
Abstract
Purpose
The maintenance of an obtained lower weight level is often found to be difficult. The aim of this study was to determine weight maintenance after an initial weight loss by consumption of a meal replacement with a vegetable-oil emulsion associated with prolonged satiety.
Methods
After a 6-week weight loss period with very low calorie diet (VLCD), subjects with >5% body weight (BW) loss were randomized to a 12-week weight maintenance follow-up period, comparing a partial meal replacement diet containing a vegetable-oil emulsion (test) or dairy fat (control). Anthropometric data and safety variables were collected at baseline and after 4, 8 and 12 weeks.
Results
A significant weight loss was observed during the 12-week weight maintenance diet in the test and control group, respectively; 1.0 ± 2.1 kg (p < 0.05) versus 1.3 ± 2.1 kg (p < 0.05) with no significant difference between the groups. Body fat mass (BFM) decreased significantly (p < 0.05) in the test group (−1.7%) compared to the control group (−0.8%).
Conclusions
Addition of a vegetable-oil emulsion to a meal replacement weight maintenance program after an initial weight loss using VLCD was associated with decreased BFM by 0.9% without any change in BW between the two groups.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Background
The prevalence of overweight and obesity is growing and has been classified as an epidemic according to the WHO (World Health Organization) [1]. Overweight/obesity is not only a health burden in itself but also linked to many metabolic diseases. Even a moderate weight loss of 5–10%, if sustained, has beneficial effects by reducing the incidence of diabetes [2]. Such a weight loss can be achieved within 6–8 weeks by using a very low calorie diet (VLCD) [3]; however, the obtained lower weight is difficult to sustain over a longer time [4]. A greater initial weight loss, induced without changes of lifestyle, has been shown to improve long-term weight maintenance [5, 6] provided that it is followed by supporting integrated weight maintenance programs [7]. One therapeutic option, which can be included in a maintenance program, is the use of a meal replacement therapy after an initial weight loss. When meal replacement therapies have been compared with traditional energy restriction diets, a greater long-term weight loss has been obtained with a partial (one) meal replacement regimen (PMR) [8]. However, compliance to the altered food intake behavior is one of the major challenges in the field of weight maintenance; hence ingredients, which affect satiety, could play a role by increasing compliance to a weight maintenance program. Satiety feelings appear as a response to ingested food, and there is good evidence supporting the existence of physiological feed back systems referred to as ileal brake, which sense the presence of nutrients in the gastrointestinal system in order to regulate appetite [9–11]. Specifically, lipids when present in the small intestine can stimulate strong feedback signals associated with satiety, slowing of gastrointestinal transit and release of satiety hormones [11, 12].
An ingredient, which has been shown to increase satiety, is a vegetable-oil emulsion (Fabuless™) of palm and oat oils in water. Previous short-term studies, all randomized, controlled, single blind or double blind trials, have demonstrated that administration of this vegetable-oil emulsion has reduced subsequent food intake and induced satiety [13–15], although two studies [16, 17] did not show any effect of this fat emulsion on food intake. The inconsistency of the results may, at least in part, be attributed to heterogeneity in statistical and methodological approaches. Further investigations to clarify the mechanism(s) behind this vegetable-oil emulsion have demonstrated a delayed transit time of a subsequent meal [18] and higher amount of lipids in the intestine compared to control treatment [19]. These findings may well be associated with ileal brake, which has been suggested as a possible mechanism behind the effect in the short-term studies. During a previous long-term study in overweight women, consumption of this vegetable-oil emulsion in a yoghurt preparation improved maintenance of body weight (BW) after an initial weight loss induced by VLCD [20]. Reduced hunger feelings most likely associated with the raised levels of satiety hormones and increased resting energy expenditure leading to a relative decrease in BFM were suggested as mechanisms explaining these findings [20].
The aim of this study was to investigate whether a PMR weight maintenance approach using this vegetable-oil emulsion may have an effect on BW and body composition after an initial weight loss with VLCD. A secondary objective was to investigate the safety relative to the effects on resting energy expenditure.
