
Abstract
Non-pathogenic mismatch repair (MMR) gene variants can be associated with decreased MMR capacity in several settings. Due to an increased mutation rate, reduced MMR capacity leads to accumulation of somatic sequence changes in tumour suppressor genes such as in the neurofibromatosis type 1 (NF1) gene. Patients with autosomal dominant NF1 typically develop neurofibromas ranging from single to thousands. Concerning the number of neurofibromas NF1 patients face a situation that is still not predictable. A few studies suggested that germline non-pathogenic MMR gene variants modify the number of neurofibromas in NF1 and by this mechanism may promote the extent of neurofibroma manifestation. This review represents first evidence that specific non-pathogenic single nucleotide variants of MMR genes act as a modifier of neurofibroma manifestation in NF1, highlighting MSH2 re4987188 as the best analysed non-pathogenic variant so far. In summary, besides MSH2 promotor methylation, specific non-pathogenic germline MSH2 variants are associated with the extent of neurofibroma manifestation. Those variants can serve as a biomarker to facilitate better mentoring of NF1 patients at risk.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Among DNA repair pathways, intact mismatch repair (MMR) is responsible for reparation of spontaneous DNA replication errors such as single-base-pair mismatches and small insertions or deletions in repetitive sequences that escape proofreading activity of DNA polymerase. In humans, mainly mutS homolog 2, mutS homolog 3, mutS homolog 6, mutL homolog 1, and post-meiotic segregation increased 2 (MSH2, MSH3, MSH6, MLH1, PMS2) interact to ensure proper repair and to maintain genomic stability. Defective MMR is associated with up to 1000‐fold increased mutation rate [1]. Additional functions of MMR were uncovered such as involvement in immunoglobulin gene hypermutation and autoimmune disorders, DNA damage surveillance, transcription coupled repair, genetic recombination and meiosis [2, 3].
Germline monoallelic or biallelic pathogenic variants (mutations) in MMR genes are involved in cancer predisposition syndromes, such as Lynch syndrome (LS) and constitutional mismatch repair deficiency (CMMRD) [4,5,6,7]. The type of sequence change in one of the MMR genes defines penetrance. More severe phenotypes result from MLH1 and MSH2 pathogenic variants in both LS and CMMRD [7, 8]. According to Knudson’s hypothesis, in autosomal dominant LS, the wild-type allele of either MLH1, MSH2, MSH6, or PMS2 is inactivated in tumours due to small sequence changes. A resulting complete gene inactivation leads to a complete loss of MMR pathway functions with subsequent hypermutability which is termed mutator phenotype. MMR deficiency induces specifically frameshifts due to slipped mispairing in susceptible repetitive mononucleotides, dinucleotides, trinucleotides, or tetranucleotides (microsatellites) and is leading to microsatellite instability (MSI) in tumours. In colon cancer cells, the percentage of instable microsatellites is used to categorize somatic MSI into low MSI (MSI-L, 10–30%), high MSI (MSI-H, ≥ 40%), or stable MSI (MSS, 0%), although only MSH-H leads to a complete MMR deficiency [9, 10]. It is important to use a defined marker panel that is reliably associated with MMR deficiency [11]. The type of pathogenic variants in repetitive sequences is associated with the extent of MSI and predicts clinical presentation, choice of conventional therapy, and outcome: Mononucleotide sequence changes are typically associated with most MSI-H tumours (often MLH1, MSH2, and MSH6 mutated) which are sensitive to 5-fluorodeoxyuracil. Dinucleotide sequence changes are mostly associated with MSI-L tumours. Tetranucleotide sequence alterations are a result of mutations in MSH3 gene (described as ‘elevated microsatellite alterations at selected tetranucleotide repeats’, EMAST) being comparable to MSI-L and sensitive to poly-ADP-ribose polymerase 1 (PARP1) inhibitors [9].
