
Abstract
Congenital heart defects (CHD) are the most common developmental errors in humans, affecting 8 out of 1,000 newborns. Clinical diagnosis and treatment of CHD has dramatically improved in the last decades. Hence, the majority of CHD patients are now reaching reproductive age. While the risk of familial recurrence has been evaluated in various population studies, little is known about the genetic pathogenesis of CHD. In recent years significant progress has been made in uncovering genetic processes during cardiac development. Data from human genetic studies in CHD patients indicate that the genetic aetiology was presumably underestimated in the past. Inherited mutations in genes encoding cardiac transcription factors and sarcomeric proteins were found as an underlying cause for familial recurrence of non-syndromic CHD in humans, in particular cardiac septal defects. Notably, the cardiac phenotypes most frequently seen in mutation carriers are ostium secundum atrial septal defects (ASDII). This review outlines experimental approaches employed for the detection of CHD-related genes in humans and summarizes recent findings in molecular genetics of congenital cardiac septal defects with an emphasis on ASDII.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Congenital heart defects (CHD) represent the most common inborn malformation and are estimated as a major cause for prenatal birth losses. Despite the progress that has been achieved in diagnostic and therapeutic strategies, CHD still cause significant morbidity and mortality in western populations. Yet, the survival of patients with CHD has dramatically increased during the last 30 years, mainly due to advances in cardiac surgery and interventional procedures. More than 80% of patients with complex inborn malformation are now reaching adulthood [38, 47]. Grown-ups with congenital heart disease (GUCH) are a heterogeneous group of patients which are also predisposed to age-related cardiac diseases such as coronary heart disease and heart failure. Therefore, adequate management of GUCH patients require sophisticated medical treatment from specialized paediatric and/or adult cardiologists. Concerns of sufficient medical treatment were met by scientific statements from cardiovascular societies [63] and by the establishment of international guidelines [21, 72].
Yet, the growing number of GUCH patients who are now reaching reproductive age also demands a better understanding of genetic transmission, disease penetrance, and risk of CHD recurrence. Recent human genetic findings have gained novel insights into genetic aetiology of cardiac malformations by identifying single gene defects as a pathogenic substrate for familial recurrence of CHD [11]. These studies have remarkably improved our understanding of gene-related regulatory processes involved in human heart development. Still, it appears that human CHD arise from a multifactorial origin with complex interactions of genetic and environmental factors. For clinicians treating patients with CHD, any knowledge about a genetic aetiology is relevant due to the following reasons: gene defects in regulatory genes may also disrupt the development of other organ systems. Moreover, the risk of disease transmission should be evaluated in GUCH patients with a desire to have children and there may be other (asymptomatic) family members warranting genetic or clinical characterization.
Cardiac development
CHD arise from perturbations of cardiac development during embryogenesis. In the past years molecular mechanisms underlying cardiac morphogenesis were studied in vivo by the generation of gene-targeted animal models [34]. The heart is composed of early progenitor cells from at least two sources located in bilateral cardiogenic fields [13]. Migration and fusion of myocardial and endocardial precursor cells form a linear heart tube on day 20 of human development. Rightward looping and chamber formation is followed by development of the cardiac conductive tissue and coronary vessels (days 28–32). During cardiac morphogenesis, different cell lineages must be tightly regulated to assure correct localization and growth [69]. In fact, regulatory genes such as transcription factors represent the key players in developmental processes. Transcription factors titrate the expression of tissue specific genes and mediate molecular responses on environmental stimuli. The important role of gene expression regulation in cardiac development was highlighted by the identification of inherited mutations in transcription factors in families with CHD [11]. Meanwhile, a large spectrum of mutations was found in a considerable number of genes expressed in early cardiogenesis. Notably, atrial septum secundum defect (ASDII) is among the most frequently noted cardiac phenotypes found in mutation carriers with non-syndromic CHD.
ASDII: a model disease for genetic CHD
The formation of atrial septa is initiated at a late stage of cardiac development (from day 60 onwards). The septum primum grows from the roof of the primordial atrium towards the endocardial cushion. Before the septum primum adnates with the cardiac atrioventricular cushion, small perforations develop in the upper part of the septum primum and subsequently fuse to the foramen secundum. The foramen secundum supplies prenatal blood flow from the right to the left atrium to bypass premature pulmonary circulation. Normally, the foramen secundum is covered by the atrial septum secundum which attaches to the septum primum after birth. ASDII are a result of an insufficient growth of the septum secundum, mostly located in the fossa ovalis of the atrial septum. The clinical appearance of patients with ASDII depends on the shunt volume. Although ASDII is considered to be a non-severe CHD, it is associated with a higher mortality when treated after the third decade of life [56]. In most clinical centres, catheter-guided device closure has become a standard treatment for ASDII [62]. Yet, aetiology of ASDII remains unclear in the majority of patients. While maternal exposure to environmental risk factors predispose to ASDII [37, 87], a familial risk is also well documented (MIM 607941) [15, 17, 59]. A recent study included more than 18,000 subjects with CHD from a Danish population and determined a recurrence risk for isolated ASDII of approximately 7% [59]. These findings are convergent with previous family studies on ASDII recurrence [17]. CHD patients and family relatives harbouring transcription factor mutations frequently present ASDII as an isolated cardiac phenotype. Intriguingly, ASDII were also found in patients with cardiomyopathies due to mutations in sarcomeric genes [14, 54] indicating that regular growth of the septum secundum may be highly vulnerable towards genetic perturbations. As most patients with ASDII reach advanced age even when left untreated [74], the clinical characterization of multiple affected family relatives is feasible in the context of a genetic workup. Given the high heterogeneity of genetic factors contributing to ASDII, this CHD subtype may well suit as a model disease for researching genetic pathogenesis of inborn developmental defects of the heart.
Approaches to study the genetic basis of CHD
In spite of the tremendous progress that has been achieved in the technology of gene analyses (including array-based technologies, whole genome sequencing, etc.), the value of a careful selection of appropriate probands cannot be overestimated. All genetic investigations start off with the identification of a well-defined phenotype in a proband or a family. While genetic results are functionally analysed and critically evaluated, the cardiac phenotypes often remain too little discussed. Although ASDII may be considered as a rather uncomplex lesion, it might be of interest if subtle anomalies are over-represented among those defects related to a genetic defect. We and others were able to show that “swiss-cheese-like” ASDII (so-called cribriform ASDII) or atrial septal aneurysms seem to be over-represented in patients harbouring single gene mutations [32, 54, 64, 65]. Genotype–phenotype associations, but among a larger number of patients, remain to be carried out in order to identify gene specific phenotypes.
Experimental settings
Genetic approaches utilized to identify CHD-associated gene defects are multifaceted and versatile. Nevertheless, one may—for comprehensible reasons—distinguish three different genetic techniques that have been successfully employed in the past to unravel the genetic aetiology of human CHD (Fig. 1). These types of analysis are now described in the following three sections.

