
Abstract
Aquaporins (AQPs) are channel-forming membrane proteins highly permeable to water. AQP4 is found in mammalian hearts; however, its expression sites, regulation and function are largely unknown. The aim was to investigate cardiac AQP4 expression in humans and mice, its regulation by ischemia and hypoxia, and in particular its role in cardiac ischemic injury using AQP4 knockout (KO) mice. Comparable levels of AQP4 were detected by Western blot and qPCR in biopsies from human donor hearts and wild type C57Bl6 mouse hearts. In mice, AQP4 was expressed on cardiomyocyte plasmalemma (qPCR, Western blot, immunogold), and its mRNA decreased following ischemia/reperfusion (isolated hearts, p = 0.02) and after normobaric hypoxia in vivo (oxygen fraction 10 % for 1 week, p < 0.001). Isolated hearts from AQP4 KO mice undergoing global ischemia and reperfusion had reduced infarct size (p = 0.05) and attenuated left ventricular end-diastolic pressure during reperfusion (p = 0.04). Infarct size was also reduced in AQP4 KO mice 24 h after left coronary artery ligation in vivo (p = 0.036). AQP4 KO hearts had no compensatory change in AQP1 protein expression. AQP4 KO cardiomyocytes were partially resisted to hypoosmotic stress in the presence of hypercontracture. AQP4 is expressed in human and mouse hearts, in the latter confined to the cardiomyocyte plasmalemma. AQP4 mRNA expression is downregulated by hypoxia and ischemia. Deletion of AQP4 is protective in acute myocardial ischemia–reperfusion, and this molecule might be a future target in the treatment of acute myocardial infarction.
Similar content being viewed by others


Introduction
There are three ways water can traverse biological membranes: by means of diffusion through the lipid bilayer, through co-transporters [23] and through the specific water channels, aquaporins (AQPs) [45]. Out of 13 AQPs identified in mammals, AQP1, -4, -7 and -11 have been detected in both human and mouse hearts at the mRNA level [7]. The corresponding proteins have been found for AQP1, -4, -6 and -7 [7, 18]. AQP4 expression in the human heart was reported to be very low [7]. Only a few studies have addressed the expression and subcellular localization of AQP4 in the heart, with inconsistent results [7, 38, 44]. Thus, it is currently unclear in which cardiac cells AQP4 is located, where in the cell it is expressed, and how its expression is regulated. AQP4 mRNA was previously found to increase during ischemia in murine hearts, although the implication of this finding was not evident [44]. A careful determination of AQP4 cellular and subcellular localization and its expression pattern will facilitate understanding the function of AQP4 in the heart.
Several studies show that AQP4 is important for ischemic injury in the brain. Mice with deletion or mislocalization of AQP4 display reduced necrosis and edema after cerebral ischemia [1, 24]. Development of tissue edema is also an integral part of ischemic damage in the heart [26]. Based on these findings, we hypothesized that cardiac ischemic injury would be attenuated in AQP4 knockout (KO) mice. Furthermore, we wanted to determine AQP4 expression in humans and mice, address its specific cellular and subcellular localization, and evaluate the effect of ischemia and hypoxia on AQP4 expression in mice.
Materials and methods
All experiments conform to the guidelines for the use and care of laboratory animals (“Principles of laboratory animal care”, NIH publication No. 86–23. Revised 1996) and the study was approved by the Norwegian Animal Health Authority. For studies of AQP4 expression, wild type (WT) C57Bl6 male mice weighing 25 ± 3 g (Scanbur AS, Oslo) were used.
We used AQP4 KO mice generated and raised in the Center for Molecular Biology and Neuroscience at the University of Oslo [42]. The gene deletion strain was generated based on the Sv129 mice and backcrossed with C57Bl6. These mice produce substantially fewer offsprings and have a lower pregnancy rate, according to a previous report by Sun et al. [41]. A detailed physiological phenotype assessment of these mice has yet to be performed.
The methods here are described in brief. For details on the procedures, see Supplementary methods.
Anesthesia during animal experiments
For anesthesia during in vivo coronary artery occlusion, please see the respective section. For the rest of our experiments we used isolated mouse hearts, where mice weighing 25 ± 3 g were injected intraperitoneally with sodium pentobarbital 60 mg/kg (Ås Produksjonslab AS, Norway) and 500 IU heparin (Leo Pharma A/S, Denmark). The adequacy of anesthesia was assessed by the pedal withdrawal reflex. We commenced surgery when there was no muscle reaction to the pinch. The animals were killed by heart excision within 30 s from the start of surgery.
Human samples
The studies were performed in accordance to the ethical standards stated in the 1964 Declaration of Helsinki, and were approved by the Regional Ethics Committee in Norway (REK nr. 07482a). Written informed consent was obtained from family members of organ donors [10]. Large biopsies (150–200 mg) were sampled from the left ventricles of hearts (n = 4) used for homograft preparation, explanted from humans deceased of non-cardiac reasons. mRNA and protein were extracted for qPCR and Western blot.
Cell fractioning
Heart tissue from six wild type (WT) mice was fractionated into crude cytosolic and membrane fractions. Their purity was assessed using Western blot with antibodies against the IkappaB kinase alpha (IκBα, cytosol) and sodium–calcium exchanger (NCX, membrane), respectively (for antibody overview, please see Supplementary methods, table ST1).
Isolation of cardiomyocytes
Cardiomyocytes from five WT mice were isolated and cultured following the procedure described by O’Connell et al. [32]. Both the cardiomyocytes and the cell population remaining after cardiomyocyte isolation were collected. To assess the cellular content of the two populations, they were analyzed by qPCR with primers specific for the proteins characteristic of different myocardial cell types: cardiomyocytes—troponin I; fibroblasts—vimentin, endothelial cells—cadherin, smooth muscle cells—smooth muscle actin (for primer sequences, please see supplementary table ST2).
