
Abstract
The benefits of remote ischaemic conditioning (RIC) have been difficult to translate to humans, when considering traditional outcome measures, such as mortality and heart failure. This paper reviews the recent literature of the anti-inflammatory effects of RIC, with a particular focus on the innate immune response and cytokine inhibition. Given the current COVID-19 pandemic, the inflammatory hypothesis of cardiac protection is an attractive target on which to re-purpose such novel therapies. A PubMed/MEDLINE™ search was performed on July 13th 2020, for the key terms RIC, cytokines, the innate immune system and inflammation. Data suggest that RIC attenuates inflammation in animals by immune conditioning, cytokine inhibition, cell survival and the release of anti-inflammatory exosomes. It is proposed that RIC inhibits cytokine release via a reduction in nuclear factor kappa beta (NF-κB)-mediated NLRP3 inflammasome production. In vivo, RIC attenuates pro-inflammatory cytokine release in myocardial/cerebral infarction and LPS models of endotoxaemia. In the latter group, cytokine inhibition is associated with a profound survival benefit. Further clinical trials should establish whether the benefits of RIC in inflammation can be observed in humans. Moreover, we must consider whether uncomplicated MI and elective surgery are the most suitable clinical conditions in which to test this hypothesis.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Background: challenges and new directions in RIC
Bringing the promise of remote ischaemic conditioning (RIC) to fruition in the clinical arena, remains a major challenge [23, 58]. RIC involves the sequential occlusion and reperfusion, of an arterial vessel distant to the target organ. It has demonstrated multi-organ benefit and cross-species cardiovascular protection in studies of ischaemia [13], and is highly effective in preventing damage in animal models of myocardial infarction [63]. However, large-scale trials in humans with ST-elevation myocardial infarction (STEMI) have proved inconclusive, with respect to traditional outcome measures of myocardial infarct size, heart failure and survival [36, 48, 58]. RIC confers cardioprotection via a combination of humoral and neuronal pathways. These link the protective, “conditioning” response to ischaemia induced in the remote vascular bed, to the target tissue at risk of severe ischaemia and reperfusion (I/R) injury [7, 80]. Whilst many potential humoral factors have been proposed such as nitric oxide (NO) and nitrite, adenosine, stromal-derived factor 1α (SDF-1α) and glucagon-like peptide-1 (GLP-1); the underlying immunological pathways remain poorly defined [8, 27, 58, 80, 118].
The effectiveness of RIC in preventing myocardial I/R injury in humans has been assessed in numerous studies, most notably the CONDI-2/ERIC-PPCI trial, an international, prospective, single-blind, randomised controlled outcome trial in 5,401 patients with ST-elevation myocardial infarction (STEMI) undergoing primary percutaneous coronary intervention (PPCI), in which no improvement in clinical outcomes (cardiac death or hospitalisation for heart failure) were seen after 12 months [48]. Importantly, however, no harmful effects were seen. Many theories have sought to explain why the success of RIC in animal models has not been directly translatable to humans [58, 64]. One important observation, highlighted in two recent articles, is that the population studied in the CONDI-2/ERIC-PPCI trial may not have been significantly ‘high-risk’ enough, to demonstrate an improvement in the primary outcome measures of infarct size and survival [11, 49, 65]. It is likely, however, that a plethora of factors make the human model of cardioprotection more complex than the animal population, who are often devoid of chronic endothelial dysfunction and medical co-morbidities [57, 65]. Moreover, the timing and size of an experimentally induced infarct in animals can be carefully predicted and is reproducible. The key to translation may lie in better understanding of the underlying mechanisms, and how these can be applied to human physiology in individual conditions.
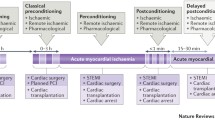
We have embarked upon a new era of inflammation in cardioprotection. With the arrival of novel diseases such as COVID-19, and its associated cardiovascular complications, there is a need to re-fashion current cardioprotective strategies. Moreover, the pandemic has identified the need to further investigate the effects of infection on the heart in addition to ischaemia. Whilst many reviews have considered RIC as an infarct limiting intervention, the effects on the innate immune system are less well documented. We present this review of the anti-inflammatory effects of RIC, and the implications for future organ-protective therapies (Fig. 1).

RIC mediates inflammation in vivo by immune conditioning and cytokine inhibition, anti-apoptotic pathways and the reduction of NLRP3 inflammasomes, (pyroptosis). Decreased oxygen tension adjusts cell metabolism and limits apoptosis. Shear stress induces flow-mediated dilatation, which enables the transfer of exosomes carrying anti-inflammatory, chemo-active compounds. RISK reperfusion injury salvage kinase, ERK extracellular signal related kinase, SAFE survivor activating factor enhancement, MTP mitochondrial transition pore, HIF-1α hypoxia inducible factor 1 alpha, NFκβ nuclear factor kappa beta
The inflammatory hypothesis of organ protection
The ‘Inflammatory Hypothesis’ is a term used to define the role of the innate immune system in I/R injury. Following reperfusion in myocardial infarction, acute inflammation contributes to endothelial dysfunction, the development of cardiac failure and poor left ventricular remodelling [99, 109, 157]. Such damage persists well after the initial ischaemic insult has ended, and the infarct related territory has been reperfused [148]. Upon reperfusion, resident immune cells detect the presence of danger-associated molecular proteins (DAMPs) and necrotic tissue in the area of infarction [157]. DAMPS combine with other ‘alarmin’ molecules, such as high mobility group box one protein (HMGB1), extracellular DNA and histones, to trigger the secretion of pro-inflammatory cytokines via the cell-mediated nuclear factor kappa beta (NF-κB) pathway (Fig. 2) [128, 157]. Macrophages, which engage with DAMPs via toll-like receptors (e.g. TLR4), are also responsible for the synthesis of pro-inflammatory molecules including cytokines and the NLRP3 inflammasome (Fig. 2).

Cytokine release in inflammation is mediated by DAMPS binding to TLR4/RAGE receptors on the cell membrane. Both pathways activate NF-κB and NLRP3 inflammasome production, resulting in secretion of pro-inflammatory cytokines [75, 150]. RAGE results in further production of HMGB1 [151]. NLRP3 inflammasome activation results in caspase mediated cell death [119]. Several studies haves demonstrated that RIC modulates NF-κB activity via in both ischaemia and endotoxaemia [74, 76, 92, 117, 130]. TLR4 Toll-like receptor 4, DAMPS damage associated molecular patterns, HMGB1 high mobility group box 1, TNF-α tumour necrosis factor alpha, LPS lipopolysaccharide, RAGE Receptor for advanced glycation end-products, ERK extracellular signal related kinase, MAPK mitogen activated protein kinase, EGR-1 early growth response 1, TAK1 transforming factor-β-activated kinase 1, Iκβ inhibitor kappa beta kinase, NF-κβ nuclear factor kappa beta
Following myocardial ischaemia, four varieties of programmed cell death are observed, including apoptosis, necrosis, necroptosis and pyroptosis [21, 61]. Apoptosis is triggered by death receptors (DR) and intracellular signals, and does not induce the release of cellular contents beyond the confines of the cell. It is largely mediated via caspase-8, 9 and Bc12. Both necroptosis and pyroptosis, enhance inflammation by facilitating the release of interleukins such as IL-1β and IL-18. [61, 135]. Pyroptosis is a type of programmed cell death (PMD) which is closely related to activity of the NLRP3 inflammasome and NF-κβ. Here, activated caspase-1 facilitates the release of interleukins via the Gasdermin (GSDMD) membrane pore. Not only does inflammasome activation contribute to cytokine release and extent of inflammation, but also to infarct size, following myocardial ischaemia [29, 135]. The latter is, therefore, an attractive target for cardioprotection [1]. Whilst there are clear associations between RIC and anti-apoptotic pathways [122], it remains unclear whether remote conditioning can directly limit pyroptosis (and at which step). There is, however, evidence for bi-directional cross-talk between caspase-1 and caspase-8 (anti-apoptotic) suggesting that inhibition of apoptosis might also influence other forms of cell death [29].
The microvasculature has a central role in mediating inflammation during I/R, and endothelial cells are notoriously more resistant to hypoxia than other cell types [109, 137]. They are, however, sensitive to the presence of reactive oxygen species (ROS) and the changes in NO metabolism that accompany an ischaemic challenge [4, 47, 125]. The coronary endothelial system has an important role in detecting mechanical and flow-mediated changes post-infarct, in addition to mediating the vascular immune response. In the case of myocardial infarction, within the coronary arteries, exposed atherothrombotic plaque causes cells of the innate immune system to migrate to the inflamed vessel, prompting further cytokine release. Increased neutrophil recruitment to the area of vascular inflammation is mediated by cytokines, the complement cascade (including IL-8; C5) and directly via ROS [109, 125]. Polymorphonuclear neutrophils (PMNs) are attracted to the endothelium via selectins and proceed to adhere and transmigrate into the microvasculature by binding to integrins and ICAM adhesion molecules [115].
As the inflammatory hypothesis has evolved, immune-modulating therapies have been extensively investigated in both myocardial and cerebral infarction, and have finally met with some recent success. The CANTOS trial demonstrated that the IL-1β blocker, Canakinumab, was able to reduce the risk of future coronary and cerebral atherosclerotic events [120]. This benefit was, however, associated with a mild increase in fatal infection, which should not be disregarded. Nevertheless, the CANTOS trial was important in measuring outcomes of inflammation in cardiac protection, and included measurements of cytokines involved in IL-6 signalling and C-reactive protein (CRP) [120]. Both biomarkers were deemed to be prognostic, with respect to the primary end-point of non-fatal atherosclerotic events. This might suggest that cytokines are valuable biomarkers in predicting future adverse events in patients with myocardial infarction [120]. As further justification to pursue cytokine inhibition in I/R, two recent clinical studies of STEMI patients have demonstrated that IL-6 and IL-8 are associated with worsening clinical outcome [42, 129].
In the VCUART3 trial, the recombinant interleukin-1 receptor antagonist, Anakinra reduced CRP levels at 14-days post STEMI, and significantly reduced mortality and re-hospitalisation. Unlike CANTOS, there was no increase in severe infection reported following administration [2]. Similarly, IL-1 inhibition was well tolerated when administered subcutaneously to patients with acute cerebral infarction, in the SCIL-STROKE trial. In this study, Anakinra significantly lowered levels of IL-6 and plasma CRP (p < 0.001) [132], suggesting that it is a key mediator of the inflammatory response in cerebral ischaemia [72]. Although experimental results are promising in both myocardial and cerebral infarction, immune modulation is not currently used as routine in STEMI patients. Inhibition of individual cytokines must be carefully risk stratified, due to the close association between innate immune suppression and the development of humoral immunity [126].