In autosomal recessive CMMRD, patients suffer from childhood haematological malignancies, brain cancer, early-onset colon cancer, and other malignancies. Patients typically show clinical features of LS and of rasopathies including neurofibromatosis type 1 (NF1). NF1-like tumours (e.g. neurofibromas) are explained by somatic NF1 pathogenic variants induced by MMR deficiency [7, 12]. Neurofibroma-like tumours were also shown in zebrafish due to MMR deficiency [13]. Although in LS pathogenesis of tumours is clearly related to MSI on the somatic level, causative genetic events for development of different tumours in CMMRD are unclear.
In general, cells with MMR deficiency experience an enormous increase of sequence alterations which explains development of both multiple cancers in tumour predisposition syndromes (such as LS and CMMRD) and in sporadic tumours. Cancer type, disease onset, and multiplicity of tumours depend on the MMR gene involved in germline. In sporadic neoplasia, MMR deficiency occurs only at the somatic level, being caused by MLH1 hypermethylation as described in endometrial and sporadic (not associated with LS) colorectal cancer [14].
Due to recent awareness the term ‘mutation’ is to be avoided and standard terminology suggests a five-tier classification system of sequence variants into ‘pathogenic’, ‘likely pathogenic’, ‘uncertain significance’, ‘likely benign’, and ‘benign’ in Mendelian disorders [15]. Historically, a mutation was interpreted as a permanent sequence and polymorphism as a variant with a frequency above 1%. To bring new guidelines and anterior literature together within this review, we call traditionally termed mutations in literature ‘pathogenic variants’ and all others such as reported polymorphisms and rare variants ‘non-pathogenic variants’. Since the term pathogenicity is complex, this approach may not be precise but helps to integrate whole literature. Besides pathogenic variants in diseases as described above, also previously considered non-pathogenic MMR variants have been associated with disease modifications such MSH3 rs26279, MLH1 rs1800734, or other MLH1, MSH3, and DHFR non-pathogenic variants [16,17,18,19,20,21]. To conclude, some missense non-pathogenic variants of MMR genes have already been extensively studied and demonstrated to produce mutator phenotypes in vitro, and therefore have been proposed to predispose humans to disease [22].
Mismatch repair and neurofibromatosis type 1
The autosomal dominant tumour predisposition syndrome neurofibromatosis type 1 (NF1; MIM# 162200) results from germline pathogenic variants (mutations) of the tumour suppressor gene NF1 on 17q11.2. NF1 is characterized by a variety of symptoms with development of multiple neurofibromas as a hallmark. The NF1 gene appears to be prone to pathogenic variants and may be specifically affected by MMR deficiency: The NF1 mutation rate is about 10-fold higher than described for other genes which has been attributed to the large size of the gene (350k bp, 61 exons). De novo germline pathogenic variants occur in about 50% of cases, and approximately 80% of those are of paternal origin. Recent studies demonstrated that mutation rates rise with increasing paternal age which also explains findings in NF1 very well [23, 24]. In general, mutation rate depends on transcription-coupled repair and the genomic context in dependence on epigenetic modifications and conserved regions: hypermutable methylated CpG dinucleotides (CpG effect) and DNase I hypersensitive sites increase mutation rate in specific regions [23, 25]. In NF1, the majority of small deletions and insertions occurs in homo-nucleotide tracts and substitutions are strongly associated with homo-nucleotide repeats and CpG/CpNpG motifs indicating a specific role of methylation [26]. Like other tumour suppressor genes, NF1 covers both CpG islands, methylated nucleotides, and repeat sequences. The high mutation rate of the NF1 gene might also be explained by gene conversion due to pseudogenes and by the high number of exons, as compared to other genes. Defective MMR also prefers specific sequences to mutate at faster rates than others such as short pair repetitive sequences, which is why it may be of interest to elucidate if the NF1 gene covers more of those sequences than other genes [27].
MMR deficiency contributes to a tissue specific tumour development due to cellular characteristics that encompass a high proliferation rate (such as gastrointestinal cells) or rapid acceleration/deceleration of proliferation (such as endometrium), a dependence on specific mutator targets, functions for immune surveillance, apoptosis, toxin exposure, and mode of inheritance [28]. Concerning development of neurofibromas that arise from skin-derived progenitor cells, more than one of these cellular characteristics is fulfilled for NF1. Accumulation of second NF1 hits occurring in rapidly proliferating cells (prone to accelerated NF1 mutation rate) may explain increased tumour load arising from a decreased MMR capacity in NF1.