Immunogold electron microscopy
Eleven WT mouse hearts were fixed by perfusion with 4 % formaldehyde with 0.1 % glutaraldehyde. One-millimeter cubic transmural tissue fragments from the left anterior ventricular wall were embedded in resin (Lowicryl HM20) and 200-nm sections were prepared and placed on the grids. The grids were then incubated with antibodies against AQP1 or AQP4 coupled to 15-nm gold particles and examined with a Tecnai 12-electron microscope. For the antibody overview, please see supplementary table ST1.
Quantification was performed as described by Bergersen et al. [6]. AQP4 KO mouse heart sections were used as negative controls for AQP4 antibody, and WT mouse brains as positive. The latter were also used as negative controls for AQP1. Quantification was performed by a person blinded to the experimental groups.
Perfusion of isolated mouse hearts and infarct size measurement
Fourteen WT mouse hearts were isolated and retrogradely buffer-perfused at a pressure of 70 mmHg as described previously [20]. They were subjected to 35 min of global ischemia followed by 60 min of reperfusion and freeze-clamped in liquid nitrogen along with 14 unperfused controls. Half of these hearts were used for protein extraction and the other half for isolation of RNA. Nine AQP4 KO and nine-matched WT mouse hearts were stabilized for 20 min followed by 35 min of global ischemia and 120 min of reperfusion. One-millimeter thick sections were incubated in 1 % triphenyl tetrazolium chloride for 15 min at 37 °C and photographed (Nikon Coolpix 5400, Nikon Corp., Japan). Infarct area was calculated by a blinded person using Adobe Photoshop CS software. The average value obtained after quantification of all images from a single heart was used for statistics.
In vivo hypoxia
Twelve WT mice were placed in a tightly sealed chamber under normobaric hypoxia (oxygen fraction 10 %) for 7 days. Another 12 mice were housed under normoxic conditions (oxygen fraction 21 %). The carbon dioxide concentration was kept <0.4 % inside the chamber. At the end of the hypoxic/normoxic protocol the animals were anesthetized, their hearts were dissected and right ventricular myocardium was freeze-clamped for Western Blot and qPCR.
In vivo myocardial ischemia
Ten AQP4 KO and 10-matched WT mice weighing 27 ± 3 g were placed in a chamber ventilated with the mixture of 5 % isoflurane and oxygen. When the animals were deeply anesthetized (no pedal withdrawal), they were intubated and connected to a rodent ventilator (Model 874 092, B. Braun, Melsungen, Germany) with 2–3 % isoflurane and oxygen for intraoperative anesthesia. The left coronary artery was permanently ligated with a silk suture and mice were extubated and given post-operative analgesia by subcutaneous injection of 0.02 ml buprenorphine (0.3 mg mL−1). The operated mice were kept in an incubator at 37 °C for 2 h to prevent heat loss. Mice displayed no signs of distress in the postoperative period.
Twenty-four hours later, the hearts were isolated for measurements of infarct size. They were first perfused with Evans blue to denote the area at risk, slices were prepared and photographed, then incubated with TTC and photographed again. Infarct size was normalized to the risk area.
Immunoblots
The protocol used for Western blot in these experiments is identical to our previous investigations [40], except that the protein samples for AQP4 analysis were not heat-denatured in Laemmli buffer to enrich the monomeric fraction of AQP4 [39] (see Supplementary methods). For determination of human cardiac AQP4 expression, extracts from four donor hearts and four mouse hearts were compared. The extracts were run on polyacrylamide gels supplemented with 3 M urea as recommended by Sorbo et al. [39], and probed using anti-AQP4 antibodies from two manufacturers: Millipore, USA and Sigma-Aldrich, USA. For controls, we used a heated extract from the donor heart, two extracts from AQP4 KO mouse hearts, wild type mouse extensor digitorum longus and soleus muscle extracts, and also the wild type mouse brain. Similar amounts of protein were loaded per lane (50 μg), except from the brain (5 μg). To evaluate a potential compensatory increase of AQP1 expression in AQP4 KO mice, protein extracts from six AQP4 KO and six WT mouse hearts were analyzed with antibodies against AQP1 and AQP4. For an overview of antibodies used see Supplementary table ST1. The protein loading was evaluated by Ponceau or Coomassie staining. To evaluate the level of expression, the ratios of antibody signal versus the non-specific protein dye were calculated.
RNA extraction and quantitative polymerase chain reaction (qPCR)
Primers were designed to span exon–exon junction areas. For primer sequences see Supplementary table ST2. The test gene was related to the expression of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) in the tissue samples and to 18S ribosomal RNA in the cell samples using commercially available primers (PE Applied Biosystems). For details, see Supplementary methods.
Optical recording and cardiomyocyte viability measurement
Cardiomyocytes were isolated from AQP4 KO and wild type hearts as described above. The cultured cells were loaded with 0.5 μmol of acetoxymethyl esther of calcein (calcein-AM) and visualized using fluorescent microscopy. The cell medium was quickly replaced by distilled water (first series) or hypotonic buffer containing 25 mM of NaCl, 1 mM EGTA and 30 mM butanedione monoxime (second series). After the application of hypotonic solution, video recording with a CCD camera was started immediately, and performed for 1,200 s (1 frame per second) to monitor calcein fluorescence quenching, and to register the time of cell bursting. In the first series with distilled water, 48 AQP4 KO and 45 WT cardiomyocytes from three AQP4 KO and three WT were filmed. In the second series with hypotonic buffer, 52 AQP4 KO and 27 WT cardiomyocytes from three more AQP4 KO and three WT were filmed. The data were quantified using MetaMorph 5.0 image-processing software (Universal Imaging Corporation, Molecular Devices, USA).