Cytokines in myocardial infarction and infection
The pro-inflammatory cytokines released in response to myocardial infarction include IL-1α, IL-1β, IL-6, TNFα, IL-8, IL-18 and small chemokine molecules such as monocyte chemoattractant protein 1 (MCP-1). These cytokines are released by a mixture of damaged cardiomyocytes, macrophages and activated endothelium [109, 115]. The primary aim of cytokine release is to activate and attract immune cells to the area of inflammation, to enable the removal of damaged products via phagocytosis [5]. In the infarcted heart, cytokine release is triggered by TLR4 signalling pathways, the activation of NF-κB in circulating macrophages and by reactive oxygen species (ROS), which interact with IL-6. The resultant release of IL-1β further stimulates additional pro-inflammatory molecules [17, 107]. Cytokine ‘cross-talk’ exists between immune cells and the activated endothelium, which maintains the amplitude of the acute inflammatory response [133].
There are two distinct phases of inflammation following myocardial infarction: an initial, pro-inflammatory phase in which damaged cells and debris are eliminated, and a second, anti-inflammatory reparative phase leading to wound healing and scar formation. Cytokines also have a key role in tissue repair. IL-6, (in a second window of cytokine release), IL-10, transforming growth factor beta (TGF-β) and a sub-population of T-lymphocytes known as ‘T Regulatory cells’ (Treg) have all been associated with supressing the pro-inflammatory response and steering the immune system towards repair and resolution following I/R [99, 107]. Macrophages expressing altered interleukin signals (such as IRAK-M) are able to downregulate other macrophages, contributing towards anti-inflammatory ‘stop’ signals [107, 155]. For suitable healing to take place, the amplitude of the initial macrophage activation syndrome must not outweigh that of regulatory immune cells. In myocardial infarction, cytokine release predominantly occurs on the border of the infarct zone, but can also be present in non-ischaemic tissue [17].
The cardiovascular system is vulnerable to inflammatory insult induced by infection and cytokine damage, including viral myocarditis, septic cardiomyopathy and recently the acute cardiovascular syndrome of COVID-19 [40, 68, 86]. The latter encompasses thrombotic acute coronary syndromes, myocarditis and pulmonary emboli, amongst other complications of pre-existing cardiac disease [40]. The cytokine response to infection is similar in the acute phase to myocardial infarction, and is triggered by DAMPS and danger signals following pathogenic invasion (stimulating IL-6, TNFα and IL-1β). In some individuals, following exposure to endotoxins or viral antigens, the innate immune system becomes hyper-active and a ‘cytokine storm’ develops. Here, further cytokines are secreted (IL-17, IL-8, G-CSF, MCP-1, CCL1-3, IFN-y) and re-circulated via the dysfunctional endothelium. The following present an in-depth review of cytokine response during infection [84, 112, 153]. This has been a topic of much importance in COVID-19 and culminates in pan-vascular and multi-organ damage [102].
Given that multiple pathologies affect the heart in this pandemic era, it is important to consider protective strategies which will target the inflammation of both I/R and pathogenic invasion. Below we will consider the evidence for RIC as a cytokine mediator in both animal and human studies.
Animal studies of RIC and inflammation
Table.1 demonstrates the animal studies of RIC and cytokine release, performed within the last 5 years, following myocardial infarction and reperfusion [9, 15, 33, 114, 136, 141, 152] Across several studies, RIC was associated with reduced levels of the pro-inflammatory cytokines, IL-1β, TNF-α and HMGB1 following reperfusion [114, 152]. Likewise, RIC applied 24h prior to myocardial I/R, appeared to increase levels of the protective cytokine IL-10 [15], which governs the amplitude of the cytokine response [34, 127]. In vivo, this increase in IL-10 was STAT5 mediated [15]. STAT5 is linked to the survivor activating factor enhancement pathway (SAFE) and operates downstream of JAK (Janus Kinase) in human myocardial injury [66]. Similarly, previous literature has discussed the protective effects of IL-10, limiting I/R injury via STAT3 [44, 79, 100]. In another study, RIC was associated with an increase in IL-6 (which the authors propose has reparative function within the infarcted myocardium) via early growth response protein 1 (EGR-1), a molecule upstream of many apoptotic pathways [9]. In animal models of myocardial infarction, RIC combined with other therapies such as sevoflurane post-conditioning (anaesthesia following ischaemia and prior to onset of reperfusion) vagal nerve stimulation or atorvastatin (HMG-CoA reductase inhibitor), provided additive organ protection and reduced inflammation [33, 141, 152].
RIC has proven effective at attenuating pro-inflammatory cytokine release in animal models of cerebral infarction, renal, pulmonary and hepatic reperfusion injury [31, 70, 83, 145, 156]. In a population of aged rats undergoing middle cerebral artery occlusion (mCAO), RIC significantly reduced levels of IL-1, IL-6 and IFN-γ in both plasma and the brain, whilst reducing the expression of hypoxia inducible factor (HIF-1α). Although HIF-1α is linked to cardioprotection by stimulating pro-survival pathways, it can equally induce a shift towards anaerobic glycolysis in macrophages resulting in increased cytokine manufacture [19]. It is previously discussed that the role of HIF-1α in cardioprotection is not fully understood, although deficiency in mice appears to dampen a reduction in infarct size [59]. In a murine model of hepatic I/R injury, RIC significantly reduced levels of intrinsic liver enzymes, IL-6 and TNF-α [150]. Furthermore, this anti-inflammatory effect was mediated by the HMGB1/TLR4/NF-κB pathway, an established mechanism of cytokine release [150]. Pro-inflammatory pathways involving NF-κB, including notch signalling [67, 121] will be considered in greater detail below.
RIC reduces inflammatory cytokine levels and improves survival in rodent models of lipopolysaccharide (LPS) induced endotoxaemia [68, 74, 76]. In mice receiving three cycles of hind limb I/R (10 min ischaemia/10 min reperfusion) prior to LPS exposure, there was a significant survival benefit from RIC (10% of the control group survived vs 60% of the intervention group; p < 0.001). In the same study, histology revealed a reduction in the diffuse parenchymal pulmonary inflammation associated with LPS-induced acute lung injury, and a reduction of cytokines in bronchoalveolar fluid (TNF-α, IL-1β and IL-6). It was further demonstrated that RIC mediates cytokine reduction via a downregulation of NF-κB and myeloperoxidase (MPO) pathways [76]. MPO is associated with increased neutrophil influx to areas of inflammation and, therefore, promotes the release of pro-inflammatory cytokines from neutrophils, which are sentinel cells in the inflammatory response [78].
LPS induces a potent inflammatory state, and causes cytokines and alarmins (e.g. HMGB1, HSP70, histones) to be released in response to infection. Bacterial DAMPs/LPS trigger NF-kB activity via TLR4, which potentiates further inflammasome and cytokine release (Fig. 2) [77]. In septic cardiomyopathy, the myocardial depressant cytokine HMBG1 is central to the stimulation of inflammation and upregulates the coagulation cascade [68, 94]. There is also evidence to suggest that the cytokines IL-1, TNF-α, and IL-6 play a pivotal inflammatory role in endotoxaemia, and this has also been observed in COVID-19 hyper-inflammation [113]. IL-6 in particular, can target the vasculature to induce vasodilatation and disruption of endothelial tight junctions, which results in capillary leak and circulatory collapse [138]. In view of the above, RIC may be a novel treatment modality with cytokine-modulating potential, without the associated side effects of immune-modulating pharmacotherapy.
Human studies of RIC and cytokine response
Contrary to the findings in animal models, the majority of recent randomised control clinical trials have been unable to demonstrate a clear effect of RIC on pro-inflammatory cytokine release (Table.2) [37, 39, 105, 106, 108, 149, 158]. However, to date, cytokines have predominantly been measured in small studies only (less than 100 participants). Despite this, the largest two trials (n = 65, n = 90 participants) demonstrated cytokine attenuation in the treatment group undergoing RIC prior to off-pump CABG and colorectal surgery, respectively [53, 140]. In the latter study, levels of IL-1β and TNF-α were significantly reduced for up to 3 days post-operatively (p < 0.01) in patients receiving RIC, compared with controls. Surgery was performed for a range of pathologies, including colorectal neoplasm [53]. Considering that RIC has conferred a profound survival benefit in animal models of intra-abdominal injury, it could be suggested that the inflammatory benefits in humans might differ between clinical conditions [110].
Humoral pathways of inflammation and cell survival
RISK, SAFE and HIF-1α
Many studies have demonstrated, that RIC reduces cardio myocyte cell death in I/R and other pathologies [31, 63, 69, 73, 154]. The reperfusion injury salvage kinase (RISK) and survivor activating factor enhancement (SAFE) pathways, are fundamental in protecting the heart from I/R injury [44, 122]. The RISK pathway acts to prevent opening of the mitochondrial permeability transition pore (MTP), when activated before reperfusion [50]. There is a clear role for RIC and protein kinase C (PKC), which has cardioprotective actions in both ischaemia and reperfusion [63]. PKC regulates the opening of the MTP by mediating KATP dependent channels and controlling calcium influx [51]. The RISK pathway is also activated by adenosine, bradykinin, and sphingosine which bind to receptors on the cell membrane [122]. This leads to upregulation of endothelial nitric oxide synthase (eNOS) and nitric oxide (NO) which induces vascular vasodilatation in the heart and vasculature. Cross-talk exists between the RISK and SAFE pathways to augment cell survival, and this has been demonstrated across different species in studies of RIC and I/R [131].
The SAFE pathway (first described by Lecour in 2009) is an alternative pro-survival axis to RISK. SAFE was found to act on the MTP when ERK/MAPK (RISK) were inactivated and thus its independent actions were demonstrated [44, 88]. SAFE describes the pathway initiated by the binding of TNF- α to the plasma membrane and the subsequent activation of JAK/STAT transcription factors [60]. In experimental studies of myocardial I/R, SAFE has upregulated STAT3, (likely via the Sphingosine Kinase 1 enzyme) [44, 79]. In human studies, RIC is associated with the upregulation of myocardial STAT5 [66]. STAT is able to activate NF-κB to influence the MTP and promote cell survival; however, NF-κB is itself pro-inflammatory, and coupled to cytokine secretion and pyroptosis/the NLRP3 inflammasome (Fig. 2). This presents somewhat of a conundrum in the treatment of inflammatory cardiac conditions. JAK/STAT can further evoke ‘notch’ signalling between local monocytes, which is directly linked to increased IL-6 manufacture and downregulation of the anti-inflammatory M2 macrophage/IL-10 [67, 121]. As the SAFE pathway can activate JAK/STAT and NF-κB, it can also promote adverse cardiac remodelling and heart failure [44]. With this in mind, other humoral factors/pathways must be triggered by RIC to account for a reduction in pro-inflammatory cytokines.