Strategies for the identification of gene mutations associated with CHD. The selection of potential gene candidates can be achieved after identification of a chromosomal locus (kindred-based haplotype mapping or cytogenetic abberations) or via a rationale based on other experimental findings (candidate gene approach). Pathogenicity of novel sequence variants should be evaluated by family study, genotyping of adequate controls and functional assays
Pedigree-based linkage analyses
Pedigree-based linkage studies rely on large “informative” families with multiple affected and unaffected relatives. Upon identification of phenotypes inherited in family relatives, the construction of a pedigree allows determination of the mode of Mendelian inheritance. Co-segregation of the respective phenotype with specific chromosomal markers is used to determine a chromosomal region in the proximity of the underlying disease gene. Fine mapping of the disease-related region may further reduce the number of potential genes. Mutational analysis of plausible gene candidates within this region may finally lead to the identification of the causative mutation. While large numbers of chromosomal markers were necessary to securely assess segregation of genetic fragments in a kindred, geneticists are now able to use genotyping arrays (the so-called “SNP-Chips”) for chromosomal mapping. Pedigree-based whole genome linkage analyses were successfully employed to identify novel genes related to familial ASDII. In fact, ASDII-associated mutations cardiac transcription factors and sarcomeric genes were first identified by use of pedigree-based haplotype mapping [18, 24, 48, 73]. However, a major obstacle of linkage analyses is the identification and recruitment of large families including compliant relatives with and without the disease phenotype.
Cytogenetic analyses
The second approach exploits chromosomal aberrations seen in patients with syndromic CHD to identify CHD-relevant genes. The most frequent syndromic disease with inborn cardiac defects is the velocardiofacial or DiGeorge syndrome with an incidence of 1 out of 4,000 live births (OMIM 188400 and 192430). More than 90% of the patients with DiGeorge syndrome show a microdeletion on chromosome 22q11 spanning 1.5–3 megabases [92]. Subsequent characterization of genes mapped to the 22q11 locus resulted in identification of Ufd1, a gene involved in the degradation of ubiquitinated proteins in neural crest cells [91]. Moreover, the cardiac transcription factor TBX1, which also maps to 22q11 was found as a key determinant for DiGeorge syndrome-associated heart defects [52]. Indeed, Yagi and colleagues [90] found TBX1 mutations in patients with DiGeorge phenotype, but without 22q11 microdeletion. The development of comparative genomic hybridization (Array-CGH) technology now allows a significantly higher resolution of chromosomal imbalances when compared to the microscopic fluorescent in situ hybridization (FISH) analyses. Genetic imbalances (duplications, deletions or inversions) of less than 5,000 base pairs can be detected by Array-CGH. In recent investigations, this technology was employed to explore sub-microscopic aberrations in patients with syndromic CHD. Thienpont et al. [86] studied 60 patients with CHD and syndromic appearance by Array-CGH and found a variety of small chromosomal imbalances related to human CHD. Moreover, sub-microscopic aberrations could also be found in a number of CHD patients with non-syndromic CHD [23]. Yet, functional consequences of a large number of aberrations are still unclear, and databases containing frequencies of non-pathogenic imbalances (copy number variations = CNVs) in a general population remain to be completed.
Candidate gene analyses
The third approach is the disease gene or candidate gene analysis. Mutational screenings of ASDII disease genes are suitable to systematically assess the frequency of mutations within one single gene in a predefined patient cohort. Given the high heterogeneity of CHD-related gene mutations (e.g. more than 50 different mutations were found in TBX5), most mutational analyses include the whole coding region of a disease gene in an attempt to find novel variants. Systematic disease gene analysis may lead to the detection of mutational hot spots within a single gene or to specific genotype–phenotype correlations. In contrast to disease gene sequencing, candidate gene analyses aim to investigate sequence alterations in genes which were not previously described in relation to human CHD. Results from gene-targeted rodent models indicate candidates that may also play a role in human heart defects. As an example Sperling and colleagues analysed the transcriptional modulator CITED2 based on the embryonic phenotype found in knock-out mice [2] and identified a considerable number of mutations in patients with cardiac septal defects [77]. The identification of the first mutations in the human transcription factor TBX20 related to CHD was also achieved by a candidate gene approach [41]. Recent advances in gene sequencing technology allow simultaneous analyses of multiple genes in hundreds of CHD patients from national registries and biobanks [76]. However, as the number of patients and genes increase, the interpretation of novel sequence variants in relation to the related phenotype becomes a major challenge.
Cardiac transcription factors and human CHD
Transcription factors regulate cell lineage-specific gene expression and are important factors in developmental processes. The current concept of transcription factor functioning is a complex regulatory meshwork of interactions to control pattern and timing of gene expression during organ development. Transcription factors are known to play a fundamental role in all levels of heart development including cardiac lineage determination, chamber formation, valvulogenesis, and septation. In this review, we focus on cardiac transcription factors involved in atrial partitioning and on those which appeared to be related to human ASDII. Therefore, we chose four transcription factor genes and outlined human genetic findings in chronological order of the identification of ASDII-related defects in men. The time point of the initial identification of a mutation should always be considered for deducting frequencies of mutations in a single gene. The total number of mutations identified is continuously growing due to mutational analyses in different patient cohorts by competitive research groups once a gene has initially been found. Regrettably, negative genetic studies are published only rarely, a fact that further biases a reliable assessment of mutation frequencies. Considering that the total number of mutations is continuously growing, the reader is encouraged to use online databases (such as OMIM) in order to remain up to date.
TBX5
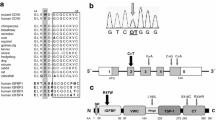
TBX5 encodes a member of the family of T-box transcription factors and is expressed in embryonic heart and limb tissues [13]. Mutations in TBX5 produce the Holt–Oram syndrome (HOS), an autosomal, dominant, highly penetrant disorder associated with malformations of the upper limb and CHD (MIM 142900) [33]. Cardiac malformations are mostly ASDII or ventricular septal defect (VSD). Genetic ablation of TBX5 in murine models imposingly recapitulates the human phenotype [12]. Human mutations in TBX5 were first identified in 1997 by two groups [3, 43] shortly after the chromosomal disease locus was mapped to 12q2 [84, 85]. Subsequently, more than 50 germline mutations were found [1, 8, 9, 16, 28, 31]. A substantial fraction of TBX5 mutations are deletions, frameshifts or premature stop codons leading to a presumably dysfunctional protein. It was proposed that the clinical phenotype depends on the genetic defect in terms that missense mutations produce less severe clinical features with accentuation of the heart (G80R) or upper limbs (R237Q and R237W), whereas mutations predicted to create dysfunctional alleles produce substantial malformations in the upper limb as well as in the heart [4]. Yet, genetic analyses of a large number of HOS patients indicated that phenotypes can vary from mild to severe, even among subjects harbouring identical mutations [10]. Notably, most mutations found in TBX5 are located in the DNA-binding T-box domain and a substantial fraction causes premature stop codons. Therefore, haploinsufficiency is generally acknowledged to be at the root of HOS. Interestingly, Postma and colleagues [66] found a gain-of-function TBX5 mutation in a family with mild limb and heart phenotypes. Most affected family relatives had atrial fibrillation in the absence of cardiac malformations, a finding not uncommon among Holt–Oram patients [49]. Other studies also found dextroposition of the heart and other extracardiac abnormalities in single patients with genetically confirmed HOS [89]. The phenotypic spectrum of inborn developmental disorders, including CHD, of HOS patients is likely to be expanded further in future studies. These findings are interesting as they allocate insights into relationships between genotypes and phenotypes. However, caution should be exercised when interpreting molecular findings in relation to a disease phenotype. An overview of mutations found in TBX5 is given in Fig. 2.