Statistical analysis
The data were analyzed using Prism 5 (GraphPad Software Inc., La Jolla, CA, USA). Cardiac function parameters and calcein fluorescent signals were analyzed with two-way analysis of variance (ANOVA) for repeated measurements. Infarct size, qPCR data and immunogold results on AQP1 were analyzed using a two-tailed Student’s t test. Immunogold results on AQP4 expression on different cardiomyocyte membranes were analyzed with one-way ANOVA followed by Bonferroni correction. Western blot results were analyzed using Mann–Whitney test due to unequal distribution. Differences were considered significant when the p value was ≤0.05. Data on cardiomyocyte survival after hypoosmotic shock were analyzed using log-rank Mantel–Cox test. Exact p values are given except when p < 0.001 or p > 0.2. All continuous data as well as infarct size and mRNA expression of cell type-specific markers are shown as mean ± SD. Expression of AQP1 and AQP4 in AQP4 KO are shown as box plots with 5, 25, 75 and 95 % percentiles and median. Infarct size, AQP1, -4 protein and mRNA expression and quantification of AQP1 immunogold results are shown as dot plots with individual values and line at median.
Results
AQP4 expression in human and WT mouse hearts
qPCR showed that AQP4 mRNA was expressed with a mean C t value of 29.8 ± 1.8 in human hearts and 28.5 ± 1.6 in hearts of mice. Normalization was not performed due to lack of a reliable housekeeping gene for both the species. Western blot results obtained with the two antibodies produced different results. The Millipore antibody detected three distinct bands in the human heart extracts, in the range of 30–45 kDa (Fig. 1a). The signal in human heart was reduced by heating the sample. With this antibody no clear signal was found in mouse heart extracts. However, the antibody detected a single band of approximately 40 kDa in the skeletal muscles and in the brain from mice. A5971 from Sigma-Aldrich, on the contrary, showed three bands in the same molecular weight range in mouse samples, similar pattern in the brain and skeletal muscle, and absence of signal in the knockouts (Fig. 1b). No signal was observed in the human hearts with the Sigma antibody.

Expression of AQP4 in the human and mouse hearts. a, b Western blots showing AQP4 expression in the human and mouse hearts. Four donor hearts were compared to four wild type mouse hearts, loading 50 μg per lane. Membrane a was incubated with the Millipore (Chemicon) anti-AQP4 antibody, while membrane b was treated with anti-AQP4 antibody from Sigma-Aldrich. Controls (inside a rectangle) included a heated sample from a donor heart 1, wild type mouse soleus and extensor digitorum longus (EDL) muscles, two AQP4 KO mouse heart samples and the wild type mouse brain. Equal protein loading was verified by Coomassie staining (below). c Western blots with AQP4 immunoreactivity in the membrane and cytoplasm fractions, n = 6. The sodium–calcium exchanger (NCX) was used as a marker of cell membrane, while inhibitory κBα (IκBα) was used as a marker of cytoplasm. d qPCR analysis of cell populations isolated from wild type mouse hearts, n = 5. Troponin I was used as a marker of cardiomyocytes, smooth muscle (SM) actin as a marker of smooth muscle cells, vimentin as a marker of fibroblasts, and cadherin as a marker of endothelial cells. Data are presented as fold change over the fraction containing the least amount of each marker. e AQP4 mRNA and protein expression in these cell fractions. qPCR data presented as mean ± SD, normalized to the mean of the mRNA content in the CM fraction (above). Western blot AQP4 immunoreactivity in these cell fractions (below). CM cardiomyocyte fraction, nCM non-cardiomyocyte fraction
Localization of AQP4 within the myocardium of WT mice
AQP4 expression in the membrane and cytoplasmic fractions The extracts of cytosolic fraction had minor contamination with membrane proteins as indicated by the presence of a weak NCX band, while the membrane fraction had no trace of the cytosol marker signal (IκBα). AQP4 immunoreactivity was detected by Western blot only in the membrane fraction. No AQP4 signal was present in the cytosolic fraction (Fig. 1c).
AQP4 expression in cultured myocardial cells The cardiomyocyte fraction was positive for troponin I and to a lesser extent for smooth muscle actin (Fig. 1d). The second fraction (non-cardiomyocytes) contained fibroblasts and endothelial cells. qPCR and Western blot demonstrated that AQP4 is expressed exclusively in the cardiomyocyte fraction (Fig. 1e).
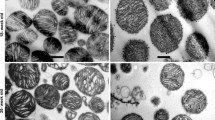
Immunogold electron microscopy Figure 2a is a representative picture of mouse myocardium. AQP4 immunoreactivity was found on cardiomyocyte membranes in junction areas (Fig. 2b) or facing adjacent cardiomyocytes (Fig. 2c) and capillaries (Fig. 2d). Quantification of the immunogold signal indicated that AQP4 is evenly distributed across these three membrane domains (Fig. 2e). The few gold particles outside these regions likely represent non-specific labeling. No AQP4 immunoreactivity was detected in cells other than cardiomyocytes.

Electron microscopy of wild type mouse hearts. Ultrathin sections were stained with anti-AQP4 antibody and labeled with gold particles, n = 11. a Representative overview with lower magnification (×18,000). b, c, d AQP4 immunoreactivity (arrowheads) detected along the cardiomyocyte membranes facing other cardiomyocytes (M), at intercalated discs (ID) and towards capillaries (C). Magnification ×25,000. e Quantification of AQP4 density (number of gold particles per μm of membrane) in three membrane locations: Myo-Myo cardiomyocyte membranes adjacent to other cardiomyocytes (excluding intercalated disks), ID cardiomyocyte membranes at the intercalated disks. Myo-Endo Cardiomyocyte membranes facing endothelial cells. Data are presented as individual values and median
Effect of ischemia on AQP4 expression
qPCR showed a decrease of AQP4 mRNA after 35 min ischemia and 60 min reperfusion in isolated WT mouse hearts, while the protein was unchanged (Fig. 3a).