Methods
Subjects
Seventy-nine female subjects aged 18–60 years with a body mass index (BMI) of 26–31 kg/m2 were screened. Subjects with a history of cardiovascular, mental or other serious diseases, or a systolic blood pressure ≥160 mm Hg or a diastolic blood pressure ≥105 mm Hg, alcohol or substance abuse, history of soy bean or milk allergy and consumption of drugs affecting BW were not eligible to enter the study. For entering the randomized phase of the 12-week weight maintenance phase, subjects should reduce BW with at least 5% at the end of a six-week VLCD period.
The local ethics committee at the Faculty of Medicine at Uppsala University approved the study. Signed informed consent was obtained from all participants.
Study design
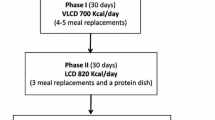
The study had a randomized, controlled, double-blind, parallel design and consisted of two phases; an initial weight reduction period of 6 weeks using a calorie-restricted diet, VLCD, followed by a random allocation to a test or control weight maintenance treatment period of 12 weeks. During the first 2 weeks of the weight reduction phase, the subjects consumed 5 units of Nutrilette Intensive daily (557 kcal Energy% C/F/P 37/21/42; Nutrilett Intensive chocolate, Axellus AB, Sweden), which gives 100% of RDI of minerals and vitamins. Nutrilette Intensive is a commercial product on the Swedish market and was purchased at a supermarket for this study. One unit of Nutrilette Intensive was added to 200 mL of cold water and immediately shaked for 10–15 s before consumption. The subjects were also strictly instructed to drink a minimum of 2.5 L of non-caloric beverage/day. No other food or drink items were allowed. During the remaining 4 weeks of the weight reduction phase, the subjects ingested ad lib a regular breakfast and 4 units of Nutrilette Intensive daily (111 kcal/unit, in total 444 kcal/day). All subjects received information and advice about a healthy breakfast meal. Subjects with a BW loss of at least 5% were randomized into the weight maintenance intervention phase of 12 weeks, during which the participants resumed to their habitual eating patterns except for the lunch, which was replaced by Nutrilette Intensive meal (111 kcal) mixed with test emulsion or control. The subject mixed one unit of Nutrilette Intensive with 200 mL of cold water as described previously and added the test or control emulsion to the mixture during stirring. The subjects were requested not to change their exercise habits during the study.
Products
The 12.5 g test vegetable-oil emulsion (Fabuless™, DSM Food specialties Delft, The Netherlands) contained 5.2 g fat and 7.3 g water (46 kcal/day), while the 13.8 g of cream, which was used as control, contained 5.2 g fat, 0.4 g protein, 0.3 g carbohydrate and 7.9 g water (49 kcal/day). The products were dispensed in a double-blind manner in ready to use portion packs for daily use. The subjects reported consumption in a diary, which together with the returned unused portion packs constituted the basis for calculation of the compliance.
Measurements
Body weight (BW), sagittal abdominal diameter SAG, waist and hip circumferences, skinfolds, heart rate and blood pressure measurements were performed every second week during the weight reduction phase and every fourth week during the weight maintenance period. The same equipments were used, and the same nurse performed the measurements during the study in order to increase the reproducibility of the measurements, and in addition, the weight loss phase was used as training of the subjects to become more comfortable with measurements (data not shown).
Anthropometric
BW was measured using a digital balance accurate (CL-300 BMI, Carl Liden, Gothenburg, Sweden) with subjects wearing indoor cloths without shoes, wallet and keys. Height was measured using a wall-mounted stadiometer at the screening visit. Body mass index (BMI) was calculated by dividing the BW (kg) by body height squared (m2). BFM and body muscle mass (BMM) were estimated by skinfold measurements at four different locations on the body: biceps, triceps, subscapular and suprailiac [21] and carried out with a Harpenden caliper (Baty Int, West Sussex, UK). The waist and hip circumferences were measured between the iliac crest and the lowest part of the lowest rib with the subjects in underwear in a standing position. The subject was asked to exhale before the waist circumference was performed. Sagittal abdominal diameter (SAG) was measured with the subject laying on the back, on a flat and hard surface and defined as the distance between the surface and the highest point of the abdomen.
Blood pressure and pulse rate
Blood pressure and pulse rate were measured oscillometrically (Omron M4-I, Omron Healthcare Europe B.V., Hoofddorp, The Netherlands) in the sitting position in the right arm after a resting period of 3–5 min. A total of three measurements with 2–5 min intervals were performed, and the average of these measurements was calculated.