Schematic presentation of mutations found in TBX5 as a cause for Holt–Oram syndrome. Mutations are described at the protein level according to reference AAC04619.1. The majority of mutations are located in the DNA-binding T-box domain of TBX5. Intronic and somatic mutations are not included
NKX2.5
NKX2.5, a vertebrate homologue of drosophila tinman, is a homeobox transcription factor highly expressed in early progenitors and in adult cardiomyocytes [44]. Targeted disruption of NKX2.5 in rodent models results in early embryonic lethality due to failed cardiac looping and defects in chamber formation [46]. Nkx2.5 was the first gene identified as a relevant disease gene for non-syndromic human ASDII. In 1998 Schott et al. [73] found two NKX2.5 mutations after mapping a chromosomal locus in families with ASDII. Most mutation carriers had ASDII associated with atrioventricular conduction defects, albeit some also had VSD, tetralogy of Fallot (TOF) or other cardiac abnormalities. Subsequently, Benson et al. [6] identified seven NKX2.5 mutations among 26 thoroughly selected patients with AV conduction disease and/or CHD. Notably, the authors found a NKX2.5 missense mutation (R25C) in a TOF patient without del22q1. A subsequent study noted the same mutation in 3 of 114 patients with TOF as well as in two African-American control subjects indicating that the variant may represent an ethnically restricted susceptibility variant over-represented in patients with TOF [26]. Future studies on large number of patients including cohorts from different ethnicities remain to be done to investigate whether R25C predisposes for CHD in a polygenic setting. A large number of candidate gene approaches resulted in identification of more than 30 NKX2.5 mutations to date [22, 25, 29, 32, 35, 36, 42, 50, 60]. It has been proposed that nonsense or frame shift mutations at any location and all mutations located in the homeodomain are associated with AV conduction disease, while patients harbouring missense mutations outside of the homeodomain show no AV conduction delay [19]. The function of NKX2.5 in atrial septal development was highlighted by the study of Biben and colleagues [7]. The authors analysed morphogenesis of the atrial septum in different mice strains harbouring one defect allele of the NKX2.5 gene. In comparison to humans harbouring NKX2.5 loss-of-function alleles, only a small subset of NKX2.5 heterozygous mice unmistakably had ASDII, a fact that may be attributed to the redundant function of murine nkx2.6. Still, subtle anomalies of the atrial septum like patent foramen ovale (PFO) and atrial septal aneurysm (ASA) were significantly over-represented among NKX2.5 mutant mice [7]. These observations and phenotypes of inbred mice with atrial septal lesions [40] suggest a continuum of ASDII to milder septal anomalies in particular PFO. Nonetheless, sequence analysis approaches of human NKX2.5 in patients with PFO have not yet resulted in any identification of pathogenic variants [5, 22].
GATA4
GATA4 encodes a member of the GATA family of zinc-finger transcription factors, which recognize (A/T)GATA(A/G) promotor motives [39]. Mice lacking both GATA4 alleles die between day 7.0 to 9.5 of embryonic development and bear no evidence of heart tube formation [53]. Crispino et al. [20] generated mice harbouring a missense mutation in a highly conserved zinc-finger domain (V217G). GATA4-V217G embryos die after heart looping and reveal cardiac malformation resembling severe CHD in humans [20]. The role of GATA4 defects in human CHD was first proposed in 1999. Pehlivan and colleagues [61] investigated patients with 8p23 syndrome and found GATA4 haploinsufficiency in those with cardiac malformation but not in a subject with 8p23.1 without CHD. In 2003, Garg et al. [24] identified the first two families with isolated congenital cardiac septal defects caused by inherited mutations in the GATA4 gene (G296S and c.1075delG). Subsequent molecular characterization of these mutations revealed a significantly impaired affinity to promotor DNA and a failure of GATA4 to interact with TBX5 resulting in a significant reduction of target gene expression in transcriptional assays [24]. One year later, Okubo and colleagues [58] reported a Japanese family with autosomal, dominant ASDII due to a heterozygous GATA4 deletion in an adjacent nucleotide upstream to the frameshift mutation reported by Garg et al. (c.1074delC). Subsequently, we and others identified several GATA4 missense mutations particularly in patients with inherited ASDII [32, 65, 68, 88] but also in consanguineous patients with TOF [57]. In an attempt to identify novel GATA4 variants in a large group of CHD patients and in control subjects without CHD Tomita-Mitchel and colleagues [88] analysed the coding region by use of denaturing high-performance liquid chromatography (WAVE). Interestingly, the authors found a considerable number of non-coding variants in patients with septal and conotruncal heart defects but not among control subjects. But whether or not GATA4 silent variants may predispose to CHD by altering exonic splice sites remains to be elucidated. Recently, a variety of GATA4 missense variants were also noted in different Chinese cohorts [83, 94, 95]. However, family and functional studies for the establishment of any pathogenic impact of these variants have not yet been performed. It is noteworthy that some candidate gene approaches which did not result in the identification of pathogenic mutations were also published [71, 93]. Given the considerable amount of genetic data including NKX2.5 and GATA4 in CHD patients, one may speculate that the diversity of CHD phenotypes is smaller in patients with GATA4 mutations when compared to those with mutations in NKX2.5. A graphical summary of ASDII-related GATA4 mutations is given in Fig. 3.