AQP4 mRNA and protein in total heart lysates following isolated heart perfusion with 35 min global ischemia and 60 min reperfusion (a) and in right ventricle lysates following 1-week-long normobaric hypoxia (b), shown as mean ± SD. Panels to the right are representative immunoblots along with Coomassie staining. Ctr controls, Isc ischemic-reperfused, Hy hypoxic
Effect of hypoxia on AQP4 expression
The hearts of mice exposed to hypoxia for 1 week had downregulation of AQP4, however, the corresponding protein only tended to decrease (Fig. 3b).
Ischemia/reperfusion in AQP4 KO and WT mouse hearts ex vivo
Infarct size was significantly smaller in hearts of AQP4 KO mice than in the WT: 26 ± 15 vs. 45 ± 22 % (Fig. 4a). Ischemia-induced increase of left ventricular end-diastolic pressure (LVEDP) was attenuated in hearts of KO mice (Fig. 4b).

Ischemia/reperfusion in AQP4 knockout (KO) hearts ex vivo and in vivo. a Infarct size in hearts from AQP4 KO (n = 10) and wild type (WT, n = 9) mice perfused in the Langendorff model with 35 min of global ischemia and 120 min reperfusion, measured with triphenyl tetrazolium chloride; presented as individual values and median. b Left ventricular end-diastolic pressure (LVEDP) in these hearts (mean ± SD), BI before ischemia. c Infarct size in AQP4 KO (n = 7) and WT (n = 10) mouse hearts evaluated 24 h following ligation of the left coronary artery (individual values and median). Expressed in percent of the area at risk
Myocardial ischemia in AQP4 KO and WT mice in vivo
Infarct size related to the area at risk 24 h after ligation of the left coronary artery was reduced in the hearts of AQP4 KO compared to WT mice hearts: 58 ± 20 vs. 77 ± 20 % (Fig. 4c). Area at risk did not differ between the two groups (not shown).
Optical recording and cardiomyocyte viability measurement
Cardiomyocytes isolated from AQP4 KO and WT mice were loaded with cell-permeable calcein-AM dye and subjected to severe hypoosmotic shock. Cell bursting and changes in fluorescence were recorded optically.
In the first series of experiments, the cardiomyocytes were treated with distilled water (Fig. 5a, Supplementary video 1). The number of cardiomyocytes from AQP4 KO mice that maintained membrane integrity was larger than the WT, resulting in longer survival time before contraction and bursting of the cells (Fig. 5b). In addition, AQP4 KO cardiomyocytes displayed lower fluorescence intensity than WT, showing that they maintained their cell volume for a longer time (Fig. 5c).

Fluorescence imaging of hypoosmotic shock in AQP4 KO and WT cardiomyocytes. a Single video frames taken 15 and 400, 800 and 1,200 s following application of distilled water on the cultured AQP4 KO and WT cardiomyocytes. * cells selected for analysis; †“ghosts” of ruptured cardiomyocytes; ¤cells with preserved membrane integrity by the end of recording. b Survival plot and cumulative distribution showing the fraction of cells that had burst throughout the 20-min recording session. c Graphs showing the mean changes of calcein fluorescence intensity in the remaining fraction of cardiomyocytes that survived (AQP4 KO, n = 18 and WT, n = 7). d Single video frames taken 15 and 400, 800 and 1,200 s following application of hypoosmotic solution (25 mM NaCl, 1 mM EGTA and 30 mM BDM) on the cultured AQP4 KO and WT cardiomyocytes. * cells selected for analysis; †“ghosts” of ruptured cardiomyocytes. e Dynamics of cell bursting throughout the 20-min recording session, and the mean changes in calcein fluorescence intensity in the cardiomyocytes surviving the initial 400 s (AQP4 KO, n = 20 and WT, n = 33)
In the second series, the cells were treated with a hypoosmotic solution designed to prevent the contracture and to inhibit the reverse mode of the Na+/Ca2+ exchanger (Fig. 5d, Supplementary video 2). The cardiomyocytes maintained initial cell length throughout the observation period and water entry was registered, but no difference in the dynamics of cell bursting was observed between the AQP4 KO and WT cardiomyocytes (Fig. 5e). All the cells burst within 1,200 s. Calcein fluorescence intensity was also similar in the cells surviving the initial 400 s (Fig. 5e).
Expression of AQP1 and AQP4 in AQP4 KO and WT mice
No quantifiable amounts of AQP4 mRNA (not shown) or AQP4 protein (Fig. 6a) were detected in AQP4 KO hearts. To check if there were compensatory changes in the expression of the other major cardiac aquaporin, AQP1, we analyzed AQP4 KO hearts with qPCR. Relative expression of AQP1 mRNA was elevated by 36 ± 24 % compared to WT, however, the corresponding protein expression was not increased (Fig. 6b). Using immunogold electron microscopy, AQP1 was found to be expressed on the endothelial cell membranes (Fig. 6c), but quantitative analysis showed no difference of AQP1 between AQP4 KO and WT mice (Fig. 6d, e).

Protein expression of AQP1 and AQP4 assessed in AQP4 KO and WT mouse hearts (n = 5–6 in each group). a, b Western blots with AQP4 and 1. Upper panels show box plots with median and 5, 25, 75 and 95 %, lower panels show representative immunoblots. Coom Coomassie staining used to evaluate protein loading. c Representative Immunogold staining showing AQP1 immunoreactivity along the endothelial cell membranes. C Capillary; M myocyte; a, b apical and basal membranes of an endothelial cell. Magnification ×25,000. d, e Quantification of AQP1 signal on along the apical and basal membranes of endothelial cells in AQP4 KO (n = 5) and WT (n = 9) mice. Shown as individual values and median
Discussion
There are two important novel observations in this study: 1. expression of AQP4 in the human heart is not negligible as previously described; and 2. cardiac ischemic injury measured as infarct size was reduced in AQP4 KO mice. The latter is in line with findings obtained in studies of the ischemic brain [24].