The molecule hypoxia inducible factor (HIF-1α) has been linked to pro-survival signalling in I/R [63]. During periods of reduced oxygen tension, HIF-1α mediates a shift in mitochondrial metabolism towards anaerobic glycolysis, which induces production of pyruvate dehydrogenase kinase 1 (PDK1) and limits entry of acetyl-CoA into the TCA cycle. This acts to preserve cellular energy and limit apoptosis [19, 90]. In differentiated macrophages, however, this metabolic change results in increased synthesis of cytokines such as IL-1β and IL-18 via the NF-κB pathway [19, 143]. This is somewhat paradoxical, as increased levels of HIF-1α have been associated with cardioprotection following RIC [59, 147]. It is possible that repeated stimulation of HIF-1α causes uncoupling of cytokine synthesis and immune tolerance; as is the case in other TLR4-dependent pathways [6]. It is already established that persistently elevated levels of HIF-1α can induce hypoxia tolerance [89]. Again, HIF-1α alone cannot explain the interaction between RIC and cytokine levels observed in animal studies [114, 152].
Stromal-derived factor (SDF-1α/CXCR4) has also been associated with HIF-1α and cardioprotection secondary to reduced apoptosis and upregulation of PI3K/ERK1/2 (RISK). Exogenous SDF-1α protects human myocardium from I/R injury [98] and is released from endothelial cells during RIC [12, 27]. In a rodent model of spinal cord injury, infused SDF-1α reduced levels of IL-1β, IL-18, TNF-α, and NLRP3 inflammasome production, suggesting that it has anti-inflammatory actions [150].
NF-κβ and associated inflammatory pathways
NF-κβ is an important molecule and transcription factor, involved in all aspects of inflammation and tumour activity. In addition to stimulation of the NLRP3 inflammasome and pyroptosis (discussed extensively above) it has many anti-apoptotic actions, making upstream inhibition problematic in inflammatory diseases [134, 146]. In response to this observation, strategies such as IL-1 inhibition and caspase-1 inhibition have been proposed in the treatment of cardiac inflammation [1]. NF-κβ is coupled to other proteins such as Iκβ (inhibitor of kappa beta) which enable self-regulation. Well-known anti-inflammatory drugs such as glucocorticoids e.g. dexamethasone can interact with inhibitors of NF-κβ, to reduce cytokines and the inflammatory response [146]. It is interesting then to note that RIC has also modified Iκβα proteins in a rodent model of acute lung injury, leading to reduced activity of NF-κβ and reduced TNF-α, IL-1β and IL-6 secretion [76].
Several other studies have proposed that RIC can suppress the TLR4/NF-κB/inflammasome axis and reduce cytokine secretion [74, 130]. Moreover, there are multiple pathways that are linked to NF-kB which are mediated by TLR receptors, and HMGB1 [3, 121]. Activity of the JAK/STAT pathway has been coupled to RIC-mediated cytokine modification as described [15, 124]. The receptor of advanced glycosylation end-products receptor (RAGE) pathway is associated with inflammasome production and HMGB1, (Fig. 2). Activation of RAGE is pro-inflammatory, and this can also downregulate RISK [124]. In mice undergoing RIC following myocardial ischaemia (RICPost), a decrease in infarct size was associated with a decrease in cardiac RAGE expression and levels of HMGB1 [142]. This may indicate that (as yet unidentified) humoral factors stimulated by RIC, can inhibit RAGE. The evidence at present for this is limited and further research is required.
To summarise, further humoral intermediaries may exist to link the vascular phenomenon of RIC to the above inflammatory pathways, and the findings of pre-clinical studies cannot be explained by RISK/SAFE alone. It seems most feasible, that such circulating anti-inflammatory factors might originate from the local trigger vessel; however, it is also recognised that there is a role of regional and distant vasculature [123]. Identifying these intermediate compounds and their mechanisms, remains a priority; as targeting both pro-survival and anti-inflammatory pathways in synergy could result in maximum cardiac protection.
Micro and macrovascular humoral factors
Following myocardial I/R, an increased number of neutrophils in the resistance vessels contribute to local vasoconstriction, microvascular obstruction and ‘no reflow’ [57]. Meanwhile, there is further immune cell influx (including mast cells), platelet activation and upregulation of the clotting cascade via tissue factor and Von Willebrand’s factor [56, 101]. Cytokines induce disruption of endothelial tight junctions and this culminates in leakage from capillaries into the extracellular space and the concurrent presence of micro-vessel haemorrhage and thrombi. Both no reflow and MVO post STEMI are considered prognostic, and this relationship is independent to infarct size [82]. It is, therefore, important to investigate ways to target this phenomenon [57]. In a large clinical trial of 696 STEMI patients (LIPSIA CONDITIONING), neither RIC alone nor in combination with post-conditioning following PPCI, demonstrated any reduction in MVO following cardiac MRI. There was, however, a significant improvement in myocardial salvage index in the cohort who received RIC and post-conditioning in combination (p = 0.02) [32].
Despite these results in larger clinical trials, RIC has increased both macro and microvascular flow on ultrasound Doppler, in two studies [85, 95] and improved forearm blood flow (venous occlusion plethysmography) in healthy male volunteers [47]. Both RIC and NO donors such as glycerin trinitrate (GTN) improve endothelial-mediated dilatation and reduce vascular reperfusion injury. Given that GTN in combination with RIC confers no additional endothelial protective benefit, it is hypothesised that conditioning utilises NO pathways to induce vasodilation and organ protection [47]. The vascular release of extracellular vesicles and exosomes in response to RIC is further considered below.
Does RIC mediate innate immune cell activity?
NETs and neutrophil recruitment
Neutrophils are a considerable driver of the inflammatory process, and are responsible for significant cytokine release and tissue damage [34, 111]. Activated neutrophils release neutrophil extracellular traps (NETs), which are structures containing DNA and histones, amongst other inflammatory molecules. Neutrophils are stimulated to release NETs following exposure to alarmins and defensins, and this is mediated by neutrophil elastase (NE), myeloperoxidase (MPO) and activated platelets, which bind to cellular HMGB1 receptors. NETs can induce host cell death in response to immune invasion, in a slow apoptotic process called NETosis [10, 104, 111]. NETs can also cause inflammatory obstruction within the microvasculature, contributing to MVO. NETosis is associated with worsening outcomes post STEMI [54].
The impact of RIC on neutrophils, adhesion molecules and enzymes such as MPO, has predominantly been investigated in animal models of I/R injury, endotoxemia and acute kidney injury [30, 69, 76]. In rats undergoing RIC by femoral artery occlusion, prior to 45 min of left renal ischaemia, RIC significantly reduced expression of ICAM-1 adhesion molecules, in addition to pro-inflammatory cytokines (TNF-α, IL-1β) [69]. In humans with ulcerative colitis and moderate disease activity, however, RIC did not reduce neutrophil infiltration or other markers of inflammation such as CRP, following rectal biopsies [39]. Conversely, in a large randomised control trial (n = 206 participants) of patients undergoing ablation for atrial fibrillation, RIC significantly reduced neutrophil–lymphocyte ratio and levels of CRP up to 48 h post-operatively (p < 0.05) [71]. A recent review has recognised neutrophils as important targets in cardioprotection [5], especially considering that these cells have the potential to polarise macrophages and enhance the acute inflammatory response.
Lymphocytes, monocytes and splenic response to RIC
In animals, RIC influences both circulating leucocytes and immune precursors in the spleen in models of cerebral ischaemia [14, 96]. In rats undergoing middle cerebral artery infarction, RIC was associated with increased splenic volume and lymphocytes, with reduced cytotoxic T cells and natural killer cells (NK) in cerebral tissue at day 3 [14]. These changes were negated when animals underwent splenectomy, suggesting an underlining mechanism of splenic conditioning. Similarly, in a second study, RIC increased a colony of non-inflammatory monocytes (CD43+/CD172a+), in addition to increasing circulating B lymphocytes [96]. This is interesting given that in infection and immunity, B lymphocytes are central to immune conditioning and ‘immunological-memory’ [77]. Others have proposed that RIC utilises a ‘splenic-vagal nerve’ axis of cardioprotection, given that the cardioprotective effects of RIC are abated in animals undergoing splenectomy and vagotomy. Splenectomy is associated with reduced amounts of STAT3 (SAFE pathway) but not all related humoral factors have been fully identified [62, 93]. It is proposed that the anti-inflammatory cytokine, IL-10 may be important in an RIC-mediated splenic axis of cardioprotection [62].
Platelets and the coagulation cascade
With respect to platelet function and the coagulation cascade, animal models have demonstrated the fibrinolytic and anticoagulant benefits of RIC [97]. However, this benefit has not been observed in humans [41, 46, 52, 116]. Following the ERIC-PPCI/CONDI-2 trial, a subsequent sub-analysis of the study population was performed to look for fibrinolysis benefit, but no firm trends were observed, with the exception of a reduction in time to thrombosis at 48 h [41]. However, a recent study of patients with underlining coronary artery disease has demonstrated that whilst RIC cannot influence platelet aggregation alone, when combined with dual anti-platelet therapy (DAPT) in vitro, there is significant de-activation of collagen-dependent, platelet glycoprotein integrin molecules [87]. Further studies showing clear fibrinolytic benefit post-RIC vs controls are, however, necessary to reaffirm this.
Extracellular vesicles (EVs) in inflammation and immunity
It has long been proposed that the organ-protective effects of RIC can be attributed to the release of humoral factors by the ‘trigger vessel’, which reach the target tissue to reduce inflammation and cell death. Recently, endogenous nanoparticles known as exosomes have been thought to facilitate this transfer, perhaps aided by an improvement in vascular flow, secondary to the release of vasoactive compounds [28, 35, 38, 139].
Exosomes represent the smallest size of extracellular vesicles (measuring 50–100 nm in diameter) and have a wide variety of functions in ischaemia and inflammation. Such nanoparticles can be derived from many types of cells including endothelium, haematopoietic cells and platelets, and their function is defined by the underlying pathology and cell of origin [22]. Exosomes carry chemokines and genetic material such as microRNA, which permits distant genetic transcription and cellular cross-talk. Such exosomes engage with target cells using a range of surface molecules expressed on their lipid bi-layer including tetraspanins, annexins, integrins and receptors of the major histocompatibility complex (MHC) [18, 26, 55]. They are distinct from other small extracellular vesicles and apoptotic bodies as they are smaller and carry different contents; which can be both anti and pro-inflammatory, depending on their stimulus [22].
It is necessary to define which contents may be most implicated in the inflammation of I/R and other conditions (Table.3). Exosomes carrying microRNA-21 (miR-21) have been identified in two recent RIC studies [35, 110] as limiting apoptosis and infarct size, respectively. In a rodent model of endotoxemia induced by LPS, mice undergoing RIC prior to caecal puncture were found to secrete organ-protective exosomes carrying miR-21, which mediated HIF-1α and led to cytokine attenuation (reduced levels of IL-6 and TNF-α) [110]. In addition to apoptosis and cytokine release, it has been demonstrated that endothelial-derived exosomes can mediate angiogenesis (via VEGF and eNOS) in response to RIC [16], and, therefore, promote cytokines and endothelial growth factors. Again, this illustrates their breadth of function, in different pathological conditions.