Schematic representation of NKX2.5, GATA4 and TBX20 protein structure with exonic germline mutations related to non-syndromic CHD indicated. All mutations related to ASDII are represented on the top. Mutations found in patients with CHD other than ASDII are shown below the structural domains. All mutations which were evaluated by family and/or functional studies are represented in bold letters. Mutations which were found in patients with dilated cardiomyopathy are marked with an asterisk. Somatic and intronic mutations are not included
TBX20
TBX20 is an ancient member of the TBX transcription factor family and is highly expressed in the forming heart tube and developing valve tissues [80, 82]. TBX20 interacts in regulatory networks with NKX2.5, GATA4 and TBX5 during embryonic heart formation [78]. In 2002 Szeto and colleagues [81] showed that TBX20 is essential for cardiac chamber formation in zebrafish. Ablation of TBX20 in murine models was independently generated by three research groups and showed similar phenotypic features [16, 75, 79]. TBX20-null embryos show small heart tubes which fail to undergo looping. While homozygous knock-out mice die at mid-gestation, heterozygous animals are viable and exhibit dilation of cardiac chambers and decreased contractility resembling human dilated cardiomyopathy [79]. Inherited TBX20 mutations in patients with CHD were first identified in 2007 [41]. Kirk et al. found two TBX20 mutations among more than 300 patients with various CHD. Interestingly, one family relative harbouring a loss-of-function allele (Q195X) had also evidence for dilated cardiomyopathy indicating that TBX20 haploinsufficiency may predispose to age-related heart failure convergent with the findings in heterozygous TBX20 mice [79]. Subsequently, Qian et al. [67] found three TBX20 missense mutations in young probands with early onset, familial DCM who showed no evidence of CHD. In the same study, the authors reported additional non-synonymous alterations (H186D, L197P) in patients with isolated ASDII and ASDII in combination with TOF [67]. Simultaneously, a number of variants were also found among Chinese patients with ASDII or TOF [45]. However, functional studies still remain to be done in order to underpin the pathogenic impact of these mutations. We have recently identified a TBX20 missense variant in a patient with cribriform ASDII which results in a gain of transcriptional function [64]. The synergistic interactions of TBX20 with co-transcription factors NKX2.5 and GATA4 were also enhanced and the mutant T-box showed a significantly increased dynamic structure [64]. Therefore, one may propose a model in which TBX20-I121M adopts a more fluid tertiary structure leading to enhanced or dysregulated interactions with cofactors and target DNA sequences, and concomitant disruption of the cardiac gene regulatory network. Notably, Hammer et al. [30] found a significant upregulation of TBX20 mRNA in myocardial samples of TOF patients. The I152M mutation found in the study by Kirk et al. [41] also revealed increased actin expression in a frog model. Future studies remain to be done so as to investigate whether a gain of transcriptional TBX20 activity may be a common finding in CHD-related mutations. It is noteworthy that family relatives positive for either gain-of-function mutations (I152M as well as I121M) had large ASDII or PFO with permanent shunt instead of ASDII [41, 64]. Therefore, one may propose that PFO shares a common genetic basis with ASDII in terms of slightly reduced, sustained or enhanced transcriptional function of TBX20.
Genes encoding sarcomeric filaments
Cardiomyocyte architecture is organized by the formation of the sarcomeres representing the molecular motor of force generation for heart muscle contraction. The pathogenicity of mutations in sarcomeric genes for human cardiomyopathies is today well accepted [55]. Still, formation of sarcomeric structures also seems to be required for cardiac development [27] and recent human genetic data indicate that (at least in some cases) cardiomyopathies and ASDII may share a common genetic aetiology [51]. Sarcomeric gene mutations were also identified as being capable of inducing ASDII with left ventricular non-compaction cardiomyopathy (LVNC) [14, 54]. Monserrat and colleagues genotyped patients with cardiomyopathies for one mutation in the alpha-actin gene (ACTC1) gene that was previously reported in patients with apical hypertrophy [54]. Surprisingly, the I101K mutation was found in five unrelated families segregating with disease status. Among 47 patients harbouring I101K 8 had ASA and ASDII in addition to cardiomyopathy [54]. The authors proposed that LVNC and hypertrophic cardiomyopathy may represent overlapping entities. Moreover, the fact that septal defects were found in five out of six families harbouring I101K suggested a relation of the mutant ACTC1 allele with ASDII although mutations in GATA4 or NKX2.5 were not excluded in ASDII probands. In 2008 Budde et al. [14] identified a large family with LVNC, Epstein anomaly and ASDII. Linkage analyses mapped the disease locus on chromosome 14q12, a region encoding two myosin heavy chain genes (MYH6 and MYH7). Subsequently, the authors identified a MYH7 missense mutation (R281T) segregating with disease status in the pedigree [14]. Four relatives harbouring R281T had ASDII in addition to cardiomyopathy. No mutation in NKX2.5 or GATA4 was found among them (unpublished data).
Mutations in sarcomeric genes were also found in families with ASDII who showed no signs of cardiomyopathy [18, 48]. The first evidence indicating that inherited sarcomeric gene mutation may cause familial ASDII arose from a linkage study in 2006. Ching et al. [18] identified a missense mutation in alpha myosin heavy chain (MYH6) in a family with autosomal, dominant ASDII. MYH6 is a cardiac-specific sarcomeric gene highly expressed in the developing atrial septum and is regulated by TBX5. Morpholino-mediated knockdown of MYH6 disrupted atrial septation in chick embryos. Moreover, the authors found the I820N mutation to specifically reduce binding affinity to regulatory light chains [18]. A study by Matsson and colleagues [48] underpinned the assumption that sarcomeric genes produce isolated ASDII. The authors presented two kindreds with familial ASDII harbouring a founder mutation in the ACTC1 gene (M123V). The M123V mutation also showed a reduction of binding properties of ACTC1 for interacting with the thick filaments. Subsequent analyses of ACTC1 in a large cohort of patients with mostly sporadic CHD revealed one additional mutation leading to a severely truncated protein (17-bpDEL c.251). Indeed, morpholino-mediated knockdown of ACTC in chick embryos revealed reduced growth of atrial septa similar to the findings for the MYH6 knockdown [48, 70].
Taken together, the above studies provide intriguing insights in the role of sarcomeric filaments in cardiac morphogenesis and atrial septal development. It is noteworthy that ASDII is by far the type of CHD which was most frequently found. The human genetic findings strongly indicate that ASDII may not only be related to defects in regulatory factors but also to mutations in their target genes.
Conclusions
In this review article we have attempted to outline previous and ongoing efforts on research regarding the genetic pathogenesis of ASDII. Though there are indeed also a number of genetic factors in ASDII aetiology which we did not mention here, we have tried to summarize the genes which were most abundantly investigated and which are, in our view, intriguing factors for future studies on human ASDII. Unequivocally, the disease pathogenesis of ASDII cannot be restricted to a single number of genes given the complex interaction of genetic environmental networks. Indeed, it is most likely that the majority of patients with ASDII have a complex, multifactorial aetiology, with underlying mechanisms that remain to be elucidated in the years to come. Large, well-characterized patient cohorts will be necessary to unravel the minor deleterious impact of more prevalent gene variants on defects in atrial partitioning. Nevertheless, we feel that the few but invaluable families harbouring single gene defect mutations related to ASDII do represent good contact points for researching the root of human septal development. Inherited, naturally occurring mutations provide us with the chance to get a glimpse into the complex genetic mechanisms involved in human heart development.
