2- and Gαi3-deficient mice display opposite severity of myocardial ischemia reperfusion injury. PLoS ONE 9:e98325. https://doi.org/10.1371/journal.pone.0098325
Köhler D, Eckle T, Faigle M, Grenz A, Mittelbronn M, Laucher S, Hart ML, Robson SC, Müller CE, Eltzschig HK (2007) CD39/ectonucleoside triphosphate diphosphohydrolase 1 provides myocardial protection during cardiac ischemia/reperfusion injury. Circulation 116:1784–1794. https://doi.org/10.1161/CIRCULATIONAHA.107.690180
Köhler D, Granja T, Volz J, Koeppen M, Langer HF, Hansmann G, Legchenko E, Geisler T, Bakchoul T, Eggstein C, Haberle HA, Nieswandt B, Rosenberger P (2020) Red blood cell-derived semaphorin 7A promotes thrombo-inflammation in myocardial ischemia-reperfusion injury through platelet GPIb. Nat Commun 11:1315. https://doi.org/10.1038/s41467-020-14958-x
Köhler D, Straub A, Weissmuller T, Faigle M, Bender S, Lehmann R, Wendel HP, Kurz J, Walter U, Zacharowski K, Rosenberger P (2011) Phosphorylation of vasodilator-stimulated phosphoprotein prevents platelet-neutrophil complex formation and dampens myocardial ischemia-reperfusion injury. Circulation 123:2579–2590. https://doi.org/10.1161/CIRCULATIONAHA.110.014555
Kologrivova I, Shtatolkina M, Suslova T, Ryabov V (2021) Cells of the immune system in cardiac remodeling: main players in resolution of inflammation and repair after myocardial infarction. Front Immunol 12:664457. https://doi.org/10.3389/fimmu.2021.664457
Kruger P, Saffarzadeh M, Weber AN, Rieber N, Radsak M, von Bernuth H, Benarafa C, Roos D, Skokowa J, Hartl D (2015) Neutrophils: between host defence, immune modulation, and tissue injury. PLoS Pathog 11:e1004651. https://doi.org/10.1371/journal.ppat.1004651
Le Y, Murphy PM, Wang JM (2002) Formyl-peptide receptors revisited. Trends Immunol 23:541–548. https://doi.org/10.1016/s1471-4906(02)02316-5
Leiss V, Flockerzie K, Novakovic A, Rath M, Schönsiegel A, Birnbaumer L, Schürmann A, Harteneck C, Nürnberg B (2014) Insulin secretion stimulated by arginine and its metabolite ornithine depends on Gαi2. Am J Physiol Endocrinol Metab. https://doi.org/10.1152/ajpendo.00337.2014
Leiss V, Schönsiegel A, Gnad T, Kerner J, Kaur J, Sartorius T, Machann J, Schick F, Birnbaumer L, Haring HU, Pfeifer A, Nürnberg B (2020) Lack of Galpha(i2) proteins in adipocytes attenuates diet-induced obesity. Mol Metab 40:101029. https://doi.org/10.1016/j.molmet.2020.101029
Leopoldt D, Harteneck C, Nürnberg B (1997) G proteins endogenously expressed in Sf 9 cells: interactions with mammalian histamine receptors. Naunyn Schmiedebergs Arch Pharmacol 356:216–224
Ley K, Laudanna C, Cybulsky MI, Nourshargh S (2007) Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol 7:678–689. https://doi.org/10.1038/nri2156
Linden MD, Furman MI, Frelinger AL 3rd, Fox ML, Barnard MR, Li Y, Przyklenk K, Michelson AD (2007) Indices of platelet activation and the stability of coronary artery disease. J Thromb Haemost 5:761–765. https://doi.org/10.1111/j.1538-7836.2007.02462.x
Lindsey ML, Bolli R, Canty JM Jr, Du XJ, Frangogiannis NG, Frantz S, Gourdie RG, Holmes JW, Jones SP, Kloner RA, Lefer DJ, Liao R, Murphy E, Ping P, Przyklenk K, Recchia FA, Schwartz Longacre L, Ripplinger CM, Van Eyk JE, Heusch G (2018) Guidelines for experimental models of myocardial ischemia and infarction. Am J Physiol Heart Circ Physiol 314:H812–H838. https://doi.org/10.1152/ajpheart.00335.2017