Regarding the anti-inflammatory actions of exosomes, (reductions in apoptosis and cytokine release), many of these benefits were negated in a study of diabetic rats vs normoglycaemic animals [144]. This suggests that animals with underlining co-morbidities and endothelial dysfunction, are unable to generate effective vesicles. However, when receiving exosomes from non-diabetic rats they can be rescued and protection is conferred [25, 144]. The authors of the latter study, make the important observation, that although RIC appears to generate cardioprotective exosomes in vivo, similar benefits can also be derived from the exosomes of control animals in absence of pre-conditioning [144]. This is re-enforced by a recent clinical study of patients undergoing RIC prior to treatment for STEMI, where no significant differences were found in the release of platelet-derived extracellular vesicles or other leucocyte derived vesicles in the intervention group [45].
The study did, however, suggest that whilst there was no increase in cardioprotective vesicles, there was also no increase in pro-inflammatory EV’s. The trial was limited by an absence of inflammatory and traditional end points in cardioprotection, such as infarct size and CRP. It is also noted that larger extracellular vesicles, as opposed to exosomes, may also be more likely to carry pro-inflammatory chemokines [22].
Does RIC reduce inflammation? Summary
There was never a more appropriate time in this COVID-19 era, to consider therapies which can treat the over-activity of the innate immune response and hyper-inflammation [112, 127]. Considering the above evidence, it is clear that RIC has anti-inflammatory benefits in vivo, across a wide range of different pathologies, at least in animal models. Perhaps the strongest evidence relates to the effects of RIC on pro-inflammatory cytokine release in endotoxaemia [68, 74, 110]; however, this has also been observed in models of myocardial and cerebral infarction (Table.1). In animals, it is proposed that RIC mediates inflammation by cytokine inhibition, regulation of anti-apoptotic pathways and possibly the reduction of NLRP3 inflammasome production and pyroptosis.
Most evidence from animal studies demonstrates that RIC is able to inhibit NF-κB related cytokine release, either by TLR4 receptor pathways, or other currently undefined mechanisms [134]. Further work is required to establish whether there is a clear link between RIC and other known mechanisms of cytokine release in inflammation, such as the RAGE pathway. It is recognised that, as a result of the nature of the NF-κB pathway, cell survival and cytokine release are closely related [99, 134] and, therefore, it can be difficult to establish if reduced cytokine concentrations are secondary to reduced cell death. Nevertheless, cytokine inhibition in cardioprotection remains a desirable goal, with prognostic value [4, 120].
Previous literature has discussed that an increase in the levels of HIF-1α in response to ischaemia, can stimulate affected tissue to maintain metabolic function upon further hypoxic insult [19]. As RIC has been shown to upregulate HIF-1α, it might, therefore, be suggested that this could induce hypoxic tolerance of both vascular endothelium and target tissue. Both HIF-1α and SDF-1 limit apoptosis following RIC [27, 63]. Shear stress and mechanical stimulation of the trigger vessel, induce flow-mediated dilatation (via NO/adenosine/COX), and stimulate the release of exosomes carrying chemo-active compounds to target tissue [38, 85, 95]. Exosomes can also carry pro-inflammatory compounds and chemokines in addition to cardioprotective substances, and are, therefore, ‘a double-edged sword’ in inflammation [45, 55]. Despite this, the aforementioned studies investigating RIC and exosome release have reported protective effects (Table.3).
Although there is some evidence that RIC can modulate immune cell response in animal studies (e.g. neutrophil/lymphocyte ratio) the authors concede that there is a lack of consistent clinical data. The role of immune cells in cardioprotection is an emerging and novel field on which to base further work, and the effects of RIC should continue to be investigated. Other potential immune targets such as fibroblasts, pericytes and mast cells have also been identified for further study [5]. Consistent with other aspects of RIC, it would be misleading to suggest that the anti-inflammatory effects have proven profound in humans, although there is a lack of large-scale focused RCT’s in patients with hyper-inflammation. Moreover, a select few studies have offered some hope that under the right circumstances, clinical translation could be achieved [53, 140]. A further step in addressing this might be to consider whether we have measured inflammatory outcomes in the correct clinical setting to date.
Future considerations: towards a higher risk patient cohort
Reflecting on clinical challenges to date, it is suggested that the impact of both baseline and peak inflammation in clinical trials of RIC has been underestimated. For example, individuals with chronic inflammatory disease and persistent low levels of inflammation at baseline, may already be resistant to remote conditioning [39]. The exact reasons for this remain elusive, but may be related to persistent endothelial activation, chronic cytokine release and defective exosome/humoral factor production, (as observed in diabetic animals) [25, 144]. With respect to the STEMI patients of CONDI-2/ERIC-PPCI, it is possible that the outcomes do not represent a failure of RIC to show significant benefit, but instead a success of modern primary percutaneous cardiovascular intervention (PPCI) in this cohort [11, 49]. I.e. it is not clear whether the ‘inflammatory peak’ following successful PCI, was significant enough to demonstrate an improvement in the primary outcome measures of the trial (cardiac death/ hospitalisation at 12 months) [48].
As proposed by several authors [11, 49, 65], higher risk patients with amplified inflammatory response to STEMI, might be proposed as the appropriate target for RIC e.g. those with large anterior infarcts who are late presenting, patients in cardiogenic shock, out of hospital cardiac arrest, those who develop angiographic no reflow of a large culprit vessel and those who are only able to receive thrombolysis and not primary PCI [49, 81]. Global inflammation is observed in patients with endotoxaemia and viral infection—these patients may also be favourable candidates for RIC [43]. Moreover, remote conditioning in combination with pharmacotherapy may be of benefit in preventing the development of a cytokine storm.
Given the challenges in clinical translation [65], RIC should be trialled as an adjunctive therapy in combination with gold-standard treatments in the above ‘high-risk’ cohort. It has been demonstrated above, that RIC can act synergistically to reduce inflammation when combined with pharmacotherapy and activation of neuronal pathways [24, 33, 141].
The difficulty in predicting the timing of a major inflammatory insult remains a significant dilemma for human interventional studies. However, given the reproducibility and extent of the survival benefits observed in animal models; the anti-inflammatory effects of RIC warrant further clinical pursuit.
References
Abbate A, Toldo S, Marchetti C, Kron J, Van Tassell BW, Dinarello CA (2020) Interleukin-1 and the inflammasome as therapeutic targets in cardiovascular disease. Circ Res 126:1260–1280. https://doi.org/10.1161/CIRCRESAHA.120.315937
Abbate A, Trankle CR, Buckley LF, Lipinski MJ, Appleton D, Kadariya D, Canada JM, Carbone S, Roberts CS, Abouzaki N, Melchior R, Christopher S, Turlington J, Mueller G, Garnett J, Thomas C, Markley R, Wohlford GF, Puckett L, Medina de Chazal H, Chiabrando JG, Bressi E, Del Buono MG, Schatz A, Vo C, Dixon DL, Biondi-Zoccai GG, Kontos MC, Van Tassell BW (2020) Interleukin-1 blockade inhibits the acute inflammatory response in patients with ST-segment-elevation myocardial infarction. J Am Heart Assoc 9:e014941. https://doi.org/10.1161/JAHA.119.014941
Abdulmahdi W, Patel D, Rabadi MM, Azar T, Jules E, Lipphardt M, Hashemiyoon R, Ratliff BB (2017) HMGB1 redox during sepsis. Redox Biol 13:600–607. https://doi.org/10.1016/j.redox.2017.08.001
Adamo L, Rocha-Resende C, Prabhu SD, Mann DL (2020) Reappraising the role of inflammation in heart failure. Nat Rev Cardiol 17:269–285. https://doi.org/10.1038/s41569-019-0315-x
Andreadou I, Cabrera-Fuentes HA, Devaux Y, Frangogiannis NG, Frantz S, Guzik T, Liehn EA, Gomes CPC, Schulz R, Hausenloy DJ (2019) Immune cells as targets for cardioprotection: new players and novel therapeutic opportunities. Cardiovasc Res 115:1117–1130. https://doi.org/10.1093/cvr/cvz050
Arts RJW, Huang PK, Yang JLAB, van der Meer JWM, Oppenheim JJ, Netea MG, Cheng SC (2018) High-mobility group nucleosome-binding protein 1 as endogenous ligand induces innate immune tolerance in a TLR4-sirtuin-1 dependent manner in human blood peripheral mononuclear cells. Front Immunol 9:526. https://doi.org/10.3389/fimmu.2018.00526
Basalay MV, Davidson SM, Gourine AV, Yellon DM (2018) Neural mechanisms in remote ischaemic conditioning in the heart and brain: mechanistic and translational aspects. Basic Res Cardiol 113:25. https://doi.org/10.1007/s00395-018-0684-z