References
Akrami SM, Winter RM, Brook JD, Armour JA (2001) Detection of a large TBX5 deletion in a family with Holt–Oram syndrome. J Med Genet 38:E44
Bamforth SD, Braganca J, Eloranta JJ, Murdoch JN, Marques FI, Kranc KR, Farza H, Henderson DJ, Hurst HC, Bhattacharya S (2001) Cardiac malformations, adrenal agenesis, neural crest defects and exencephaly in mice lacking Cited2, a new Tfap2 co-activator. Nat Genet 29:469–474
Basson CT, Bachinsky DR, Lin RC, Levi T, Elkins JA, Soults J, Grayzel D, Kroumpouzou E, Traill TA, Leblanc-Straceski J, Renault B, Kucherlapati R, Seidman JG, Seidman CE (1997) Mutations in human TBX5 cause limb and cardiac malformation in Holt–Oram syndrome. Nat Genet 15:30–35
Basson CT, Huang T, Lin RC, Bachinsky DR, Weremowicz S, Vaglio A, Bruzzone R, Quadrelli R, Lerone M, Romeo G, Silengo M, Pereira A, Krieger J, Mesquita SF, Kamisago M, Morton CC, Pierpont ME, Muller CW, Seidman JG, Seidman CE (1999) Different TBX5 interactions in heart and limb defined by Holt–Oram syndrome mutations. Proc Natl Acad Sci USA 96:2919–2924
Belvis R, Tizzano EF, Marti-Fabregas J, Leta RG, Baena M, Carreras F, Pons-Llado G, Baiget M, Marti-Vilalta JL (2009) Mutations in the NKX2–5 gene in patients with stroke and patent foramen ovale. Clin Neurol Neurosurg 111:574–578
Benson DW, Silberbach GM, Kavanaugh-McHugh A, Cottrill C, Zhang Y, Riggs S, Smalls O, Johnson MC, Watson MS, Seidman JG, Seidman CE, Plowden J, Kugler JD (1999) Mutations in the cardiac transcription factor NKX2.5 affect diverse cardiac developmental pathways. J Clin Invest 104:1567–1573
Biben C, Weber R, Kesteven S, Stanley E, McDonald L, Elliott DA, Barnett L, Koentgen F, Robb L, Feneley M, Harvey RP (2000) Cardiac septal and valvular dysmorphogenesis in mice heterozygous for mutations in the homeobox gene Nkx2–5. Circ Res 87:888–895
Borozdin W, Bravo-Ferrer Acosta AM, Seemanova E, Leipoldt M, Bamshad MJ, Unger S, Kohlhase J (2006) Contiguous hemizygous deletion of TBX5, TBX3, and RBM19 resulting in a combined phenotype of Holt–Oram and ulnar-mammary syndromes. Am J Med Genet A 140A:1880–1886
Borozdin W, Bravo Ferrer Acosta AM, Bamshad MJ, Botzenhart EM, Froster UG, Lemke J, Schinzel A, Spranger S, McGaughran J, Wand D, Chrzanowska KH, Kohlhase J (2006) Expanding the spectrum of TBX5 mutations in Holt–Oram syndrome: detection of two intragenic deletions by quantitative real-time PCR, and report of eight novel point mutations. Hum Mutat 27:975–976
Brassington AM, Sung SS, Toydemir RM, Le T, Roeder AD, Rutherford AE, Whitby FG, Jorde LB, Bamshad MJ (2003) Expressivity of Holt–Oram syndrome is not predicted by TBX5 genotype. Am J Hum Genet 73:74–85
Bruneau BG (2008) The developmental genetics of congenital heart disease. Nature 451:943–948
Bruneau BG, Nemer G, Schmitt JP, Charron F, Robitaille L, Caron S, Conner DA, Gessler M, Nemer M, Seidman CE, Seidman JG (2001) A murine model of Holt–Oram syndrome defines roles of the T-box transcription factor Tbx5 in cardiogenesis and disease. Cell 106:709–721
Buckingham M, Meilhac S, Zaffran S (2005) Building the mammalian heart from two sources of myocardial cells. Nat Rev Genet 6:826–835
Budde BS, Binner P, Waldmuller S, Hohne W, Blankenfeldt W, Hassfeld S, Bromsen J, Dermintzoglou A, Wieczorek M, May E, Kirst E, Selignow C, Rackebrandt K, Muller M, Goody RS, Vosberg HP, Nurnberg P, Scheffold T (2007) Noncompaction of the ventricular myocardium is associated with a de novo mutation in the beta-myosin heavy chain gene. PLoS One 2:e1362
Burn J, Brennan P, Little J, Holloway S, Coffey R, Somerville J, Dennis NR, Allan L, Arnold R, Deanfield JE, Godman M, Houston A, Keeton B, Oakley C, Scott O, Silove E, Wilkinson J, Pembrey M, Hunter AS (1998) Recurrence risks in offspring of adults with major heart defects: results from first cohort of British collaborative study. Lancet 351:311–316
Cai CL, Zhou W, Yang L, Bu L, Qyang Y, Zhang X, Li X, Rosenfeld MG, Chen J, Evans S (2005) T-box genes coordinate regional rates of proliferation and regional specification during cardiogenesis. Development 132:2475–2487
Caputo S, Capozzi G, Russo MG, Esposito T, Martina L, Cardaropoli D, Ricci C, Argiento P, Pacileo G, Calabro R (2005) Familial recurrence of congenital heart disease in patients with ostium secundum atrial septal defect. Eur Heart J 26:2179–2184
Ching YH, Ghosh TK, Cross SJ, Packham EA, Honeyman L, Loughna S, Robinson TE, Dearlove AM, Ribas G, Bonser AJ, Thomas NR, Scotter AJ, Caves LS, Tyrrell GP, Newbury-Ecob RA, Munnich A, Bonnet D, Brook JD (2005) Mutation in myosin heavy chain 6 causes atrial septal defect. Nat Genet 37:423–428
Clark KL, Yutzey KE, Benson DW (2006) Transcription factors and congenital heart defects. Annu Rev Physiol 68:97–121
Crispino JD, Lodish MB, Thurberg BL, Litovsky SH, Collins T, Molkentin JD, Orkin SH (2001) Proper coronary vascular development and heart morphogenesis depend on interaction of GATA-4 with FOG cofactors. Genes Dev 15:839–844
Deanfield J, Thaulow E, Warnes C, Webb G, Kolbel F, Hoffman A, Sorenson K, Kaemmer H, Thilen U, Bink-Boelkens M, Iserin L, Daliento L, Silove E, Redington A, Vouhe P, Priori S, Alonso MA, Blanc JJ, Budaj A, Cowie M, Deckers J, Fernandez Burgos E, Lekakis J, Lindahl B, Mazzotta G, Morais J, Oto A, Smiseth O, Trappe HJ, Klein W, Blomstrom-Lundqvist C, de Backer G, Hradec J, Mazzotta G, Parkhomenko A, Presbitero P, Torbicki A (2003) Management of grown up congenital heart disease. Eur Heart J 24:1035–1084
Elliott DA, Kirk EP, Yeoh T, Chandar S, McKenzie F, Taylor P, Grossfeld P, Fatkin D, Jones O, Hayes P, Feneley M, Harvey RP (2003) Cardiac homeobox gene NKX2–5 mutations and congenital heart disease: associations with atrial septal defect and hypoplastic left heart syndrome. J Am Coll Cardiol 41:2072–2076