Lindsey ML, Brunt KR, Kirk JA, Kleinbongard P, Calvert JW, de Castro Bras LE, DeLeon-Pennell KY, Del Re DP, Frangogiannis NG, Frantz S, Gumina RJ, Halade GV, Jones SP, Ritchie RH, Spinale FG, Thorp EB, Ripplinger CM, Kassiri Z (2021) Guidelines for in vivo mouse models of myocardial infarction. Am J Physiol Heart Circ Physiol 321:H1056–H1073. https://doi.org/10.1152/ajpheart.00459.2021
Lindsey ML, Kassiri Z, Virag JAI, de Castro Bras LE, Scherrer-Crosbie M (2018) Guidelines for measuring cardiac physiology in mice. Am J Physiol Heart Circ Physiol 314:H733–H752. https://doi.org/10.1152/ajpheart.00339.2017
Lisman T (2018) Platelet-neutrophil interactions as drivers of inflammatory and thrombotic disease. Cell Tissue Res 371:567–576. https://doi.org/10.1007/s00441-017-2727-4
Lobo-Gonzalez M, Galan-Arriola C, Rossello X, Gonzalez-Del-Hoyo M, Vilchez JP, Higuero-Verdejo MI, Garcia-Ruiz JM, Lopez-Martin GJ, Sanchez-Gonzalez J, Oliver E, Pizarro G, Fuster V, Ibanez B (2020) Metoprolol blunts the time-dependent progression of infarct size. Basic Res Cardiol 115:55. https://doi.org/10.1007/s00395-020-0812-4
Ma Y (2021) Role of neutrophils in cardiac injury and repair following myocardial infarction. Cells 10:1676. https://doi.org/10.3390/cells10071676
Maugeri N, Baldini M, Ramirez GA, Rovere-Querini P, Manfredi AA (2012) Platelet-leukocyte deregulated interactions foster sterile inflammation and tissue damage in immune-mediated vessel diseases. Thromb Res 129:267–273. https://doi.org/10.1016/j.thromres.2011.12.001
Mauler M, Herr N, Schoenichen C, Witsch T, Marchini T, Hardtner C, Koentges C, Kienle K, Ollivier V, Schell M, Dorner L, Wippel C, Stallmann D, Normann C, Bugger H, Walther P, Wolf D, Ahrens I, Lammermann T, Ho-Tin-Noe B, Ley K, Bode C, Hilgendorf I, Duerschmied D (2019) Platelet serotonin aggravates myocardial ischemia/reperfusion injury via neutrophil degranulation. Circulation 139:918–931. https://doi.org/10.1161/CIRCULATIONAHA.118.033942
Migeotte I, Communi D, Parmentier M (2006) Formyl peptide receptors: a promiscuous subfamily of G protein-coupled receptors controlling immune responses. Cytokine Growth Factor Rev 17:501–519. https://doi.org/10.1016/j.cytogfr.2006.09.009
Niccoli G, Kharbanda RK, Crea F, Banning AP (2010) No-reflow: again prevention is better than treatment. Eur Heart J 31:2449–2455. https://doi.org/10.1093/eurheartj/ehq299
Noble PW, Bernatsky S, Clarke AE, Isenberg DA, Ramsey-Goldman R, Hansen JE (2016) DNA-damaging autoantibodies and cancer: the lupus butterfly theory. Nat Rev Rheumatol 12:429–434. https://doi.org/10.1038/nrrheum.2016.23
Nürnberg B (2000) Pertussis toxin as a pharmacological tool. In: Aktories K, Just I (eds) Bacterial protein toxins. Springer, Berlin, Heidelberg, pp 187–206
Nürnberg B (2000) Pertussis toxins as a pharmacological tool. In: Aktories K, Just I (eds) Bacterial protein toxins. Handbook of experimental pharmacology. Springer, Berlin, pp 187–206
Nürnberg B, Beer-Hammer S, Reisinger E, Leiss V (2024) Non-canonical G protein signaling. Pharmacol Ther 255:108589. https://doi.org/10.1016/j.pharmthera.2024.108589