Body temperature
Body temperature was measured rectal by Terumo Electronic Fever Thermometer C402. The same thermometer was used during the study.
Biochemical analyses
The subjects arrived at the study site after 12-h fasting. Plasma and serum were obtained from blood samples via venapuncture in the forearm for determination of hemoglobin (Hb), glycosylated hemoglobin (HbA1c), alanine aminotransferase (ALAT), triglycerides (TG), apolipoprotein A1 (ApoA1), apolipoprotein B (ApoB), gamma glutamyl transpeptidase (GGT), glucose (Glc), potassium (K), sodium (Na), creatinine (Crea) and thyroid-stimulating hormone (TSH). Urine dipstick analyses were carried out as well. The biochemical analyses were performed before the weight reduction phase, at baseline and after 12-week weight maintenance treatment and carried out at the Department of Clinical Chemistry, University Hospital, Uppsala.
Adverse events
All adverse events defined as any untoward occurrence in a subject in the study, whether considered related to the study treatment, were collected, documented and reported. An adverse event form was filled in for each separate adverse event.
Power calculation
Study group sizes were determined to detect a difference of 1.8 kg between test and control groups; with a power of 80% and a standard deviation (SD) of 2.0 and a type I error of 5%, a sample size of 21 subjects in each group was required, with a total of 42 subjects (Sample power, SPSS, Chicago, Illinois, USA). Drop out rate for the initial weight loss period as well as for the intervention period was set at 30% leading to a total number of 60 subjects to include in the weight reduction period (Fig. 1).

Overview and disposition of subjects
Statistics
Data are presented as mean and standard deviation (SD) or 95% confidence intervals (CI). Differences between test treatment and control treatment groups for baseline characteristics were analyzed with an unpaired t-test using SPSS version 15.0 (Chicago, Illinois, USA). The study of data evolution throughout the follow-up was performed with the analysis of variance for repeated measurements (ANOVA). For inferring a treatment effect from the difference between test group and control group on a quantitative outcome measured before and after treatment, analysis of covariance (ANCOVA) of the outcome with the baseline as covariate was performed (Univariate Mixed Effect Model Approach). Statistical analysis of variance as well as analysis of covariance was performed using SAS software (SAS Institute, Cary, NC, USA). p values <0.05 were considered statistically significant. Subjects were included in the statistical evaluation if compliance to treatment was at least 70%.
Results
Of 79 screened subjects, 54 were found eligible to the weight reduction phase, after which 46 subjects were randomized into the weight maintenance intervention phase based on the body weight loss. One subject withdrew for personal reasons; thus, 45 subjects completed the study. Of these 45 subjects who completed the entire study, two subjects were excluded from the statistical analysis due to violation of the study protocol and poor compliance, respectively. The relative compliance was 90.8 ± 7.8 for the test treatment and 91.4 ± 8.5% for the control treatment. The two excluded subjects had a compliance of 66.7 and 64.3%, respectively, hence well below the level in the protocol. Thus, 43 subjects were included in the statistical calculations (Fig. 1).
Initial characteristics
Table 1 shows the characteristics of the study participants at the start of the weight reduction phase, and the two groups did not differ in characteristics. As a consequence of the restricted energy intake, BW was significantly lowered at the end of the weight reduction phase compared to the initial values. The test treatment group decreased the mean body weight by 6.7 ± 1.5 kg, and the corresponding weight loss for the control group was 7.5 ± 1.7 kg with no significant difference between the two groups.
Characteristics at randomization
Table 2 shows the characteristics of the study participants during weight maintenance phase. At baseline, after randomization, systolic and diastolic blood pressures were found to be significantly higher in the test treatment group (121/80 mm Hg) when compared to the control group (116/75 mm Hg) (p < 0.05). No other significant differences were observed between the two groups.