Basalay MV, Mastitskaya S, Mrochek A, Ackland GL, Del Arroyo AG, Sanchez J, Sjoquist PO, Pernow J, Gourine AV, Gourine A (2016) Glucagon-like peptide-1 (GLP-1) mediates cardioprotection by remote ischaemic conditioning. Cardiovasc Res 112:669–676. https://doi.org/10.1093/cvr/cvw216
Billah M, Ridiandries A, Rayner BS, Allahwala UK, Dona A, Khachigian LM, Bhindi R (2019) Egr-1 functions as a master switch regulator of remote ischemic preconditioning-induced cardioprotection. Basic Res Cardiol 115:3–3. https://doi.org/10.1007/s00395-019-0763-9
Bonaventura A, Vecchie A, Abbate A, Montecucco F (2020) Neutrophil Extracellular traps and cardiovascular diseases: an update. Cells 9:231. https://doi.org/10.3390/cells9010231
Botker HE (2020) The future of cardioprotection-pointing toward patients at elevated risk as the target populations. J Cardiovasc Pharmacol Ther. https://doi.org/10.1177/1074248420937871
Bromage DI, Davidson SM, Yellon DM (2014) Stromal derived factor 1alpha: a chemokine that delivers a two-pronged defence of the myocardium. Pharmacol Ther 143:305–315. https://doi.org/10.1016/j.pharmthera.2014.03.009
Bromage DI, Pickard JM, Rossello X, Ziff OJ, Burke N, Yellon DM, Davidson SM (2017) Remote ischaemic conditioning reduces infarct size in animal in vivo models of ischaemia-reperfusion injury: a systematic review and meta-analysis. Cardiovasc Res 113:288–297. https://doi.org/10.1093/cvr/cvw219
Chen C, Jiang W, Liu Z, Li F, Yang J, Zhao Y, Ran Y, Meng Y, Ji X, Geng X, Du H, Hu X (2018) Splenic responses play an important role in remote ischemic preconditioning-mediated neuroprotection against stroke. J Neuroinflammation 15:167. https://doi.org/10.1186/s12974-018-1190-9
Chen H, Jing X-Y, Shen Y-J, Wang T-L, Ou C, Lu S-F, Cai Y, Li Q, Chen X, Ding Y-J, Yu X-C, Zhu B-M (2018) Stat5-dependent cardioprotection in late remote ischaemia preconditioning. Cardiovasc Res 114:679–689
Chen Q, Huang M, Wu J, Jiang Q, Zheng X (2020) Exosomes isolated from the plasma of remote ischemic conditioning rats improved cardiac function and angiogenesis after myocardial infarction through targeting Hsp70. Aging (Albany NY) 12:3682–3693. https://doi.org/10.18632/aging.102837
Christia P, Frangogiannis NG (2013) Targeting inflammatory pathways in myocardial infarction. Eur J Clin Invest 43:986–995. https://doi.org/10.1111/eci.12118
Console L, Scalise M, Indiveri C (2019) Exosomes in inflammation and role as biomarkers. Clin Chim Acta 488:165–171. https://doi.org/10.1016/j.cca.2018.11.009
Corcoran SE, O’Neill LA (2016) HIF1alpha and metabolic reprogramming in inflammation. J Clin Invest 126:3699–3707. https://doi.org/10.1172/JCI84431
Cui J, Liu N, Chang Z, Gao Y, Bao M, Xie Y, Xu W, Liu X, Jiang S, Liu Y, Shi R, Xie W, Jia X, Shi J, Ren C, Gong K, Zhang C, Bade R, Shao G, Ji X (2020) Exosomal MicroRNA-126 from RIPC serum is involved in hypoxia tolerance in SH-SY5Y cells by downregulating DNMT3B. Mol Ther Nucleic Acids 20:649–660. https://doi.org/10.1016/j.omtn.2020.04.008
Davidson SM, Adameova A, Barile L, Cabrera-Fuentes HA, Lazou A, Pagliaro P, Stenslokken KO, Garcia-Dorado D, Action E-CC (2020) Mitochondrial and mitochondrial-independent pathways of myocardial cell death during ischaemia and reperfusion injury. J Cell Mol Med 24:3795–3806. https://doi.org/10.1111/jcmm.15127
Davidson SM, Andreadou I, Barile L, Birnbaum Y, Cabrera-Fuentes HA, Cohen MV, Downey JM, Girao H, Pagliaro P, Penna C, Pernow J, Preissner KT, Ferdinandy P (2019) Circulating blood cells and extracellular vesicles in acute cardioprotection. Cardiovasc Res 115:1156–1166. https://doi.org/10.1093/cvr/cvy314
Davidson SM, Arjun S, Basalay MV, Bell RM, Bromage DI, Botker HE, Carr RD, Cunningham J, Ghosh AK, Heusch G, Ibanez B, Kleinbongard P, Lecour S, Maddock H, Ovize M, Walker M, Wiart M, Yellon DM (2018) The 10th Biennial Hatter Cardiovascular Institute workshop: cellular protection-evaluating new directions in the setting of myocardial infarction, ischaemic stroke, and cardio-oncology. Basic Res Cardiol 113:43. https://doi.org/10.1007/s00395-018-0704-z
Davidson SM, Ferdinandy P, Andreadou I, Botker HE, Heusch G, Ibanez B, Ovize M, Schulz R, Yellon DM, Hausenloy DJ, Garcia-Dorado D, Action CC (2019) Multitarget strategies to reduce myocardial ischemia/reperfusion injury: JACC review topic of the week. J Am Coll Cardiol 73:89–99. https://doi.org/10.1016/j.jacc.2018.09.086
Davidson SM, Riquelme JA, Takov K, Vicencio JM, Boi-Doku C, Khoo V, Doreth C, Radenkovic D, Lavandero S, Yellon DM (2018) Cardioprotection mediated by exosomes is impaired in the setting of type II diabetes but can be rescued by the use of non-diabetic exosomes in vitro. J Cell Mol Med 22:141–151. https://doi.org/10.1111/jcmm.13302
Davidson SM, Riquelme JA, Zheng Y, Vicencio JM, Lavandero S, Yellon DM (2018) Endothelial cells release cardioprotective exosomes that may contribute to ischaemic preconditioning. Sci Rep 8:15885. https://doi.org/10.1038/s41598-018-34357-z
Davidson SM, Selvaraj P, He D, Boi-Doku C, Yellon RL, Vicencio JM, Yellon DM (2013) Remote ischaemic preconditioning involves signalling through the SDF-1alpha/CXCR4 signalling axis. Basic Res Cardiol 108:377. https://doi.org/10.1007/s00395-013-0377-6
Davidson SM, Takov K, Yellon DM (2017) Exosomes and cardiovascular protection. Cardiovasc Drugs Ther 31:77–86. https://doi.org/10.1007/s10557-016-6698-6
Denes A, Lopez-Castejon G, Brough D (2012) Caspase-1: is IL-1 just the tip of the ICEberg? Cell Death Dis 3:e338. https://doi.org/10.1038/cddis.2012.86
Dong W, Yu P, Zhang T, Zhu C, Qi J, Liang J (2018) Adrenomedullin serves a role in the humoral pathway of delayed remote ischemic preconditioning via a hypoxia-inducible factor-1alpha-associated mechanism. Mol Med Rep 17:4547–4553. https://doi.org/10.3892/mmr.2018.8450
Du X, Yang J, Liu C, Wang S, Zhang C, Zhao H, Du H, Geng X (2020) Hypoxia-inducible factor 1α and 2α have beneficial effects in remote ischemic preconditioning against stroke by modulating inflammatory responses in aged rats. Front Ageing Neurosci 12:54–54
Eitel I, Stiermaier T, Rommel KP, Fuernau G, Sandri M, Mangner N, Linke A, Erbs S, Lurz P, Boudriot E, Mende M, Desch S, Schuler G, Thiele H (2015) Cardioprotection by combined intrahospital remote ischaemic perconditioning and postconditioning in ST-elevation myocardial infarction: the randomized LIPSIA CONDITIONING trial. Eur Heart J 36:3049–3057. https://doi.org/10.1093/eurheartj/ehv463
El Desoky ES, Hassan AKM, Salem SY, Fadil SA, Taha AF (2016) Cardioprotective effect of atorvastatin alone or in combination with remote ischemic preconditioning on the biochemical changes induced by ischemic/reperfusion injury in a mutual prospective study with a clinical and experimental animal arm. Int J Cardiol 222:866–873. https://doi.org/10.1016/j.ijcard.2016.07.178
Frangogiannis NG, Smith CW, Entman ML (2002) The inflammatory response in myocardial infarction. Cardiovasc Res 53:31–47. https://doi.org/10.1016/s0008-6363(01)00434-5
Frey UH, Klaassen M, Ochsenfarth C, Murke F, Thielmann M, Kottenberg E, Kleinbongard P, Klenke S, Engler A, Heusch G, Giebel B, Peters J (2019) Remote ischaemic preconditioning increases serum extracellular vesicle concentrations with altered micro-RNA signature in CABG patients. Acta Anaesth Scand 63:483–492. https://doi.org/10.1111/aas.13296
Gaspar A, Lourenco AP, Pereira MA, Azevedo P, Roncon-Albuquerque R Jr, Marques J, Leite-Moreira AF (2018) Randomized controlled trial of remote ischaemic conditioning in ST-elevation myocardial infarction as adjuvant to primary angioplasty (RIC-STEMI). Basic Res Cardiol 113:14. https://doi.org/10.1007/s00395-018-0672-3
Gedik N, Kottenberg E, Thielmann M, Frey UH, Jakob H, Peters J, Heusch G, Kleinbongard P (2017) Potential humoral mediators of remote ischemic preconditioning in patients undergoing surgical coronary revascularization. Sci Rep 7:12660. https://doi.org/10.1038/s41598-017-12833-2
Giricz Z, Varga ZV, Baranyai T, Sipos P, Paloczi K, Kittel A, Buzas EI, Ferdinandy P (2014) Cardioprotection by remote ischemic preconditioning of the rat heart is mediated by extracellular vesicles. J Mol Cell Cardiol 68:75–78. https://doi.org/10.1016/j.yjmcc.2014.01.004
Godskesen LE, Lassen TR, Jespersen NR, Siersbaek M, Yan Y, Nielsen MM, Tjonnfjord SK, Grontved L, Madsen G, Kjems J, Botker HE, Schmidt MR, Krag A, Kjeldsen J (2020) Remote ischemic conditioning in active ulcerative colitis: an explorative randomized clinical trial. Sci Rep 10:9537. https://doi.org/10.1038/s41598-020-65692-9
Gori T, Lelieveld J, Munzel T (2020) Perspective: cardiovascular disease and the Covid-19 pandemic. Basic Res Cardiol 115:32. https://doi.org/10.1007/s00395-020-0792-4
Gorog DA, Farag M, Spinthakis N, Yellon DM, Botker HE, Kharbanda RK, Hausenloy DJ (2020) Effect of remote ischaemic conditioning on platelet reactivity and endogenous fibrinolysis in ST-elevation myocardial infarction- a substudy of the CONDI-2/ERIC-PPCI randomised controlled trial. Cardiovasc Res. https://doi.org/10.1093/cvr/cvaa061
Groot HE, Al Ali L, van der Horst ICC, Schurer RAJ, van der Werf HW, Lipsic E, van Veldhuisen DJ, Karper JC, van der Harst P (2019) Plasma interleukin 6 levels are associated with cardiac function after ST-elevation myocardial infarction. Clin Res Cardiol 108:612–621. https://doi.org/10.1007/s00392-018-1387-z
Guillon A, Preau S, Aboab J, Azabou E, Jung B, Silva S, Textoris J, Uhel F, Vodovar D, Zafrani L, de Prost N, Radermacher P, Soc FIC, Francaise SRL (2019) Preclinical septic shock research: why we need an animal ICU. Ann Intensive Care 9:66. https://doi.org/10.1186/s13613-019-0543-6