AQP4 expression in the human heart
An important question regarding the relevance of the present study is whether AQP4 really exists in human hearts. Previous studies failed to demonstrate appreciable protein expression, although the corresponding mRNA was reported [7, 11]. Using qPCR, we found that the C t values obtained for AQP4 in mice and humans were comparable when the reaction was performed with the same amounts of cardiac RNA in μg. Western blot showed different species-specificity of the available antibodies. The Millipore antibody used on protein extracts from human hearts produced three distinct bands, possibly corresponding to the three prevalent isoforms of AQP4—M1, M23 and Mz [30, 39]. Similar bands were detected by the Sigma antibody in mouse hearts. Murine positive controls worked for both antibodies, reflecting a known high level of AQP4 expression in the brain and in the fast-twitch muscle extensor digitorum longus [31]. However, the Millipore antibody did not detect AQP4 in mouse hearts at the tested dilution, while Sigma antibody did not reveal AQP4 in human hearts. It is striking that both antibodies detect AQP4 in mouse brain and skeletal muscle, but differ when applied to extracts from mouse and human hearts. Both are rabbit polyclonals raised against amino acids 249–323 of rat AQP4, the conserved C-terminus. According to the UniProt sequence database, the human C-terminus (the last 75 residues) differs from the rat sequence by six amino acids, while the mouse differs by one. The signal in mouse skeletal muscle was weaker for the Millipore antibody, indicating its generally low affinity for the murine AQP4. Possibly, the selective affinity of these antibodies for human and mouse AQP4 may reflect a yet-unknown variation in the C-terminus of AQP4.
Although the quantitative comparison of AQP4 expression is not possible either for the PCR data or the Western blot results, we may conclude that AQP4 expression in the human heart is present at both the mRNA and protein level, supporting the clinical relevance of our findings regarding AQP4 and its function as assessed by studies in AQP4 KO mice.
AQP4 localization in the mouse heart
With AQP4 being the most abundant aquaporin in the brain, the number of mRNA copies in murine cardiomyocytes has been estimated at around 11 % of that in the fast-twitch skeletal muscle and only 2.5 % of that in the cerebellum [38]. However, to evaluate the importance of function, cellular localization may be as significant as quantity. In most tissues, AQP4 is located on the plasmalemma of cells, which would be essential to its function as a water channel. The shorter isoforms—products of alternative splicing—are found in the cytoplasm [28]. By means of a crude fractionation experiment, we show that AQP4 is only expressed on the membranes of murine myocardial cells.
Cell type specificity of cardiac AQP4 has not been established so far. In 2007, Wakayama et al. [43] used immunofluorescence and freeze-fracture electron microscopy to demonstrate expression of AQP4 in mouse cardiomyocytes; however, no data from the other cell types in the heart were presented. We separated myocardial cells in two fractions, and found AQP4 signal in the cardiomyocyte fraction, which was also positive for smooth muscle actin. Analysis of primer specificity using NCBI primer-BLAST submission form revealed negligible probability of off-target binding, therefore indicating possible presence of smooth muscle cells in the cardiomyocyte fraction. In addition, smooth muscle actin is also expressed by pericytes [16]. At the same time, neither mRNA nor protein was detected in significant amounts in the fraction containing fibroblasts and endothelial cells.
AQP4 is identified as a building block of supramolecular structures called square arrays [47], known to occur in the mouse heart [25]. Square arrays are best studied with freeze-fracture electron microscopy, while regular electron microscopy gives access to the cell domains that are difficult to run a fracture through. A distinctive feature of cardiomyocytes is to form a syncytium, where the cells are connected end-to-end at intercalated disks, in order to allow the transfer of action potential. The plasmalemma of cardiomyocytes either faces adjacent cardiomyocytes or other neighboring cells (fibroblasts, endothelial cells), or forms a part of an intercalated disk. We found equal density of AQP4 in these membrane domains. The presence of AQP4 in the membranes of intercalated disks may suggest a function other than water transport between the cell and the interstitium. Possibly it affects transfer of action potential from one cardiomyocyte to another. This transfer is carried out by connexins, which also appear to play a significant role in endogenous cardioprotection [17]. A recent study showed a decreased expression of connexin 43 in AQP4 KO mouse hearts [9], possibly suggesting a functional link between the two proteins.
Regulation of AQP4 in ischemia and hypoxia
A number of AQPs, including AQP4, change expression in response to ischemia and hypoxia in various organs. In particular, downregulation of AQP4 was observed in ischemic and anoxic brain [22], retina [19] and spinal cord [46]. We found that cardiac AQP4 mRNA was downregulated by short-term ischemia ex vivo and prolonged hypoxia in vivo, though the decrease of corresponding protein was not detected. In contrast, Warth et al. [44] in a different timeframe found upregulation of murine cardiac AQP4 mRNA and protein after ischemia in vivo. What signaling mechanisms may cause AQP4 regulation is at present unknown, but there is evidence that microRNA-320 may downregulate AQP4 in the brain [37]. The effects of ischemia and hypoxia on AQP4 expression may potentially be confounded by an aseptic inflammation caused by these processes [13].
Role of AQP4 in ischemia
Water accumulation in the myocardium occurs as a result of ischemia [2, 12], when ischemic tissue becomes hyperosmolar and attracts water from the capillary lumen. This water is transported through inter-endothelial clefts, as well as across the endothelial cells, potentially facilitated by AQP1 [3, 12, 34] and shedding of endothelial glycocalyx [4, 8]. Then, it is transported into the myocardial cells, most importantly, cardiomyocytes. Reperfusion delivers normoosmolar blood to the hyperosmolar cells, which leads to further cell swelling [14, 15], which may even involve cells outside the risk area [27]. This water accumulation leads to a pronounced depression of cardiac function [21], and aggravates effects of shortage in oxygen and nutrient supply [26].