Treatment effects during the weight maintenance
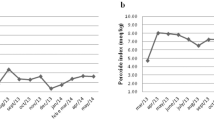
After 12 weeks of weight maintenance by partial meal replacement regimen (PMR), body weight was significantly reduced in both groups (test group: 1.0 ± 2.1 kg, p < 0.05), (control group: 1.3 ± 2.1 kg, p < 0.05) with no difference between the groups. BFM was significantly reduced in the test group from baseline to 12-week follow-up (p < 0.05) (Fig. 2), whereas the reduction in the control group was not significant. A significant over time difference (analysis of variance (ANOVA) two-factor repeated measures) was obtained between the treatments (p < 0.01). Furthermore, correction for baseline measurement did not alter the results (difference between test-control = 0.57, p < 0.01, repeated measurement ANCOVA). Furthermore, waist circumference decreased by 1.2 cm in the test group (p < 0.05, t-student), while the decrease by 0.6 cm in the control group was not significant, with no significant difference between the two treatment regimens. The muscle mass increased by 3.6% (p < 0.05), from 39.2 to 40.6%, in the test treatment group and by 2.8% (p < 0.05), from 39.8 to 40.9%, in the control group, however, with no statistical difference between the groups. No other significant intergroup differences for other parameters listed in Table 2 were observed. There were no significant group–time interactions (data not shown). The reduction of BFM showed a nadir with both treatment regimens at week 8, the level tended to stabilize, which could be noticed on the body weight reduction (Fig. 2).

Change in body fat mass (% of body weight for the test and control groups during treatment). Values are means. p < 0.05 over time difference from baseline compared to week 12 (repeated measurement ANOVA)
Safety and adverse effects
The safety and metabolic parameters (data not shown) as well as blood pressure, pulse rate, and body temperature (data not shown) were within the normal range and unaltered during the study. No intra- or intergroup changes were observed regarding the metabolic and lipid variables. The ALAT levels, which increase in both groups during the weight reduction phase, were normalized during the 12-week weight maintenance phase. The frequency of adverse events collected during the different phases of the study showed no differences between treatments. Thirteen adverse events were reported in each treatment group. The incidence of adverse events by class is summarized in Table 3. In the test group, three adverse events were reported as possibly related whereof all were resolved at the end of the study. In the control group, two adverse events were reported as possibly related, and both of them were resolved at the end of the study. One of the subjects in the control group noted two adverse events. Seven and six subjects, respectively, in the test and control group, reported changes in background medication during the study; however, the results observed in this study remained when the participants reporting changes in background medication were excluded from the statistical calculation.
Discussion
Our study demonstrated that a 12-week partial meal replacement with a vegetable-oil emulsion regimen performed after an initial body weight loss with VLCD significantly lowered the BFM when compared to a control group. This alteration was independent of baseline values. However, regarding changes in BW, there was no difference between the groups treated with the vegetable-oil emulsion compared to control. Since the anthropometric measures are subjective, steps were taken to ensure that these measurements were reproducible by using the same nurse and equipments as well as allow subjects to get accustomed to the procedure.
There were no significant differences between the test and control groups on waist circumference and SAG; nevertheless, a significant decrease in waist circumference was obtained from baseline in the test group, which might support the change in BFM. Waist circumference has been shown to predict BFM [22] and obesity-related metabolic [23] and cardiovascular risk factors [24]. In a study, Ross et al. [25] showed that a reduction in waist circumference by 1 cm is equivalent to a loss of abdominal fat of 0.33 kg. If applied to our study, the subjects in the test group treatment would have lost 0.40 kg of abdominal fat versus 0.20 kg in the control group during the follow-up phase. Given the importance of abdominal fat in the pathogenesis of the metabolic syndrome [26], it could be speculated that the observed reduction in body fat mass is an important finding with respect to its impact on health.
In our study, overweight subjects were recruited, but by reducing body weight, BMI decreased accordingly leading to a BMI close to normal at the end of the study with a major reduction during the initial weight loss. A BMI close to the normal range during the treatment period could explain the relatively small effect on BW, BFM and waist circumference and lack of significant effect on SAG. In contrast to the BFM, which decreased over time and between treatments, the BW was only slightly reduced. The BMM increased 1.4% at the 12-week follow-up compared to baseline and not over time between treatments. Hence, this reciprocal relationship between BMM and BFM might help to explain the limited decrease in total BW. The wide interindividual variations might also help to explain why changes in BW did not reach statistical significance.