Hadebe N, Cour M, Lecour S (2018) The SAFE pathway for cardioprotection: is this a promising target? Basic Res Cardiol 113:9. https://doi.org/10.1007/s00395-018-0670-5
Haller PM, Jager B, Piackova E, Sztulman L, Wegberger C, Wojta J, Gyongyosi M, Kiss A, Podesser BK, Spittler A, Huber K (2020) Changes in circulating extracellular vesicles in patients with ST-elevation myocardial infarction and potential effects of remote ischemic conditioning-a randomized controlled trial. Biomedicines 8:218. https://doi.org/10.3390/biomedicines8070218
Han R, Liu X, Yin X, Zheng M, Sun K, Liu X, Tian Y, Yang X (2016) Effect of remote ischemic preconditioning on myocardial injury and inflammatory response induced by ablation for atrial fibrillation: a randomized controlled trial. Int J Cardiol 222:396–400. https://doi.org/10.1016/j.ijcard.2016.07.229
Hauerslev M, Mork SR, Pryds K, Contractor H, Hansen J, Jespersen NR, Johnsen J, Heusch G, Kleinbongard P, Kharbanda R, Botker HE, Schmidt MR (2018) Influence of long-term treatment with glyceryl trinitrate on remote ischemic conditioning. Am J Physiol Heart Circ Physiol 315:H150–H158. https://doi.org/10.1152/ajpheart.00114.2018
Hausenloy DJ, Kharbanda RK, Moller UK, Ramlall M, Aaroe J, Butler R, Bulluck H, Clayton T, Dana A, Dodd M, Engstrom T, Evans R, Lassen JF, Christensen EF, Garcia-Ruiz JM, Gorog DA, Hjort J, Houghton RF, Ibanez B, Knight R, Lippert FK, Lonborg JT, Maeng M, Milasinovic D, More R, Nicholas JM, Jensen LO, Perkins A, Radovanovic N, Rakhit RD, Ravkilde J, Ryding AD, Schmidt MR, Riddervold IS, Sorensen HT, Stankovic G, Varma M, Webb I, Terkelsen CJ, Greenwood JP, Yellon DM, Botker HE, Investigators C-E-P (2019) Effect of remote ischaemic conditioning on clinical outcomes in patients with acute myocardial infarction (CONDI-2/ERIC-PPCI): a single-blind randomised controlled trial. Lancet 394:1415–1424. https://doi.org/10.1016/S0140-6736(19)32039-2
Hausenloy DJ, Ntsekhe M, Yellon DM (2020) A future for remote ischaemic conditioning in high-risk patients. Basic Res Cardiol 115:35. https://doi.org/10.1007/s00395-020-0794-2
Hausenloy DJ, Yellon DM (2016) Ischaemic conditioning and reperfusion injury. Nat Rev Cardiol 13:193–209. https://doi.org/10.1038/nrcardio.2016.5
Hausenloy DJ, Yellon DM (2007) Reperfusion injury salvage kinase signalling: taking a RISK for cardioprotection. Heart Fail Rev 12:217–234. https://doi.org/10.1007/s10741-007-9026-1
He YD, Guo ZN, Qin C, Jin H, Zhang P, Abuduxukuer R, Yang Y (2020) Remote ischemic conditioning combined with intravenous thrombolysis for acute ischemic stroke. Ann Clin Transl Neurol 7:972–979. https://doi.org/10.1002/acn3.51063
He Z, Xu N, Qi S (2017) Remote ischemic preconditioning improves the cognitive function of elderly patients following colon surgery: a randomized clinical trial. Medicine (Baltimore) 96:e6719. https://doi.org/10.1097/MD.0000000000006719
Helseth R, Shetelig C, Andersen GO, Langseth MS, Limalanathan S, Opstad TB, Arnesen H, Hoffmann P, Eritsland J, Seljeflot I (2019) Neutrophil extracellular trap components associate with infarct size, ventricular function, and clinical outcome in STEMI. Mediators Inflamm 2019:7816491. https://doi.org/10.1155/2019/7816491
Henning RJ (2020) Cardiovascular exosomes and microRNAs in cardiovascular physiology and pathophysiology. J Cardiovasc Transl Res. https://doi.org/10.1007/s12265-020-10040-5
Heusch G (2016) The coronary circulation as a target of cardioprotection. Circ Res 118:1643–1658. https://doi.org/10.1161/CIRCRESAHA.116.308640
Heusch G (2019) Coronary microvascular obstruction: the new frontier in cardioprotection. Basic Res Cardiol 114:45. https://doi.org/10.1007/s00395-019-0756-8
Heusch G (2017) Critical issues for the translation of cardioprotection. Circ Res 120:1477–1486. https://doi.org/10.1161/circresaha.117.310820
Heusch G (2012) HIF-1alpha and paradoxical phenomena in cardioprotection. Cardiovasc Res 96:214–215. https://doi.org/10.1093/cvr/cvs145 (discussion 216-219)
Heusch G (2015) Molecular basis of cardioprotection: signal transduction in ischemic pre-, post-, and remote conditioning. Circ Res 116:674–699. https://doi.org/10.1161/CIRCRESAHA.116.305348
Heusch G (2020) Myocardial ischaemia-reperfusion injury and cardioprotection in perspective. Nat Rev Cardiol 17:773–789. https://doi.org/10.1038/s41569-020-0403-y
Heusch G (2019) The spleen in myocardial infarction. Circ Res 124:26–28. https://doi.org/10.1161/CIRCRESAHA.118.314331
Heusch G, Botker HE, Przyklenk K, Redington A, Yellon D (2015) Remote ischemic conditioning. J Am Coll Cardiol 65:177–195. https://doi.org/10.1016/j.jacc.2014.10.031
Heusch G, Gersh BJ (2016) ERICCA and RIPHeart: two nails in the coffin for cardioprotection by remote ischemic conditioning? Probably not! Eur Heart J 37:200–202. https://doi.org/10.1093/eurheartj/ehv606
Heusch G, Gersh BJ (2020) Is Cardioprotection salvageable? Circulation 141:415–417. https://doi.org/10.1161/CIRCULATIONAHA.119.044176
Heusch G, Musiolik J, Kottenberg E, Peters J, Jakob H, Thielmann M (2012) STAT5 activation and cardioprotection by remote ischemic preconditioning in humans: short communication. Circ Res 110:111–115. https://doi.org/10.1161/CIRCRESAHA.111.259556
Hildebrand D, Uhle F, Sahin D, Krauser U, Weigand MA, Heeg K (2018) The interplay of notch signaling and STAT3 in TLR-activated human primary monocytes. Front Cell Infect Microbiol 8:241. https://doi.org/10.3389/fcimb.2018.00241
Honda T, He Q, Wang F, Redington AN (2019) Acute and chronic remote ischemic conditioning attenuate septic cardiomyopathy, improve cardiac output, protect systemic organs, and improve mortality in a lipopolysaccharide-induced sepsis model. Basic Res Cardiol 114:15. https://doi.org/10.1007/s00395-019-0724-3
Hussein AM, Harraz AM, Awadalla A, Barakat N, Khater S, Shokeir AA (2016) Remote limb ischemic preconditioning (rIPC) activates antioxidant and antiapoptotic genes and inhibits proinflammatory cytokine genes in renal ischemia/reperfusion injury. Gen Physiol Biophys 35:77–86. https://doi.org/10.4149/gpb_2015035
Hussein AM, Sakr HF, Alenzi FQ (2016) Possible underlying mechanisms of the renoprotective effect of remote limb ischemic preconditioning against renal ischemia/reperfusion injury: a role of osteopontin, transforming growth factor-beta and survivin. Nephron 134:117–129. https://doi.org/10.1159/000447953
Jiang Q, Xiang B, Wang H, Huang K, Kong H, Hu S (2019) Remote ischaemic preconditioning ameliorates sinus rhythm restoration rate through Cox maze radiofrequency procedure associated with inflammation reaction reduction. Basic Res Cardiol 114:14. https://doi.org/10.1007/s00395-019-0723-4
Jin R, Liu L, Zhang S, Nanda A, Li G (2013) Role of inflammation and its mediators in acute ischemic stroke. J Cardiovasc Transl Res 6:834–851. https://doi.org/10.1007/s12265-013-9508-6
Jing L, Guan W, You Q, Deng L, Zhu Y, Guo K, Gao X, Kong J, Yang C (2020) RIPC provides neuroprotection against ischemic stroke by suppressing apoptosis via the mitochondrial pathway. Sci Rep 10:5361. https://doi.org/10.1038/s41598-020-62336-w
Joseph B, Khalil M, Hashmi A, Hecker L, Kulvatunyou N, Tang A, Friese RS, Rhee P (2017) Survival benefits of remote ischemic conditioning in sepsis. J Surg Res 213:131–137. https://doi.org/10.1016/j.jss.2016.01.033
Kierdorf K, Fritz G (2013) RAGE regulation and signaling in inflammation and beyond. J Leukocyte Biol 94:55–68. https://doi.org/10.1189/jlb.1012519
Kim YH, Kim YS, Kim BH, Lee KS, Park HS, Lim CH (2019) Remote ischemic preconditioning ameliorates indirect acute lung injury by modulating phosphorylation of IkappaBalpha in mice. J Int Med Res 47:936–950. https://doi.org/10.1177/0300060518818300
Kim YK, Shin JS, Nahm MH (2016) NOD-like receptors in infection, immunity, and diseases. Yonsei Med J 57:5–14. https://doi.org/10.3349/ymj.2016.57.1.5
Klebanoff SJ (2005) Myeloperoxidase: friend and foe. J Leukoc Biol 77:598–625. https://doi.org/10.1189/jlb.1204697
Kleinbongard P, Amanakis G, Skyschally A, Heusch G (2018) Reflection of cardioprotection by remote ischemic perconditioning in attenuated ST-segment elevation during ongoing coronary occlusion in pigs: evidence for cardioprotection from ischemic injury. Circ Res 122:1102–1108. https://doi.org/10.1161/CIRCRESAHA.118.312784
Kleinbongard P, Skyschally A, Heusch G (2017) Cardioprotection by remote ischemic conditioning and its signal transduction. Pflugers Arch 469:159–181. https://doi.org/10.1007/s00424-016-1922-6
Kloner RA (2017) The importance of no-reflow/microvascular obstruction in the STEMI patient. Eur Heart J 38:3511–3513. https://doi.org/10.1093/eurheartj/ehx288
Kloner RA, King KS, Harrington MG (2018) No-reflow phenomenon in the heart and brain. Am J Physiol Heart Circ Physiol 315:H550–H562. https://doi.org/10.1152/ajpheart.00183.2018
Koh WU, Kim J, Lee J, Song GW, Hwang GS, Tak E, Song JG (2019) Remote ischemic preconditioning and diazoxide protect from hepatic ischemic reperfusion injury by inhibiting HMGB1-induced TLR4/MyD88/NF-kappaB signaling. Int J Mol Sci. https://doi.org/10.3390/ijms20235899
Koyama S, Ishii KJ, Coban C, Akira S (2008) Innate immune response to viral infection. Cytokine 43:336–341. https://doi.org/10.1016/j.cyto.2008.07.009
Lang JA, Kim J, Franke WD, Vianna LC (2019) Seven consecutive days of remote ischaemic preconditioning improves cutaneous vasodilatory capacity in young adults. J Physiol 597:757–765. https://doi.org/10.1113/JP277185