AQP4 KO mice had reduced infarct size both after ex vivo ischemia–reperfusion and after in vivo infarction without reperfusion. Reduction of necrosis in a Langendorff model was associated with attenuated increase of left ventricular diastolic pressure. This parameter in isolated hearts characterizes the degree of contracture following ischemia and reperfusion, which is one of the major determinants of functional impairment. Cardioprotective effect of AQP4 ablation in the heart is a novel observation, which opens a new field of therapeutic measures. The fact that cardioprotection was observed in two distinctly different models presents strong evidence that the AQP4 KO genotype confers increased tolerance to ischemic injury.
Furthermore, we sought to demonstrate that the effects of AQP4 KO may be attributed to AQP4, and not to compensatory changes of AQP1. We investigated AQP1 expression in AQP4 KO hearts, and no change in protein expression was observed in AQP4 KO hearts compared to the wild types.
AQP4 and cardiomyocyte water permeability
To further determine if AQP4 plays a role in osmotic swelling, optical recording was used to study cell survival and volume changes following acute hypoosmotic stress in cardiomyocytes loaded with calcein-AM dye, a derivative of calcein that penetrates the cytoplasmatic membrane and fluoresces proportionally to the cell volume [29]. The bursting cells immediately lose their fluorescence.
When cardiomyocytes were placed in distilled water, cells from AQP4 KO mice showed a higher resistance to hypoosmolarity, however, they developed a significant degree of contracture (see Supplementary video 1), possibly due to Ca2+ overload in the cytoplasm. In subsequent experiments we excluded hypercontracture by adding NaCl (to block Na+/Ca2+ exchange), EGTA (to buffer extracellular calcium) and 30 mM butanedione monoxime (to block contracture on the level of myofibrils). As a result, the cardiomyocytes swelled and burst, while maintaining the cell length (see Supplementary video 2). This time, no effect of AQP4 KO was observed. Our data suggest that AQP4 KO influences hypercontracture-induced cell rupture. The newly published paper by Cheng et al. [9] shows that AQP4 KO cardiomyocytes have higher Ca2+ content in the cytoplasm and a decreased expression of calcium-handling proteins, which could potentially underlie reduced cell rupture in connection with contracture in our experiments. In addition, recent studies show that AQP4 may mediate the regulatory volume changes [42], possibly via its interaction with the transient receptor potential vanilloid 4 [5]. The effect of AQP4 KO may also be associated with osmotic fragility of cardiomyocytes, which is known to be increased by ischemia [35, 36] due to a change in membrane lipid composition [33]. All these hypotheses require further investigation.
Conclusion
AQP4 is expressed in the plasmalemma of murine cardiomyocytes, influences cardiomyocyte resistance to hypoosmotic shock with hypercontracture and has detrimental effects during ischemic injury. AQP4 cardiac expression is comparable between humans and mice, thus the observed effect of AQP4 knockout on ischemic tolerance may be relevant to humans. Cardiac AQPs can become a novel therapeutic target for combatting ischemic injury of the heart.
References
Amiry-Moghaddam M, Otsuka T, Hurn PD, Traystman RJ, Haug FM, Froehner SC, Adams ME, Neely JD, Agre P, Ottersen OP, Bhardwaj A (2003) An alpha-syntrophin-dependent pool of AQP4 in astroglial end-feet confers bidirectional water flow between blood and brain. Proc Natl Acad Sci USA 100(4):2106–2111. doi:10.1073/pnas.0437946100
Askenasy N, Navon G (1997) Continuous monitoring of intracellular volumes in isolated rat hearts during normothermic perfusion and ischemia. J Magn Reson 124(1):42–50. doi:10.1006/jmre.1996.1026
Au CG, Cooper ST, Lo HP, Compton AG, Yang N, Wintour EM, North KN, Winlaw DS (2004) Expression of aquaporin 1 in human cardiac and skeletal muscle. J Mol Cell Cardiol 36(5):655–662. doi:10.1016/j.yjmcc.2004.01.009
Becker BF, Chappell D, Jacob M (2010) Endothelial glycocalyx and coronary vascular permeability: the fringe benefit. Basic Res Cardiol 105(6):687–701. doi:10.1007/s00395-010-0118-z
Benfenati V, Caprini M, Dovizio M, Mylonakou MN, Ferroni S, Ottersen OP, Amiry-Moghaddam M (2011) An aquaporin-4/transient receptor potential vanilloid 4 (AQP4/TRPV4) complex is essential for cell-volume control in astrocytes. Proc Natl Acad Sci USA 108(6):2563–2568. doi:10.1073/pnas.1012867108
Bergersen LH, Storm-Mathisen J, Gundersen V (2008) Immunogold quantification of amino acids and proteins in complex subcellular compartments. Nat Protoc 3(1):144–152. doi:10.1038/nprot.2007.525
Butler TL, Au CG, Yang B, Egan JR, Tan YM, Hardeman EC, North KN, Verkman AS, Winlaw DS (2006) Cardiac aquaporin expression in humans, rats, and mice. Am J Physiol Heart Circ Physiol 291(2):H705–H713. doi:10.1152/ajpheart.00090.2006