There is a growing body of literature that supports the effectiveness of meal replacement programs [8, 27, 28]. Subjects using these strategies were shown to improve behavioral compliance, increase their nutritional knowledge and had more regular meals and snacked less [28]. Accordingly, VLCD and PMR programs have beneficial effect on parameters linked to the metabolic syndrome [29, 30], and a 12% reduction in the prevalence of the metabolic syndrome was recently shown in a study [31]. Thus, the suppressive effect on food intake and appetite observed previously with this vegetable-oil emulsion in combination with the obtained effect on body fat mass in this study with a partial meal replacement therapy could be an important tool to facilitate compliance to a weight maintenance program regimen, especially since the test product was well tolerated. A plausible explanation to the decreased body fat mass is the effect on resting energy expenditure, which was found in the previous long-term study, though further investigations are necessary to confirm the data obtained on this [20].
Conclusion
A diet using meal replacements containing this stable emulsion was safe and yielded a significantly greater body fat loss with no differences in weight change after an initial weight loss compared to the control group. This may facilitate longer-term compliance with a weight loss program and suggests that the program is nutritionally sound if applied appropriately.
References
World Health Organ Tech Rep Ser (2000) Obesity: preventing and managing the global epidemic. Report of a WHO consultation. WHO: Geneva 894:i–xii, 1–253
Ditschuneit HH, Flechtner-Mors M (2001) Value of structured meals for weight management: risk factors and long-term weight maintenance. Obes Res 4(Suppl):284S–289S
Anderson JW, Vitchitbandra S, Oian W, Kryscio RJ (1999) Long-term weight maintenance after an intensive weight-loss program. J Am Coll Nutr 18:620–627
Wadden TA, Sternberg JA, Letizia KA, Stunkard AJ, Foster GD (1989) Treatment of obesity by very low calorie diet, behavior therapy, and their combination: a five-year perspective. Int J Obes 2(Suppl):39–46
Saris WH (2001) Very-low-calorie diets and sustained. Weight loss 4(Suppl):295S–301S
Astrup A, Rossner S (2000) Lessons from obesity management: greater initial weight loss improves long-term maintenance. Obes Res 1:17–19
Ayyad C, Andersen T (2001) Long-term efficacy of dietary treatment of obesity: a systematic review of studies published between 1931 and 1999. Obes Rev 2:113–119
Heymsfield SB, Mierlo CA, van der Knaap HC, Heo M, Frier HI (2003) Weight management using a meal replacement strategy: meta and pooling analysis from six studies. Int J Obes Relat Metab Disord 27:537–549
Read NW, McFarlane A, Kinsman RI, Bates TE, Blackhall NW, Farrar GB et al (1984) Effect of infusion of nutrient solutions into the ileum on gastrointestinal transit and plasma levels of neurotensin and entroglucagon. Gastroenterology 86:274–280
Spiller RC, Trotman IF, Higgins BE, Ghatei MA, Grimble GK, Lee YC et al (1984) The ileal brake: inhibition of jejunal motility after ileal fat perfusion in man. Gut 25:365–374
Maljaars PW, Peters HP, Mela DJ, Masclee AA (2008) Ileal brake: a sensible food target for appetite control. A rev Physiol Beh 95:271–281
Van Citters GW, Lin HC (1999) The ileal brake: a fifteen-year progress report. Curr Gastroenterol Rep 1:404–409
Burns AA, Livingstone MB, Welch RW, Dunne A, Robson PJ, Lindmark L, Reid CA, Mullaney U, Rowland IR (2000) Short-term effects of yoghurt containing a novel fat emulsion on energy and macronutrient intakes in non-obese subjects. Int J Obes Relat Metab Disord 24:1419–1425
Burns AA, Livingstone MB, Welch RW, Dunne A, Reid CA, Rowland IR (2001) The effects of yoghurt containing a novel fat emulsion on energy and macronutrient intakes in non-overweight, overweight and obese subjects. Int J Obes Relat Disord 25:1487–1496