Nürnberg B, Spicher K, Harhammer R, Bosserhoff A, Frank R, Hilz H, Schultz G (1994) Purification of a novel G-protein alpha 0-subtype from mammalian brain. Biochem J 300(Pt 2):387–394. https://doi.org/10.1042/bj3000387
Ott I, Neumann FJ, Gawaz M, Schmitt M, Schomig A (1996) Increased neutrophil-platelet adhesion in patients with unstable angina. Circulation 94:1239–1246. https://doi.org/10.1161/01.cir.94.6.1239
Page C, Pitchford S (2013) Neutrophil and platelet complexes and their relevance to neutrophil recruitment and activation. Int Immunopharmacol 17:1176–1184. https://doi.org/10.1016/j.intimp.2013.06.004
Peters MJ, Dixon G, Kotowicz KT, Hatch DJ, Heyderman RS, Klein NJ (1999) Circulating platelet-neutrophil complexes represent a subpopulation of activated neutrophils primed for adhesion, phagocytosis and intracellular killing. Br J Haematol 106:391–399. https://doi.org/10.1046/j.1365-2141.1999.01553.x
Pischke SE, Gustavsen A, Orrem HL, Egge KH, Courivaud F, Fontenelle H, Despont A, Bongoni AK, Rieben R, Tonnessen TI, Nunn MA, Scott H, Skulstad H, Barratt-Due A, Mollnes TE (2017) Complement factor 5 blockade reduces porcine myocardial infarction size and improves immediate cardiac function. Basic Res Cardiol 112:20. https://doi.org/10.1007/s00395-017-0610-9
Puhl SL, Steffens S (2019) Neutrophils in post-myocardial infarction inflammation: damage vs. resolution? Front Cardiovasc Med 6:25. https://doi.org/10.3389/fcvm.2019.00025
Reed GW, Rossi JE, Cannon CP (2017) Acute myocardial infarction. Lancet 389:197–210. https://doi.org/10.1016/S0140-6736(16)30677-8
Reffelmann T, Kloner RA (2002) Microvascular reperfusion injury: rapid expansion of anatomic no reflow during reperfusion in the rabbit. Am J Physiol Heart Circ Physiol 283:H1099–H1107. https://doi.org/10.1152/ajpheart.00270.2002
Reffelmann T, Kloner RA (2002) The “no-reflow” phenomenon: basic science and clinical correlates. Heart 87:162–168. https://doi.org/10.1136/heart.87.2.162
Romson JL, Hook BG, Kunkel SL, Abrams GD, Schork MA, Lucchesi BR (1983) Reduction of the extent of ischemic myocardial injury by neutrophil depletion in the dog. Circulation 67:1016–1023. https://doi.org/10.1161/01.cir.67.5.1016
Rossaint J, Margraf A, Zarbock A (2018) Role of platelets in leukocyte recruitment and resolution of inflammation. Front Immunol 9:2712. https://doi.org/10.3389/fimmu.2018.02712
Rossello X, Raposeiras-Roubin S, Latini R, Dominguez-Rodriguez A, Barrabes JA, Sanchez PL, Anguita M, Fernandez-Vazquez F, Pascual-Figal D, De la Torre Hernandez JM, Ferraro S, Vetrano A, Perez-Rivera JA, Prada-Delgado O, Escalera N, Staszewsky L, Pizarro G, Aguero J, Pocock S, Ottani F, Fuster V, Ibanez B, REBOOT-CNIC investigators (2022) Rationale and design of the pragmatic clinical trial tREatment with Beta-blockers after myOcardial infarction withOut reduced ejection fracTion (REBOOT). Eur Heart J Cardiovasc Pharmacother 8:291–301. https://doi.org/10.1093/ehjcvp/pvab060