Chappell D, Hofmann-Kiefer K, Jacob M, Rehm M, Briegel J, Welsch U, Conzen P, Becker BF (2009) TNF-alpha induced shedding of the endothelial glycocalyx is prevented by hydrocortisone and antithrombin. Basic Res Cardiol 104(1):78–89. doi:10.1007/s00395-008-0749-5
Cheng YS, Tang YQ, Dai DZ, Dai Y (2012) AQP4 knockout mice manifest abnormal expressions of calcium handling proteins possibly due to exacerbating pro-inflammatory factors in the heart. Biochem Pharmacol 83(1):97–105. doi:10.1016/j.bcp.2011.10.006
Dahl CP, Husberg C, Gullestad L, Waehre A, Damas JK, Vinge LE, Finsen AV, Ueland T, Florholmen G, Aakhus S, Halvorsen B, Aukrust P, Oie E, Yndestad A, Christensen G (2009) Increased production of CXCL16 in experimental and clinical heart failure: a possible role in extracellular matrix remodeling. Circ Heart Fail 2(6):624–632. doi:10.1161/CIRCHEARTFAILURE.108.821074
Egan JR, Butler TL, Au CG, Tan YM, North KN, Winlaw DS (2006) Myocardial water handling and the role of aquaporins. Biochim Biophys Acta 1758(8):1043–1052. doi:10.1016/j.bbamem.2006.05.021
Egan JR, Butler TL, Cole AD, Aharonyan A, Baines D, Street N, Navaratnam M, Biecker O, Zazulak C, Au CG, Tan YM, North KN, Winlaw DS (2008) Myocardial ischemia is more important than the effects of cardiopulmonary bypass on myocardial water handling and postoperative dysfunction: a pediatric animal model. J Thorac Cardiovasc Surg 136(5):1265–1273. doi:10.1016/j.jtcvs.2008.04.002
Eltzschig HK, Carmeliet P (2011) Hypoxia and inflammation. N Engl J Med 364(7):656–665. doi:10.1056/NEJMra0910283
Garcia-Dorado D, Oliveras J, Gili J, Sanz E, Perez-Villa F, Barrabes J, Carreras MJ, Solares J, Soler-Soler J (1993) Analysis of myocardial oedema by magnetic resonance imaging early after coronary artery occlusion with or without reperfusion. Cardiovasc Res 27(8):1462–1469. doi:10.1093/cvr/27.8.1462
Garcia-Dorado D, Theroux P, Munoz R, Alonso J, Elizaga J, Fernandez-Aviles F, Botas J, Solares J, Soriano J, Duran JM (1992) Favorable effects of hyperosmotic reperfusion on myocardial edema and infarct size. Am J Physiol 262(1 Pt 2):H17–H22
He Q, Spiro MJ (1995) Isolation of rat heart endothelial cells and pericytes: evaluation of their role in the formation of extracellular matrix components. J Mol Cell Cardiol 27(5):1173–1183. doi:10.1016/0022-2828(95)90053-5
Heusch G, Boengler K, Schulz R (2008) Cardioprotection: nitric oxide, protein kinases, and mitochondria. Circulation 118(19):1915–1919. doi:10.1161/CIRCULATIONAHA.108.805242
Hibuse T, Maeda N, Nakatsuji H, Tochino Y, Fujita K, Kihara S, Funahashi T, Shimomura I (2009) The heart requires glycerol as an energy substrate through aquaporin 7, a glycerol facilitator. Cardiovasc Res 83(1):34–41. doi:10.1093/cvr/cvp095
Iandiev I, Pannicke T, Biedermann B, Wiedemann P, Reichenbach A, Bringmann A (2006) Ischemia-reperfusion alters the immunolocalization of glial aquaporins in rat retina. Neurosci Lett 408(2):108–112. doi:10.1016/j.neulet.2006.08.084
Kaljusto ML, Mori T, Mohammad Husain Rizvi S, Galagudza M, Frantzen ML, Valen G, Vaage J (2006) Postconditioning in rats and mice. Scand Cardiovasc J 40(6):334–341. doi:10.1080/14017430601007587
Laine GA, Allen SJ (1991) Left ventricular myocardial edema. Lymph flow, interstitial fibrosis, and cardiac function. Circ Res 68(6):1713–1721
Lee M, Lee SJ, Choi HJ, Jung YW, Frokiaer J, Nielsen S, Kwon TH (2008) Regulation of AQP4 protein expression in rat brain astrocytes: role of P2X7 receptor activation. Brain Res 1195:1–11. doi:10.1016/j.brainres.2007.12.023
Loo DD, Wright EM, Zeuthen T (2002) Water pumps. J Physiol 542(Pt 1):53–60. doi:PHY_18713
Manley GT, Fujimura M, Ma T, Noshita N, Filiz F, Bollen AW, Chan P, Verkman AS (2000) Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat Med 6(2):159–163. doi:10.1038/72256
McNutt NS (1975) Ultrastructure of the myocardial sarcolemma. Circ Res 37(1):1–13
Mehlhorn U, Geissler HJ, Laine GA, Allen SJ (2001) Myocardial fluid balance. Eur J Cardiothorac Surg 20(6):1220–1230. doi:S1010-7940(01)01031-4
Mewton N, Rapacchi S, Augeul L, Ferrera R, Loufouat J, Boussel L, Micolich A, Rioufol G, Revel D, Ovize M, Croisille P (2011) Determination of the myocardial area at risk with pre- versus post-reperfusion imaging techniques in the pig model. Basic Res Cardiol 106(6):1247–1257. doi:10.1007/s00395-011-0214-8
Moe SE, Sorbo JG, Sogaard R, Zeuthen T, Petter Ottersen O, Holen T (2008) New isoforms of rat Aquaporin-4. Genomics 91(4):367–377. doi:10.1016/j.ygeno.2007.12.003
Mola MG, Nicchia GP, Svelto M, Spray DC, Frigeri A (2009) Automated cell-based assay for screening of aquaporin inhibitors. Anal Chem 81(19):8219–8229. doi:10.1021/ac901526k
Neely JD, Christensen BM, Nielsen S, Agre P (1999) Heterotetrameric composition of aquaporin-4 water channels. Biochemistry 38(34):11156–11163. doi:10.1021/bi990941s