Lang JP, Wang X, Moura FA, Siddiqi HK, Morrow DA, Bohula EA (2020) A current review of COVID-19 for the cardiovascular specialist. Am Heart J 226:29–44. https://doi.org/10.1016/j.ahj.2020.04.025
Lau JK, Pennings GJ, Reddel CJ, Campbell H, Liang HPH, Traini M, Gardiner EE, Yong AS, Chen VM, Kritharides L (2020) Remote ischemic preconditioning inhibits platelet alphaIIb beta3 activation in coronary artery disease patients receiving dual antiplatelet therapy: A randomized trial. J Thromb Haemost 18:1221–1232. https://doi.org/10.1111/jth.14763
Lecour S (2009) Activation of the protective Survivor Activating Factor Enhancement (SAFE) pathway against reperfusion injury: Does it go beyond the RISK pathway? J Mol Cell Cardiol 47:32–40. https://doi.org/10.1016/j.yjmcc.2009.03.019
Lee JW, Ko J, Ju C, Eltzschig HK (2019) Hypoxia signaling in human diseases and therapeutic targets. Exp Mol Med 51:1–13. https://doi.org/10.1038/s12276-019-0235-1
Lee P, Chandel NS, Simon MC (2020) Cellular adaptation to hypoxia through hypoxia inducible factors and beyond. Nat Rev Mol Cell Biol 21:268–283. https://doi.org/10.1038/s41580-020-0227-y
Li Y, Ren C, Li H, Jiang F, Wang L, Xia C, Ji X (2019) Role of exosomes induced by remote ischemic preconditioning in neuroprotection against cerebral ischemia. NeuroReport 30:834–841. https://doi.org/10.1097/WNR.0000000000001280
Liang W, Lin C, Yuan L, Chen L, Guo P, Li P, Wang W, Zhang X (2019) Preactivation of Notch1 in remote ischemic preconditioning reduces cerebral ischemia-reperfusion injury through crosstalk with the NF-κB pathway. J Neuroinflammation 16:181. https://doi.org/10.1186/s12974-019-1570-9
Lieder HR, Kleinbongard P, Skyschally A, Hagelschuer H, Chilian WM, Heusch G (2018) Vago-splenic axis in signal transduction of remote ischemic preconditioning in pigs and rats. Circ Res 123:1152–1163. https://doi.org/10.1161/CIRCRESAHA.118.313859
Lin H, Wang W, Lee M, Meng Q, Ren H (2020) Current status of septic cardiomyopathy: basic science and clinical progress. Front Pharmacol 11:210. https://doi.org/10.3389/fphar.2020.00210
Liu ZB, Yang WX, Fu XH, Zhao LF, Gao JL (2015) Remote ischemic precondition prevents radial artery endothelial dysfunction induced by ischemia and reperfusion based on a cyclooxygenase-2-dependent mechanism. Int J Clin Exp Med 8:20946–20952
Liu ZJ, Chen C, Li XR, Ran YY, Xu T, Zhang Y, Geng XK, Zhang Y, Du HS, Leak RK, Ji XM, Hu XM (2016) Remote ischemic preconditioning-mediated neuroprotection against stroke is associated with significant alterations in peripheral immune responses. CNS Neurosci Ther 22:43–52. https://doi.org/10.1111/cns.12448
Ma F, Liu H, Shen Y, Zhang Y, Pan S (2015) Platelet-derived microvesicles are involved in cardio-protective effects of remote preconditioning. Int J Clin Exp Pathol 8:10832–10839
Malik A, Bromage DI, He Z, Candilio L, Hamarneh A, Taferner S, Davidson SM, Yellon DM (2015) Exogenous SDF-1alpha protects human myocardium from hypoxia-reoxygenation injury via CXCR4. Cardiovasc Drugs Ther 29:589–592. https://doi.org/10.1007/s10557-015-6622-5
Mann DL (2015) Innate immunity and the failing heart: the cytokine hypothesis revisited. Circ Res 116:1254–1268. https://doi.org/10.1161/CIRCRESAHA.116.302317
Manukyan MC, Alvernaz CH, Poynter JA, Wang Y, Brewster BD, Weil BR, Abarbanell AM, Herrmann JL, Crowe BJ, Keck AC, Meldrum DR (2011) Interleukin-10 protects the ischemic heart from reperfusion injury via the STAT3 pathway. Surgery 150:231–239. https://doi.org/10.1016/j.surg.2011.05.017
Marti CN, Gheorghiade M, Kalogeropoulos AP, Georgiopoulou VV, Quyyumi AA, Butler J (2012) Endothelial dysfunction, arterial stiffness, and heart failure. J Am Coll Cardiol 60:1455–1469. https://doi.org/10.1016/j.jacc.2011.11.082
McGonagle D, Sharif K, O’Regan A, Bridgewood C (2020) The role of cytokines including interleukin-6 in COVID-19 induced pneumonia and macrophage activation syndrome-like disease. Autoimmun Rev 19:102537. https://doi.org/10.1016/j.autrev.2020.102537
Minghua W, Zhijian G, Chahua H, Qiang L, Minxuan X, Luqiao W, Weifang Z, Peng L, Biming Z, Lingling Y, Zhenzhen W, Jianqing X, Huihui B, Xiaozhong W, Xiaoshu C (2018) Plasma exosomes induced by remote ischaemic preconditioning attenuate myocardial ischaemia/reperfusion injury by transferring miR-24. Cell Death Dis 9:320. https://doi.org/10.1038/s41419-018-0274-x
Montecucco F, Liberale L, Bonaventura A, Vecchie A, Dallegri F, Carbone F (2017) The role of inflammation in cardiovascular outcome. Curr Atheroscler Rep 19:11. https://doi.org/10.1007/s11883-017-0646-1
Nederlof R, Weber NC, Juffermans NP, de Mol BA, Hollmann MW, Preckel B, Zuurbier CJ (2017) A randomized trial of remote ischemic preconditioning and control treatment for cardioprotection in sevoflurane-anesthetized CABG patients. BMC Anesthesiol 17:51. https://doi.org/10.1186/s12871-017-0330-6
Ney J, Hoffmann K, Meybohm P, Goetzenich A, Kraemer S, Benstom C, Weber NC, Bickenbach J, Rossaint R, Marx G, Zacharowski K, Bernhagen J, Stoppe C (2018) Remote ischemic preconditioning does not affect the release of humoral factors in propofol-anesthetized cardiac surgery patients: a secondary analysis of the RIPHeart study. Int J Mol Sci 19:1094. https://doi.org/10.3390/ijms19041094
Nian M, Lee P, Khaper N, Liu P (2004) Inflammatory cytokines and postmyocardial infarction remodeling. Circ Res 94:1543–1553. https://doi.org/10.1161/01.RES.0000130526.20854.fa
Oh CS, Sa M, Park HJ, Piao L, Oh KS, Kim SH (2020) Effects of remote ischemic preconditioning on regional cerebral oxygen saturation in patients in the beach chair position during shoulder surgery: A double-blind randomized controlled trial. J Clin Anesth 61:109661. https://doi.org/10.1016/j.jclinane.2019.109661
Ong SB, Hernandez-Resendiz S, Crespo-Avilan GE, Mukhametshina RT, Kwek XY, Cabrera-Fuentes HA, Hausenloy DJ (2018) Inflammation following acute myocardial infarction: Multiple players, dynamic roles, and novel therapeutic opportunities. Pharmacol Ther 186:73–87. https://doi.org/10.1016/j.pharmthera.2018.01.001
Pan T, Jia P, Chen N, Fang Y, Liang Y, Guo M, Ding X (2019) Delayed remote ischemic preconditioning confersrenoprotection against septic acute kidney injury via exosomal miR-21. Theranostics 9:405–423. https://doi.org/10.7150/thno.29832
Papayannopoulos V (2018) Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol 18:134–147. https://doi.org/10.1038/nri.2017.105
Pearce L, Davidson SM, Yellon DM (2020) The cytokine storm of COVID-19: a spotlight on prevention and protection. Expert Opin Ther Targets 24:723–730. https://doi.org/10.1080/14728222.2020.1783243
Pedersen SF, Ho YC (2020) SARS-CoV-2: a storm is raging. J Clin Invest 130:2202–2205. https://doi.org/10.1172/JCI137647
Pilz PM, Hamza O, Gidlöf O, Gonçalves IF, Tretter EV, Trojanek S, Abraham D, Heber S, Haller PM, Podesser BK, Kiss A (2019) Remote ischemic perconditioning attenuates adverse cardiac remodeling and preserves left ventricular function in a rat model of reperfused myocardial infarction. Int J Cardiol 285:72–79. https://doi.org/10.1016/j.ijcard.2019.03.003
Prabhu SD, Frangogiannis NG (2016) The biological basis for cardiac repair after myocardial infarction: from inflammation to fibrosis. Circ Res 119:91–112. https://doi.org/10.1161/CIRCRESAHA.116.303577
Pryds K, Nielsen RR, Jorsal A, Hansen MS, Ringgaard S, Refsgaard J, Kim WY, Petersen AK, Botker HE, Schmidt MR (2017) Effect of long-term remote ischemic conditioning in patients with chronic ischemic heart failure. Basic Res Cardiol 112:67. https://doi.org/10.1007/s00395-017-0658-6
Qi W, Zhou F, Li S, Zong Y, Zhang M, Lin Y, Zhang X, Yang H, Zou Y, Qi C, Wang T, Hu X (2016) Remote ischemic postconditioning protects ischemic brain from injury in rats with focal cerebral ischemia/reperfusion associated with suppression of TLR4 and NF-small ka, CyrillicB expression. NeuroReport 27:469–475. https://doi.org/10.1097/WNR.0000000000000553
Rassaf T, Totzeck M, Hendgen-Cotta UB, Shiva S, Heusch G, Kelm M (2014) Circulating nitrite contributes to cardioprotection by remote ischemic preconditioning. Circ Res 114:1601–1610. https://doi.org/10.1161/CIRCRESAHA.114.303822
Rauf A, Shah M, Yellon DM, Davidson SM (2019) Role of caspase 1 in ischemia/reperfusion injury of the myocardium. J Cardiovasc Pharmacol 74:194–200. https://doi.org/10.1097/FJC.0000000000000694
Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, Kastelein JJP, Cornel JH, Pais P, Pella D, Genest J, Cifkova R, Lorenzatti A, Forster T, Kobalava Z, Vida-Simiti L, Flather M, Shimokawa H, Ogawa H, Dellborg M, Rossi PRF, Troquay RPT, Libby P, Glynn RJ, Krum H, Varigos J, Siostrzonek P, Sinnaeve P, Gotcheva N, Yong H, Urina-Triana M, Milicic D, Vettus R, Manolis AJ, Wyss F, Sigurdsson A, Fucili A, Veze I, Petrauskiene B, Salvador L, Klemsdal TO, Medina F, Budaj A, Otasevic P, Lainscak M, Seung KB, Commerford P, Donath M, Hwang JJ, Kultursay H, Bilazarian S, East C, Forgosh L, Harris B, Ligueros M, Bohula E, Charmarthi B, Cheng S, Chou S, Danik J, McMahon G, Maron B, Ning M, Olenchock B, Pande R, Perlstein T, Pradhan A, Rost N, Singhal A, Taqueti V, Wei N, Burris H, Cioffi A, Dalseg AM, Ghosh N, Gralow J, Mayer T, Rugo H, Fowler V, Limaye AP, Cosgrove S, Levine D, Lopes R, Scott J, Hilkert R, Tamesby G, Mickel C, Manning B, Woelcke J, Tan M, Manfreda S, Ponce T, Kam J, Saini R, Banker K, Salko T et al (2017) Antiinflammatory therapy with canakinumab for atherosclerotic disease. New Engl J Med 377:1119–1131. https://doi.org/10.1056/NEJMoa1707914