Erdogan F, Larsen LA, Zhang L, Tumer Z, Tommerup N, Chen W, Jacobsen JR, Schubert M, Jurkatis J, Tzschach A, Ropers HH, Ullmann R (2008) High frequency of submicroscopic genomic aberrations detected by tiling path array comparative genome hybridisation in patients with isolated congenital heart disease. J Med Genet 45:704–709
Garg V, Kathiriya IS, Barnes R, Schluterman MK, King IN, Butler CA, Rothrock CR, Eapen RS, Hirayama-Yamada K, Joo K, Matsuoka R, Cohen JC, Srivastava D (2003) GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5. Nature 424:443–447
Gioli-Pereira L, Pereira AC, Mesquita SM, Xavier-Neto J, Lopes AA, Krieger JE (2008) NKX2.5 mutations in patients with non-syndromic congenital heart disease. Int J Cardiol (in press). doi:10.1016/j.ijcard.2008.08.035
Goldmuntz E, Geiger E, Benson DW (2001) NKX2.5 mutations in patients with tetralogy of fallot. Circulation 104:2565–2568
Gramlich M, Michely B, Krohne C, Heuser A, Erdmann B, Klaassen S, Hudson B, Magarin M, Kirchner F, Todiras M, Granzier H, Labeit S, Thierfelder L, Gerull B (2009) Stress-induced dilated cardiomyopathy in a knock-in mouse model mimicking human titin-based disease. J Mol Cell Cardiol 47:352–358
Gruenauer-Kloevekorn C, Froster UG (2003) Holt–Oram syndrome: a new mutation in the TBX5 gene in two unrelated families. Ann Genet 46:19–23
Gutierrez-Roelens I, Sluysmans T, Gewillig M, Devriendt K, Vikkula M (2002) Progressive AV-block and anomalous venous return among cardiac anomalies associated with two novel missense mutations in the CSX/NKX2–5 gene. Hum Mutat 20:75–76
Hammer S, Toenjes M, Lange M, Fischer JJ, Dunkel I, Mebus S, Grimm CH, Hetzer R, Berger F, Sperling S (2008) Characterization of TBX20 in human hearts and its regulation by TFAP2. J Cell Biochem 104:1022–1033
Heinritz W, Moschik A, Kujat A, Spranger S, Heilbronner H, Demuth S, Bier A, Tihanyi M, Mundlos S, Gruenauer-Kloevekorn C, Froster UG (2005) Identification of new mutations in the TBX5 gene in patients with Holt–Oram syndrome. Heart 91:383–384
Hirayama-Yamada K, Kamisago M, Akimoto K, Aotsuka H, Nakamura Y, Tomita H, Furutani M, Imamura S, Takao A, Nakazawa M, Matsuoka R (2005) Phenotypes with GATA4 or NKX2.5 mutations in familial atrial septal defect. Am J Med Genet A 135:47–52
Holt M, Oram S (1960) Familial heart disease with skeletal malformations. Br Heart J 22:236–242
Horsthuis T, Christoffels VM, Anderson RH, Moorman AF (2009) Can recent insights into cardiac development improve our understanding of congenitally malformed hearts? Clin Anat 22:4–20
Hosoda T, Komuro I, Shiojima I, Hiroi Y, Harada M, Murakawa Y, Hirata Y, Yazaki Y (1999) Familial atrial septal defect and atrioventricular conduction disturbance associated with a point mutation in the cardiac homeobox gene CSX/NKX2–5 in a Japanese patient. Jpn Circ J 63:425–426
Ikeda Y, Hiroi Y, Hosoda T, Utsunomiya T, Matsuo S, Ito T, Inoue J, Sumiyoshi T, Takano H, Nagai R, Komuro I (2002) Novel point mutation in the cardiac transcription factor CSX/NKX2.5 associated with congenital heart disease. Circ J 66:561–563
Jenkins KJ, Correa A, Feinstein JA, Botto L, Britt AE, Daniels SR, Elixson M, Warnes CA, Webb CL (2007) Noninherited risk factors and congenital cardiovascular defects: current knowledge: a scientific statement from the American Heart Association Council on Cardiovascular Disease in the Young: endorsed by the American Academy of Pediatrics. Circulation 115:2995–3014
Kaemmerer H, Hess J (2005) Adult patients with congenital heart abnormalities: present and future. Dtsch Med Wochenschr 130:97–101
Kelley C, Blumberg H, Zon LI, Evans T (1993) GATA-4 is a novel transcription factor expressed in endocardium of the developing heart. Development 118:817–827
Kirk EP, Hyun C, Thomson PC, Lai D, Castro ML, Biben C, Buckley MF, Martin IC, Moran C, Harvey RP (2006) Quantitative trait loci modifying cardiac atrial septal morphology and risk of patent foramen ovale in the mouse. Circ Res 98:651–658
Kirk EP, Sunde M, Costa MW, Rankin SA, Wolstein O, Castro ML, Butler TL, Hyun C, Guo G, Otway R, Mackay JP, Waddell LB, Cole AD, Hayward C, Keogh A, Macdonald P, Griffiths L, Fatkin D, Sholler GF, Zorn AM, Feneley MP, Winlaw DS, Harvey RP (2007) Mutations in cardiac T-box factor gene TBX20 are associated with diverse cardiac pathologies, including defects of septation and valvulogenesis and cardiomyopathy. Am J Hum Genet 81:280–291
Konig K, Will JC, Berger F, Muller D, Benson DW (2006) Familial congenital heart disease, progressive atrioventricular block and the cardiac homeobox transcription factor gene NKX2.5: identification of a novel mutation. Clin Res Cardiol 95:499–503
Li QY, Newbury-Ecob RA, Terrett JA, Wilson DI, Curtis AR, Yi CH, Gebuhr T, Bullen PJ, Robson SC, Strachan T, Bonnet D, Lyonnet S, Young ID, Raeburn JA, Buckler AJ, Law DJ, Brook JD (1997) Holt–Oram syndrome is caused by mutations in TBX5, a member of the Brachyury (T) gene family. Nat Genet 15:21–29
Lints TJ, Parsons LM, Hartley L, Lyons I, Harvey RP (1993) Nkx-2.5: a novel murine homeobox gene expressed in early heart progenitor cells and their myogenic descendants. Development 119:419–431
Liu C, Shen A, Li X, Jiao W, Zhang X, Li Z (2008) T-box transcription factor TBX20 mutations in Chinese patients with congenital heart disease. Eur J Med Genet 51:580–587
Lyons I, Parsons LM, Hartley L, Li R, Andrews JE, Robb L, Harvey RP (1995) Myogenic and morphogenetic defects in the heart tubes of murine embryos lacking the homeo box gene Nkx2–5. Genes Dev 9:1654–1666
Marelli AJ, Mackie AS, Ionescu-Ittu R, Rahme E, Pilote L (2007) Congenital heart disease in the general population: changing prevalence and age distribution. Circulation 115:163–172