Burns AA, Livingstone MB, Welch RW, Dunne A, Rowland IR (2002) Dose-response effects of a novel fat emulsion (Olibra) on energy and macronutrient intakes up to 36 h post-consumption. Eur J Clin Nutr 56:368–377
Logan CM, McCaffrey TA, Wallace JM, Robson PJ, Welch RW, Dunne A, Livingstone MB (2006) Investigation of the medium-term effects of Olibra fat emulsion on food intake in non-oese subjects. Eur J Clin Nutr 60:1081–1091
Diepvens K, Steijns J, Zuurendonk P, Westerterp-Plantenga MS (2008) Short-term effects of a novel fat emulsion on appetite and food intake. Physiol Behav 95:114–117
Haenni A, Sundberg B, Yazdanpandah N, Viberg A, Olsson J (2009) Effect of fat emulsion (Fabuless) on orocecal transit time in healthy men. Scand J Gastroenterol 44:1186–1190
Knutson L, Koenders DJPC, Fridblom H, Viberg A, Sein A, Lennernäs H (2010) Gastrointestinal metabolism of a vegetable-oil emulsion in healthy subjects. Am J Clin Nutr. doi:10.3945/ajcn.2009.28941
Diepvens K, Soenen S, Steijns J, Arnold M, Westerterp-Plantenga M (2007) Long term effects of consumption of a novel fat emulsion in relation to body-weight management. In J Obes 31:942–949
Durnin JVGA, Womersley J (1974) Body fat assessed from total body density and its estimation from skinfold thickness: measurements on 481 men and women aged from 16 to 72 years. Br J Nutr 32:77–97
Ketel IJ, Volman MN, Seidell JC, Stehouwer CD, Twisk JW, Lambalk CB (2007) Superiority of skindfold measurements and waist over waist-to-hip ratio for the determination of body fat distribution in a population-based cohort of Caucasian Dutch adults. Eur J Endocrinol 156:655–661
Bosy-Westphal A, Geisler C, Onur S, Korth O, Selberg O, Schrezenmeir J, Muller MJ (2006) Value of body fat mass vs. anthropometric obesity indices in the assessment of metabolic risk factors. Int J Obes 30:475–483
Li C, Engstrom G, Hedblad B, Calling S, Berglund G, Janzon L (2006) Sex differences in the relationships between BMI, WHR and incidence of cardiovascular disease: a population-based cohort study. Int J Obes 30:1775–1781
Ross R, Dagnone D, Jones PJ, Smith H, Paddags A, Hudson R, Janssen I (2000) Reduction in obesity and related comorbid conditions after diet-induced weight loss or exercise-induced weight loss in men. A randomized, controlled trial. Ann Intern Med 133:92–103
Waichenberg BL (2000) Subcutaneous and visceral adipose tissue: their relation to the metabolic syndrome. Endocr Rev 21:697–738
Wing RR, Jeffery RW, Burton LR, Thorson C, Nissinoff KS, Baxter JE (1996) Food provision vs. structured meal plans in the behavioral treatment of obesity. Int J Obes Relat Metab Disord 20:56–62
Wing RR, Jeffery RW (2001) Food provision as a strategy to promote weight loss. Obes Res 9:271S–275S
Volek JS, Fernandez ML, Feinman RD, Phinney SD (2008) Dietary carbohydrate restriction induces a unique metabolic state positively affecting atherogenic dyslipidemia, fatty acid partitioning, and metabolic syndrome. Prog Lidip Res 47:307–318
Foster GD, Wyatt HR, Hill JO, MCGuckin BG, Brill C, Mohammed BS, Szapary PO, Rader DJ, Edman JS, Klein S (2003) A randomized trial of a low-carbohydrate diet for obesity. N Engl J Med 348:2048–2090
Konig D, Deibert P, Frey I, Landmann U, Berg A (2008) Effect of meal replacement on metabolic risk factors in overweigh and obese subjects. Ann Nutr Metab 52:74–78
Acknowledgments
The study was funded by DSM Food Specialties and conducted by Good Food Practice independently but on commission by the sponsor.
Competing interests
None.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Olsson, J., Sundberg, B., Viberg, A. et al. Effect of a vegetable-oil emulsion on body composition; a 12-week study in overweight women on a meal replacement therapy after an initial weight loss: a randomized controlled trial. Eur J Nutr 50, 235–242 (2011). https://doi.org/10.1007/s00394-010-0131-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00394-010-0131-x