Rizzo P, Vieceli Dalla Sega F, Fortini F, Marracino L, Rapezzi C, Ferrari R (2020) COVID-19 in the heart and the lungs: could we “Notch” the inflammatory storm? Basic Res Cardiol 115:31. https://doi.org/10.1007/s00395-020-0791-5
Rossello X, Yellon DM (2018) The RISK pathway and beyond. Basic Res Cardiol 113:2. https://doi.org/10.1007/s00395-017-0662-x
Rubino A, Yellon DM (2000) Ischaemic preconditioning of the vasculature: an overlooked phenomenon for protecting the heart? Trends Pharmacol Sci 21:225–230. https://doi.org/10.1016/s0165-6147(00)01483-8
Sawashita Y, Hirata N, Yoshikawa Y, Terada H, Tokinaga Y, Yamakage M (2020) Remote ischemic preconditioning reduces myocardial ischemia-reperfusion injury through unacylated ghrelin-induced activation of the JAK/STAT pathway. Basic Res Cardiol 115:50. https://doi.org/10.1007/s00395-020-0809-z
Saxena A, Russo I, Frangogiannis NG (2016) Inflammation as a therapeutic target in myocardial infarction: learning from past failures to meet future challenges. Transl Res 167:152–166. https://doi.org/10.1016/j.trsl.2015.07.002
Schett G, Elewaut D, McInnes IB, Dayer JM, Neurath MF (2013) How cytokine networks fuel inflammation: toward a cytokine-based disease taxonomy. Nat Med 19:822–824. https://doi.org/10.1038/nm.3260
Schett G, Sticherling M, Neurath MF (2020) COVID-19: risk for cytokine targeting in chronic inflammatory diseases? Nat Rev Immunol 20:271–272. https://doi.org/10.1038/s41577-020-0312-7
Shah M, Yellon DM, Davidson SM (2020) The role of extracellular DNA and histones in ischaemia-reperfusion injury of the myocardium. Cardiovasc Drugs Ther 34:123–131. https://doi.org/10.1007/s10557-020-06946-6
Shetelig C, Limalanathan S, Hoffmann P, Seljeflot I, Gran JM, Eritsland J, Andersen GO (2018) Association of IL-8 with infarct size and clinical outcomes in patients with STEMI. J Am Coll Cardiol 72:187–198. https://doi.org/10.1016/j.jacc.2018.04.053
Shin HJ, Won NH, Lee HW (2014) Remote ischemic preconditioning prevents lipopolysaccharide-induced liver injury through inhibition of NF-kappaB activation in mice. J Anesth 28:898–905. https://doi.org/10.1007/s00540-014-1850-6
Skyschally A, Gent S, Amanakis G, Schulte C, Kleinbongard P, Heusch G (2015) Across-species transfer of protection by remote ischemic preconditioning with species-specific myocardial signal transduction by reperfusion injury salvage kinase and survival activating factor enhancement pathways. Circ Res 117:279–288. https://doi.org/10.1161/CIRCRESAHA.117.306878
Smith CJ, Hulme S, Vail A, Heal C, Parry-Jones AR, Scarth S, Hopkins K, Hoadley M, Allan SM, Rothwell NJ, Hopkins SJ, Tyrrell PJ (2018) SCIL-STROKE (subcutaneous interleukin-1 receptor antagonist in ischemic stroke): a randomized controlled phase 2 trial. Stroke 49:1210–1216. https://doi.org/10.1161/STROKEAHA.118.020750
Stanley AC, Lacy P (2010) Pathways for cytokine secretion. Physiology (Bethesda) 25:218–229. https://doi.org/10.1152/physiol.00017.2010
Toldo S, Abbate A (2018) The NLRP3 inflammasome in acute myocardial infarction. Nat Rev Cardiol 15:203–214. https://doi.org/10.1038/nrcardio.2017.161
Toldo S, Mauro AG, Cutter Z, Abbate A (2018) Inflammasome, pyroptosis, and cytokines in myocardial ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol 315:H1553–H1568. https://doi.org/10.1152/ajpheart.00158.2018
Tolkmitt KJ, Simsekyilmaz S, Schipke J, Muhlfeld C, Preissner KT, Boning A (2019) Influence of medication-induced preconditioning or remote ischemic preconditioning on the intrinsic vascular extracellular rna/ribonuclease system in cardioprotection. Thorac Cardiovasc Surg 67:494–501. https://doi.org/10.1055/s-0038-1675398
Tretyakov AV, Farber HW (1995) Endothelial cell tolerance to hypoxia. Potential role of purine nucleotide phosphates. J Clin Invest 95:738–744. https://doi.org/10.1172/JCI117721
Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel AS, Mehra MR, Schuepbach RA, Ruschitzka F, Moch H (2020) Endothelial cell infection and endotheliitis in COVID-19. Lancet 395:1417–1418. https://doi.org/10.1016/S0140-6736(20)30937-5
Vicencio JM, Yellon DM, Sivaraman V, Das D, Boi-Doku C, Arjun S, Zheng Y, Riquelme JA, Kearney J, Sharma V, Multhoff G, Hall AR, Davidson SM (2015) Plasma exosomes protect the myocardium from ischemia-reperfusion injury. J Am Coll Cardiol 65:1525–1536. https://doi.org/10.1016/j.jacc.2015.02.026
Wang H, Lyu Y, Liao Q, Jin L, Xu L, Hu Y, Yu Y, Guo K (2019) Effects of remote ischemic preconditioning in patients undergoing off-pump coronary artery bypass graft surgery. Front Physiol 10:495. https://doi.org/10.3389/fphys.2019.00495
Wang Q, Liu G-P, Xue F-S, Wang S-Y, Cui X-L, Li R-P, Yang G-Z, Sun C, Liao X (2015) Combined vagal stimulation and limb remote ischemic perconditioning enhances cardioprotection via an anti-inflammatory pathway. Inflammation 38:1748–1760. https://doi.org/10.1007/s10753-015-0152-y
Wang X, Wang J, Tu T, Iyan Z, Mungun D, Yang Z, Guo Y (2018) Remote ischemic postconditioning protects against myocardial ischemia-reperfusion injury by inhibition of the RAGE-HMGB1 pathway. Biomed Res Int 2018:4565630. https://doi.org/10.1155/2018/4565630
Weidemann A, Johnson RS (2008) Biology of HIF-1alpha. Cell Death Differ 15:621–627. https://doi.org/10.1038/cdd.2008.12
Wider J, Undyala VVR, Whittaker P, Woods J, Chen XQ, Przyklenk K (2018) Remote ischemic preconditioning fails to reduce infarct size in the Zucker fatty rat model of type-2 diabetes: role of defective humoral communication. Basic Res Cardiol 113:16. https://doi.org/10.1007/s00395-018-0674-1
Xia M, Ding Q, Zhang Z, Feng Q (2017) Remote limb ischemic preconditioning protects rats against cerebral ischemia via HIF-1alpha/AMPK/HSP70 pathway. Cell Mol Neurobiol 37:1105–1114. https://doi.org/10.1007/s10571-016-0444-2
Yamamoto Y, Gaynor RB (2001) Therapeutic potential of inhibition of the NF-kappaB pathway in the treatment of inflammation and cancer. J Clin Invest 107:135–142. https://doi.org/10.1172/JCI11914
Yang J, Liu C, Du X, Liu M, Ji X, Du H, Zhao H (2018) Hypoxia inducible factor 1alpha plays a key role in remote ischemic preconditioning against stroke by modulating inflammatory responses in rats. J Am Heart Assoc 7:e007589. https://doi.org/10.1161/JAHA.117.007589
Yellon DM, Hausenloy DJ (2007) Myocardial reperfusion injury. N Engl J Med 357:1121–1135. https://doi.org/10.1056/NEJMra071667
Zapata-Chavira H, Hernandez-Guedea M, Jimenez-Perez JC, Perez-Rodriguez E, Munoz-Espinosa L, Munoz-Maldonado G, Cordero-Perez P (2019) Modulation of remote ischemic preconditioning by proinflammatory cytokines in renal transplant recipients. J Invest Surg 32:63–71. https://doi.org/10.1080/08941939.2017.1375052
Zendedel A, Johann S, Mehrabi S, Mt J, Hassanzadeh G, Kipp M, Beyer C (2016) Activation and regulation of NLRP3 inflammasome by intrathecal application of SDF-1a in a spinal cord injury model. Mol Neurobiol 53:3063–3075. https://doi.org/10.1007/s12035-015-9203-5
Zhang C, Dong H, Chen F, Wang Y, Ma J, Wang G (2019) The HMGB1-RAGE/TLR-TNF-alpha signaling pathway may contribute to kidney injury induced by hypoxia. Exp Ther Med 17:17–26. https://doi.org/10.3892/etm.2018.6932
Zhang J, Zhang J, Yu P, Chen M, Peng Q, Wang Z, Dong N (2017) Remote ischaemic preconditioning and sevoflurane postconditioning synergistically protect rats from myocardial injury induced by ischemia and reperfusion partly via inhibition TLR4/MyD88/NF-κB signaling pathway. Cell Physiol Biochem 41:22–32. https://doi.org/10.1159/000455815
Zhang JM, An J (2007) Cytokines, inflammation, and pain. Int Anesthesiol Clin 45:27–37. https://doi.org/10.1097/AIA.0b013e318034194e
Zhang W, Chen C, Jing R, Liu T, Liu B (2019) Remote ischemic preconditioning protects cisplatin-induced acute kidney injury through the PTEN/AKT signaling pathway. Oxid Med Cell Longev 2019:7629396. https://doi.org/10.1155/2019/7629396
Zhou H, Yu M, Fukuda K, Im J, Yao P, Cui W, Bulek K, Zepp J, Wan Y, Kim TW, Yin W, Ma V, Thomas J, Gu J, Wang JA, DiCorleto PE, Fox PL, Qin J, Li X (2013) IRAK-M mediates Toll-like receptor/IL-1R-induced NFkappaB activation and cytokine production. EMBO J 32:583–596. https://doi.org/10.1038/emboj.2013.2
Zhou X, Jiang R, Dong Y, Wang L (2017) Remote ischemic preconditioning attenuates cardiopulmonary bypass-induced lung injury. PLoS ONE 12:e0189501. https://doi.org/10.1371/journal.pone.0189501
Zuurbier CJ, Abbate A, Cabrera-Fuentes HA, Cohen MV, Collino M, De Kleijn DPV, Downey JM, Pagliaro P, Preissner KT, Takahashi M, Davidson SM (2019) Innate immunity as a target for acute cardioprotection. Cardiovasc Res 115:1131–1142. https://doi.org/10.1093/cvr/cvy304
Zwaag J, Beunders R, Warle MC, Kellum JA, Riksen NP, Pickkers P, Kox M (2019) Remote ischaemic preconditioning does not modulate the systemic inflammatory response or renal tubular stress biomarkers after endotoxaemia in healthy human volunteers: a single-centre, mechanistic, randomised controlled trial. Br J Anaesth 123:177–185. https://doi.org/10.1016/j.bja.2019.03.037
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pearce, L., Davidson, S.M. & Yellon, D.M. Does remote ischaemic conditioning reduce inflammation? A focus on innate immunity and cytokine response. Basic Res Cardiol 116, 12 (2021). https://doi.org/10.1007/s00395-021-00852-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00395-021-00852-0