Matsson H, Eason J, Bookwalter CS, Klar J, Gustavsson P, Sunnegardh J, Enell H, Jonzon A, Vikkula M, Gutierrez I, Granados-Riveron J, Pope M, Bu’Lock F, Cox J, Robinson TE, Song F, Brook DJ, Marston S, Trybus KM, Dahl N (2008) Alpha-cardiac actin mutations produce atrial septal defects. Hum Mol Genet 17:256–265
McDermott DA, Hatcher CJ, Basson CT (2008) Atrial fibrillation and other clinical manifestations of altered TBX5 dosage in typical Holt–Oram syndrome. Circ Res 103:e96
McElhinney DB, Geiger E, Blinder J, Benson DW, Goldmuntz E (2003) NKX2.5 mutations in patients with congenital heart disease. J Am Coll Cardiol 42:1650–1655
McNally E, Dellefave L (2009) Sarcomere mutations in cardiogenesis and ventricular noncompaction. Trends Cardiovasc Med 19:17–21
Merscher S, Funke B, Epstein JA, Heyer J, Puech A, Lu MM, Xavier RJ, Demay MB, Russell RG, Factor S, Tokooya K, Jore BS, Lopez M, Pandita RK, Lia M, Carrion D, Xu H, Schorle H, Kobler JB, Scambler P, Wynshaw-Boris A, Skoultchi AI, Morrow BE, Kucherlapati R (2001) TBX1 is responsible for cardiovascular defects in velo-cardio-facial/DiGeorge syndrome. Cell 104:619–629
Molkentin JD, Lin Q, Duncan SA, Olson EN (1997) Requirement of the transcription factor GATA4 for heart tube formation and ventral morphogenesis. Genes Dev 11:1061–1072
Monserrat L, Hermida-Prieto M, Fernandez X, Rodriguez I, Dumont C, Cazon L, Cuesta MG, Gonzalez-Juanatey C, Peteiro J, Alvarez N, Penas-Lado M, Castro-Beiras A (2007) Mutation in the alpha-cardiac actin gene associated with apical hypertrophic cardiomyopathy, left ventricular non-compaction, and septal defects. Eur Heart J 28:1953–1961
Morimoto S (2008) Sarcomeric proteins and inherited cardiomyopathies. Cardiovasc Res 77:659–666
Murphy JG, Gersh BJ, McGoon MD, Mair DD, Porter CJ, Ilstrup DM, McGoon DC, Puga FJ, Kirklin JW, Danielson GK (1990) Long-term outcome after surgical repair of isolated atrial septal defect. Follow-up at 27 to 32 years. N Engl J Med 323:1645–1650
Nemer G, Fadlalah F, Usta J, Nemer M, Dbaibo G, Obeid M, Bitar F (2006) A novel mutation in the GATA4 gene in patients with tetralogy of fallot. Hum Mutat 27:293–294
Okubo A, Miyoshi O, Baba K, Takagi M, Tsukamoto K, Kinoshita A, Yoshiura K, Kishino T, Ohta T, Niikawa N, Matsumoto N (2004) A novel GATA4 mutation completely segregated with atrial septal defect in a large Japanese family. J Med Genet 41:e97
Oyen N, Poulsen G, Boyd HA, Wohlfahrt J, Jensen PK, Melbye M (2009) Recurrence of congenital heart defects in families. Circulation 120:295–301
Pabst S, Wollnik B, Rohmann E, Hintz Y, Glanzer K, Vetter H, Nickenig G, Grohe C (2008) A novel stop mutation truncating critical regions of the cardiac transcription factor NKX2–5 in a large family with autosomal-dominant inherited congenital heart disease. Clin Res Cardiol 97:39–42
Pehlivan T, Pober BR, Brueckner M, Garrett S, Slaugh R, Van Rheeden R, Wilson DB, Watson MS, Hing AV (1999) GATA4 haploinsufficiency in patients with interstitial deletion of chromosome region 8p23.1 and congenital heart disease. Am J Med Genet 83:201–206
Peters B, Ewert P, Schubert S, Abdul-Khaliq H, Schmitt B, Nagdyman N, Berger F (2006) Self-fabricated fenestrated Amplatzer occluders for transcatheter closure of atrial septal defect in patients with left ventricular restriction: midterm results. Clin Res Cardiol 95:88–92
Pierpont ME, Basson CT, Benson DW Jr, Gelb BD, Giglia TM, Goldmuntz E, McGee G, Sable CA, Srivastava D, Webb CL (2007) Genetic basis for congenital heart defects: current knowledge: a scientific statement from the American Heart Association Congenital Cardiac Defects Committee, Council on Cardiovascular Disease in the Young: endorsed by the American Academy of Pediatrics. Circulation 115:3015–3038
Posch MG, Gramlich M, Sunde M, Schmitt KR, Lee SHY, Richter S, Perrot A, Panek AN, Al Khatib IH, Nemer G, Megarbane A, Dietz R, Stiller B, Berger F, Harvey RP, Ozcelik C (2009) A gain-of-function TBX20 mutation causes congenital atrial septal defects, patent foramen ovale and cardiac valve defects. J Med Genet (in press). doi:10.1136/jmg.2009.069997
Posch MG, Perrot A, Schmitt K, Mittelhaus S, Esenwein EM, Stiller B, Geier C, Dietz R, Gessner R, Ozcelik C, Berger F (2008) Mutations in GATA4, NKX2.5, CRELD1, and BMP4 are infrequently found in patients with congenital cardiac septal defects. Am J Med Genet A 146A:251–253
Postma AV, van de Meerakker JB, Mathijssen IB, Barnett P, Christoffels VM, Ilgun A, Lam J, Wilde AA, Lekanne Deprez RH, Moorman AF (2008) A gain-of-function TBX5 mutation is associated with atypical Holt–Oram syndrome and paroxysmal atrial fibrillation. Circ Res 102:1433–1442
Qian L, Mohapatra B, Akasaka T, Liu J, Ocorr K, Towbin JA, Bodmer R (2008) Transcription factor neuromancer/TBX20 is required for cardiac function in Drosophila with implications for human heart disease. Proc Natl Acad Sci USA 105:19833–19838
Rajagopal SK, Ma Q, Obler D, Shen J, Manichaikul A, Tomita-Mitchell A, Boardman K, Briggs C, Garg V, Srivastava D, Goldmuntz E, Broman KW, Benson DW, Smoot LB, Pu WT (2007) Spectrum of heart disease associated with murine and human GATA4 mutation. J Mol Cell Cardiol 43:677–685
Rochais F, Mesbah K, Kelly RG (2009) Signaling pathways controlling second heart field development. Circ Res 104:933–942
Rutland C, Warner L, Thorpe A, Alibhai A, Robinson T, Shaw B, Layfield R, Brook JD, Loughna S (2009) Knockdown of alpha myosin heavy chain disrupts the cytoskeleton and leads to multiple defects during chick cardiogenesis. J Anat 214:905–915
Sarkozy A, Esposito G, Conti E, Digilio MC, Marino B, Calabro R, Pizzuti A, Dallapiccola B (2005) CRELD1 and GATA4 gene analysis in patients with nonsyndromic atrioventricular canal defects. Am J Med Genet A 139:236–238