Roth GA, Abate D, Abate KH, Abay SM, Abbafati C, Abbasi N, Abbastabar H, Abd-Allah F, Abdela J, Abdelalim A, Abdollahpour I, Abdulkader RS, Abebe HT, Abebe M, Abebe Z, Abejie AN, Abera SF, Abil OZ, Abraha HN, Abrham AR, Abu-Raddad LJ, Accrombessi MMK, Acharya D, Adamu AA, Adebayo OM, Adedoyin RA, Adekanmbi V, Adetokunboh OO, Adhena BM, Adib MG, Admasie A, Afshin A, Agarwal G, Agesa KM, Agrawal A, Agrawal S, Ahmadi A, Ahmadi M, Ahmed MB, Ahmed S, Aichour AN, Aichour I, Aichour MTE, Akbari ME, Akinyemi RO, Akseer N, Al-Aly Z, Al-Eyadhy A, Al-Raddadi RM, Alahdab F, Alam K, Alam T, Alebel A, Alene KA, Alijanzadeh M, Alizadeh-Navaei R, Aljunid SM, Aa A, Alla F, Allebeck P, Alonso J, Altirkawi K, Alvis-Guzman N, Amare AT, Aminde LN, Amini E, Ammar W, Amoako YA, Anber NH, Andrei CL, Androudi S, Animut MD, Anjomshoa M, Ansari H, Ansha MG, Antonio CAT, Anwari P, Aremu O, Ärnlöv J, Arora A, Arora M, Artaman A, Aryal KK, Asayesh H, Asfaw ET, Ataro Z, Atique S, Atre SR, Ausloos M, Avokpaho EFGA, Awasthi A, Quintanilla BPA, Ayele Y, Ayer R, Azzopardi PS, Babazadeh A, Bacha U, Badali H, Badawi A, Bali AG et al (2018) Global, regional, and national age-sex-specific mortality for 282 causes of death in 195 countries and territories, 1980–2017: a systematic analysis for the Global Burden of Disease Study 2017. The Lancet 392:1736–1788. https://doi.org/10.1016/S0140-6736(18)32203-7
Ruiz-Arguelles A, Alarcon-Segovia D (2001) Novel facts about an old marker: the LE cell. Scand J Clin Lab Invest Suppl 235:31–37. https://doi.org/10.1080/003655101753352022
Rusnak JM, Kopecky SL, Clements IP, Gibbons RJ, Holland AE, Peterman HS, Martin JS, Saoud JB, Feldman RL, Breisblatt WM, Simons M, Gessler CJ Jr, Yu AS (2001) An anti-CD11/CD18 monoclonal antibody in patients with acute myocardial infarction having percutaneous transluminal coronary angioplasty (the FESTIVAL study). Am J Cardiol 88:482–487. https://doi.org/10.1016/s0002-9149(01)01723-4
Saxena A, Russo I, Frangogiannis NG (2016) Inflammation as a therapeutic target in myocardial infarction: learning from past failures to meet future challenges. Transl Res 167:152–166. https://doi.org/10.1016/j.trsl.2015.07.002
Schanze N, Hamad MA, Nuhrenberg TG, Bode C, Duerschmied D (2023) Platelets in myocardial ischemia/reperfusion injury. Hamostaseologie 43:110–121. https://doi.org/10.1055/a-1739-9351
Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A (2012) Fiji: an open-source platform for biological-image analysis. Nat Methods 9:676–682. https://doi.org/10.1038/nmeth.2019
Schofield ZV, Woodruff TM, Halai R, Wu MC, Cooper MA (2013) Neutrophils–a key component of ischemia-reperfusion injury. Shock 40:463–470. https://doi.org/10.1097/SHK.0000000000000044
Silvestre-Roig C, Braster Q, Ortega-Gomez A, Soehnlein O (2020) Neutrophils as regulators of cardiovascular inflammation. Nat Rev Cardiol 17:327–340. https://doi.org/10.1038/s41569-019-0326-7
Slastnikova TA, Ulasov AV, Rosenkranz AA, Sobolev AS (2018) Targeted intracellular delivery of antibodies: the state of the art. Front Pharmacol 9:1208. https://doi.org/10.3389/fphar.2018.01208
Spangrude GJ, Sacchi F, Hill HR, Van Epps DE, Daynes RA (1985) Inhibition of lymphocyte and neutrophil chemotaxis by pertussis toxin. J Immunol 135:4135–4143
Stojkov D, Gigon L, Peng S, Lukowski R, Ruth P, Karaulov A, Rizvanov A, Barlev NA, Yousefi S, Simon HU (2022) Physiological and pathophysiological roles of metabolic pathways for NET formation and other neutrophil functions. Front Immunol 13:826515. https://doi.org/10.3389/fimmu.2022.826515