Nicchia GP, Mola MG, Pisoni M, Frigeri A, Svelto M (2007) Different pattern of aquaporin-4 expression in extensor digitorum longus and soleus during early development. Muscle Nerve 35(5):625–631
O’Connell TD, Rodrigo MC, Simpson PC (2007) Isolation and culture of adult mouse cardiac myocytes. Methods Mol Biol 357:271–296. doi:10.1385/1-59745-214-9:271
Piper HM, Garcia-Dorado D (1999) Prime causes of rapid cardiomyocyte death during reperfusion. Ann Thorac Surg 68(5):1913–1919. doi:10.1016/S0003-4975(99)01025-5
Ran X, Wang H, Chen Y, Zeng Z, Zhou Q, Zheng R, Sun J, Wang B, Lv X, Liang Y, Zhang K, Liu W (2010) Aquaporin-1 expression and angiogenesis in rabbit chronic myocardial ischemia is decreased by acetazolamide. Heart Vessels 25(3):237–247. doi:10.1007/s00380-009-1179-5
Ruiz-Meana M, Garcia-Dorado D, Gonzalez MA, Barrabes JA, Soler-Soler J (1995) Effect of osmotic stress on sarcolemmal integrity of isolated cardiomyocytes following transient metabolic inhibition. Cardiovasc Res 30(1):64–69. doi:10.1016/S0008-6363(95)00008-9
Schluter KD, Jakob G, Ruiz-Meana M, Garcia-Dorado D, Piper HM (1996) Protection of reoxygenated cardiomyocytes against osmotic fragility by nitric oxide donors. Am J Physiol 271(2 Pt 2):H428–H434
Sepramaniam S, Armugam A, Lim KY, Karolina DS, Swaminathan P, Tan JR, Jeyaseelan K (2010) MicroRNA 320a functions as a novel endogenous modulator of aquaporins 1 and 4 as well as a potential therapeutic target in cerebral ischemia. J Biol Chem 285(38):29223–29230. doi:10.1074/jbc.M110.144576
Shibuya S, Hara H, Wakayama Y, Inoue M, Jimi T, Matsuzaki Y (2008) Aquaporin 4 mRNA levels in neuromuscular tissues of wild-type and dystrophin-deficient mice. Tohoku J Exp Med 215(4):313–319. doi:10.1620/tjem.215.313
Sorbo JG, Moe SE, Ottersen OP, Holen T (2008) The molecular composition of square arrays. Biochemistry 47(8):2631–2637. doi:10.1021/bi702146k
Stenslokken KO, Rutkovskiy A, Kaljusto ML, Hafstad AD, Larsen TS, Vaage J (2009) Inadvertent phosphorylation of survival kinases in isolated perfused hearts: a word of caution. Basic Res Cardiol 104(4):412–423. doi:10.1007/s00395-009-0780-1
Sun XL, Zhang J, Fan Y, Ding JH, Sha JH, Hu G (2009) Aquaporin-4 deficiency induces subfertility in female mice. Fertil Steril 92(5):1736–1743. doi:10.1016/j.fertnstert.2008.07.1785
Thrane AS, Rappold PM, Fujita T, Torres A, Bekar LK, Takano T, Peng W, Wang F, Thrane VR, Enger R, Haj-Yasein NN, Skare O, Holen T, Klungland A, Ottersen OP, Nedergaard M, Nagelhus EA (2011) Critical role of aquaporin-4 (AQP4) in astrocytic Ca2+ signaling events elicited by cerebral edema. Proc Natl Acad Sci USA 108(2):846–851. doi:10.1073/pnas.1015217108
Wakayama Y, Takahashi J, Shibuya S, Inoue M, Kojima H, Oniki H, Arata S, Hara H, Jimi T, Shioda S, Sunada Y, Ohi H, Shimizu T (2007) Generation of muscle aquaporin 4 overexpressing transgenic mouse: its characterization at RNA and protein levels including freeze-fracture study. Micron 38(3):257–267. doi:10.1016/j.micron.2006.05.001
Warth A, Eckle T, Kohler D, Faigle M, Zug S, Klingel K, Eltzschig HK, Wolburg H (2007) Upregulation of the water channel aquaporin-4 as a potential cause of postischemic cell swelling in a murine model of myocardial infarction. Cardiology 107(4):402–410. doi:10.1159/000099060
Wright AR, Rees SA (1998) Cardiac cell volume: crystal clear or murky waters? A comparison with other cell types. Pharmacol Ther 80(1):89–121. doi:10.1016/S0163-7258(98)00025-4
Xu WB, Gu YT, Wang YF, Lu XH, Jia LS, Lv G (2008) Bradykinin preconditioning modulates aquaporin-4 expression after spinal cord ischemic injury in rats. Brain Res 1246:11–18. doi:10.1016/j.brainres.2008.09.087
Yang B, Brown D, Verkman AS (1996) The mercurial insensitive water channel (AQP-4) forms orthogonal arrays in stably transfected Chinese hamster ovary cells. J Biol Chem 271(9):4577–4580. doi:10.1074/jbc.271.9.4577
Acknowledgments
Mubashar Amin and Bjørg Riber helped with quantitative immunogold, Torgeir Holen gave valuable advice regarding PCR technique and Western blot, Anna Thoren provided animals, and Torun Flatebø provided excellent technical assistance, all of which is gratefully acknowledged. This work was supported by Gjensidigestiftelsen, The Southeastern Regional Health Trust, The Jahre Foundation, The University of Oslo, and Oslo University Hospital—Ullevål, The Nansen Foundation, and the Norwegian Health Association. Lars Mariero is the recipient of a medical student’s scholarship from the University of Oslo and the Norwegian Research Council.
Conflict of interests
None declared.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary material 1 (MPG 12319 kb)
Supplementary material 2 (MPG 6794 kb)
Rights and permissions
About this article
Cite this article
Rutkovskiy, A., Stensløkken, KO., Mariero, L.H. et al. Aquaporin-4 in the heart: expression, regulation and functional role in ischemia. Basic Res Cardiol 107, 280 (2012). https://doi.org/10.1007/s00395-012-0280-6
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00395-012-0280-6