Schmaltz AA, Bauer U, Baumgartner H, Cesnjevar R, de Haan F, Franke C, Gabriel H, Gohlke-Barwolf C, Hagl S, Hess J, Hofbeck M, Kaemmerer H, Kallfelz HC, Lange PE, Nock H, Oechslin E, Schirmer KR, Tebbe U, Trindade PT, Weyand M, Breithardt G (2008) Medical guideline for the treatment of adults with congenital heart abnormalities of the German-Austrian-Swiss Cardiology Specialty Society. Clin Res Cardiol 97:194–214
Schott JJ, Benson DW, Basson CT, Pease W, Silberbach GM, Moak JP, Maron BJ, Seidman CE, Seidman JG (1998) Congenital heart disease caused by mutations in the transcription factor NKX2-5. Science 281:108–111
Shah D, Azhar M, Oakley CM, Cleland JG, Nihoyannopoulos P (1994) Natural history of secundum atrial septal defect in adults after medical or surgical treatment: a historical prospective study. Br Heart J 71:224–227 discussion 228
Singh MK, Christoffels VM, Dias JM, Trowe MO, Petry M, Schuster-Gossler K, Burger A, Ericson J, Kispert A (2005) Tbx20 is essential for cardiac chamber differentiation and repression of Tbx2. Development 132:2697–2707
Smith KA, Joziasse IC, Chocron S, van Dinther M, Guryev V, Verhoeven MC, Rehmann H, van der Smagt JJ, Doevendans PA, Cuppen E, Mulder BJ, Ten Dijke P, Bakkers J (2009) Dominant-negative ALK2 allele associates with congenital heart defects. Circulation 119:3062–3069
Sperling S, Grimm CH, Dunkel I, Mebus S, Sperling HP, Ebner A, Galli R, Lehrach H, Fusch C, Berger F, Hammer S (2005) Identification and functional analysis of CITED2 mutations in patients with congenital heart defects. Hum Mutat 26:575–582
Stennard FA, Costa MW, Elliott DA, Rankin S, Haast SJ, Lai D, McDonald LP, Niederreither K, Dolle P, Bruneau BG, Zorn AM, Harvey RP (2003) Cardiac T-box factor Tbx20 directly interacts with Nkx2–5, GATA4, and GATA5 in regulation of gene expression in the developing heart. Dev Biol 262:206–224
Stennard FA, Costa MW, Lai D, Biben C, Furtado MB, Solloway MJ, McCulley DJ, Leimena C, Preis JI, Dunwoodie SL, Elliott DE, Prall OW, Black BL, Fatkin D, Harvey RP (2005) Murine T-box transcription factor Tbx20 acts as a repressor during heart development, and is essential for adult heart integrity, function and adaptation. Development 132:2451–2462
Stennard FA, Harvey RP (2005) T-box transcription factors and their roles in regulatory hierarchies in the developing heart. Development 132:4897–4910
Szeto DP, Griffin KJ, Kimelman D (2002) HrT is required for cardiovascular development in zebrafish. Development 129:5093–5101
Takeuchi JK, Mileikovskaia M, Koshiba-Takeuchi K, Heidt AB, Mori AD, Arruda EP, Gertsenstein M, Georges R, Davidson L, Mo R, Hui CC, Henkelman RM, Nemer M, Black BL, Nagy A, Bruneau BG (2005) Tbx20 dose-dependently regulates transcription factor networks required for mouse heart and motoneuron development. Development 132:2463–2474
Tang ZH, Xia L, Chang W, Li H, Shen F, Liu JY, Wang Q, Liu MG (2006) Two novel missense mutations of GATA4 gene in Chinese patients with sporadic congenital heart defects. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 23:134–137
Terrett JA, Newbury-Ecob R, Cross GS, Fenton I, Raeburn JA, Young ID, Brook JD (1994) Holt–Oram syndrome is a genetically heterogeneous disease with one locus mapping to human chromosome 12q. Nat Genet 6:401–404
Terrett JA, Newbury-Ecob R, Smith NM, Li QY, Garrett C, Cox P, Bonnet D, Lyonnet S, Munnich A, Buckler AJ, Brook JD (1996) A translocation at 12q2 refines the interval containing the Holt–Oram syndrome 1 gene. Am J Hum Genet 59:1337–1341
Thienpont B, Mertens L, de Ravel T, Eyskens B, Boshoff D, Maas N, Fryns JP, Gewillig M, Vermeesch JR, Devriendt K (2007) Submicroscopic chromosomal imbalances detected by array-CGH are a frequent cause of congenital heart defects in selected patients. Eur Heart J 28:2778–2784
Tikkanen J, Heinonen OP (1992) Risk factors for atrial septal defect. Eur J Epidemiol 8:509–515
Tomita-Mitchell A, Maslen CL, Morris CD, Garg V, Goldmuntz E (2007) GATA4 sequence variants in patients with congenital heart disease. J Med Genet 44:779–783
Tseng YR, Su YN, Lu FL, Jeng SF, Hsieh WS, Chen CY, Chou HC, Peng SS (2007) Holt–Oram syndrome with right lung agenesis caused by a de novo mutation in the TBX5 gene. Am J Med Genet A 143A:1012–1014
Yagi H, Furutani Y, Hamada H, Sasaki T, Asakawa S, Minoshima S, Ichida F, Joo K, Kimura M, Imamura S, Kamatani N, Momma K, Takao A, Nakazawa M, Shimizu N, Matsuoka R (2003) Role of TBX1 in human del22q11.2 syndrome. Lancet 362:1366–1373
Yamagishi H, Garg V, Matsuoka R, Thomas T, Srivastava D (1999) A molecular pathway revealing a genetic basis for human cardiac and craniofacial defects. Science 283:1158–1161
Yamagishi H, Srivastava D (2003) Unraveling the genetic and developmental mysteries of 22q11 deletion syndrome. Trends Mol Med 9:383–389
Zhang L, Tumer Z, Jacobsen JR, Andersen PS, Tommerup N, Larsen LA (2006) Screening of 99 Danish patients with congenital heart disease for GATA4 mutations. Genet Test 10:277–280
Zhang W, Li X, Shen A, Jiao W, Guan X, Li Z (2008) GATA4 mutations in 486 Chinese patients with congenital heart disease. Eur J Med Genet 51:527–535
Zhang WM, Li XF, Ma ZY, Zhang J, Zhou SH, Li T, Shi L, Li ZZ (2009) GATA4 and NKX2.5 gene analysis in Chinese Uygur patients with congenital heart disease. Chin Med J (Engl) 122:416–419
Acknowledgments
This work was supported by Deutsche Forschungsgemeinschaft (DFG: OE276/2-1) and the Competence Network for Heart Failure (CNHF) funded by the Ministry of research and Education (01GI0205).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Posch, M.G., Perrot, A., Berger, F. et al. Molecular genetics of congenital atrial septal defects. Clin Res Cardiol 99, 137–147 (2010). https://doi.org/10.1007/s00392-009-0095-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00392-009-0095-0