The top 10 causes of death. https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death. Accessed 3 Apr 2024
Vakeva AP, Agah A, Rollins SA, Matis LA, Li L, Stahl GL (1998) Myocardial infarction and apoptosis after myocardial ischemia and reperfusion: role of the terminal complement components and inhibition by anti-C5 therapy. Circulation 97:2259–2267. https://doi.org/10.1161/01.cir.97.22.2259
Vinten-Johansen J (2004) Involvement of neutrophils in the pathogenesis of lethal myocardial reperfusion injury. Cardiovasc Res 61:481–497. https://doi.org/10.1016/j.cardiores.2003.10.011
Wan Q, Xu C, Zhu L, Zhang Y, Peng Z, Chen H, Rao H, Zhang E, Wang H, Chu F, Ning X, Yang X, Yuan J, Wu Y, Huang Y, Hu S, Liu DP, Wang M (2022) Targeting PDE4B (Phosphodiesterase-4 Subtype B) for cardioprotection in acute myocardial infarction via neutrophils and microcirculation. Circ Res 131:442–455. https://doi.org/10.1161/CIRCRESAHA.122.321365
Wiege K, Ali SR, Gewecke B, Novakovic A, Konrad FM, Pexa K, Beer-Hammer S, Reutershan J, Piekorz RP, Schmidt RE, Nürnberg B, Gessner JE (2013) Gαi2 is the essential Gαi protein in immune complex-induced lung disease. J Immunol 190:324–333. https://doi.org/10.4049/jimmunol.1201398
Ye RD, Boulay F, Wang JM, Dahlgren C, Gerard C, Parmentier M, Serhan CN, Murphy PM (2009) International union of basic and clinical pharmacology. LXXIII. Nomenclature for the formyl peptide receptor (FPR) family. Pharmacol Rev 61:119–161. https://doi.org/10.1124/pr.109.001578
Yellon DM, Hausenloy DJ (2007) Myocardial reperfusion injury. N Engl J Med 357:1121–1135. https://doi.org/10.1056/NEJMra071667
Zarbock A, Deem TL, Burcin TL, Ley K (2007) Galphai2 is required for chemokine-induced neutrophil arrest. Blood 110:3773–3779. https://doi.org/10.1182/blood-2007-06-094565
Acknowledgements
We thank Sandra Schwegmann and Renate Riehle for technical assistance. This work was supported by grants to D.K., R.L., B.S., P.R., B.N. and S.B.-H. of the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation). Further support came from the DFG-funded RTG2816 to B.S., B.W., B.N. and S.B.-H., from the Clusters of Excellence CMFI and iFIT (EXC2124 and EXC2180) to B.S., S.B.-H. and B.W., University of Tübingen, Germany, funded by the DFG under Germany’s Excellence Strategies—EXC2124—390838134 and EXC2180—390900677, by TR156 to B.S., by ICEPHA Research Training Group grants to S.B.-H., B.N. and R.L., by a IZKF Junior Research Group grant (2561-0-0) of the Medical Faculty Tübingen (MFT) to B.W., and by MFT start-up funding to establish RTG2816 to B.N.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflict of interest.
Supplementary Information
Below is the link to the electronic supplementary material.
Supplementary file3 (MP4 1840 KB)
Supplementary file4 (MP4 2942 KB)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Köhler, D., Leiss, V., Beichert, L. et al. Targeting Gαi2 in neutrophils protects from myocardial ischemia reperfusion injury. Basic Res Cardiol (2024). https://doi.org/10.1007/s00395-024-01057-x
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00395-024-01057-x