Whether source of increased somatic mutation rates is different compared to germline mutation rates in NF1 still remains unknown.
In neurofibromatosis patients, benign neurofibromas develop when the NF1 gene is completely inactivated. But besides the first/germline and a second/somatic hit of the wildtype NF1 allele, other factors modify tumour development [29].
Somatic NF1 pathogenic mutations in tumours occur in several settings: In NF1 patients, a second NF1 hit occurs in NF1 associated nerve sheath tumours besides a germline NF1 pathogenic variant (first hit) and leads to biallelic inactivation of NF1 following the Knudson two-hit hypothesis. In sporadic malignant tumours such as melanomas, lung and breast cancer, and many others, a novel second NF1 hit occurs. In this setting, the NF1 hit act solely or together with other tumour suppressor gene alterations and is interpreted to be a driver mutation and even predict resistance to therapy [30, 31]. In CMMRD associated tumours and other features, such café-au-lait spots, a second double NF1 hit leads to a biallelic NF1 inactivation which may be difficult to distinguish from mosaic or segmental NF1 [12].
In NF1, malignant peripheral nerve sheath tumours and benign neurofibromas differ in their mutational spectrum of the second hit. Neurofibromas show an increased proportion of small sequence changes [32]. This indicates that different mutational mechanisms are involved in tumorigenesis and suggest that a reduced MMR capacity probably due to minor alterations in MMR genes may influence neurofibroma development. One should necessarily not mix up that constitutional MMR pathogenic gene variants such as in CMMRD are associated with a distinct tumour syndrome that only focally resembles NF1 [33].
Investigating the function and mutational spectrum of NF1 consistently leads to a reflection of genotype-phenotype-correlations. Several single correlations have already emerged, but the question why patients may exhibit only very few or thousands of neurofibromas is not yet clarified. Only microdeletion NF1 patients are known to suffer from a higher tumour burden and more severe phenotype. Nevertheless, patients without microdeletions may also suffer from a multiplicity of neurofibromas.
MMR capacity and manifestation of cutaneous neurofibromas in NF1
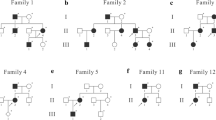
Modifiers of the NF1 phenotype have been discussed and proposed [34,35,36,37]. In 2003, Wiest and co-workers suggested that MMR genes may be modifiers since they lead to accumulation of second hits in NF1 [38]. Further evidence came from a mhl1-deficient mouse model [39]. In 2009, non-pathogenic variants in MMR genes were shown to be associated with neurofibroma load, possibly explaining variability in the number of neurofibromas and indicating a role of a biomarker to identify patients at risk for a high neurofibroma load (Fig. 1) [40].

How MMR capacity may impact somatic NF1 mutation rate and may modulate neurofibroma manifestation in proliferating cells.
CMMRD patients show a distinct reduction of MMR activity that can be measured even by in vitro assays in blood lymphocytes to diagnose the disease with 100% specificity and sensitivity [12, 41, 42]. In contrast, the key mechanism in NF1 patients might be a reduced but not absent MMR capacity. In the study by Shuen and co-workers, MMR activity in lymphoblastoid cell lines of patients with NF1 was comparable to controls [42]. Similar results were obtained by another study that did not detected MSI in lymphocytes of patients with de novo NF1 mutations [43]. This finding is not unexpected because in NF1, the reduced MMR activity should be mild and only be associated with a specific NF1 phenotype, namely the one with a higher frequency of second NF1 hits and subsequent biallelic inactivation of NF1 on the somatic level. Such a mild event unfortunately implicates that every germline MMR variant in NF1 needs to be examined in detail and for segregation. Interestingly, a general finding fits very well to this hypothesis: a small to moderate reduction of the DNA repair capacity was proposed to affect cancer predisposition and to be more difficult to prove (Mohrenweiser et al., 2003).
In 2014, a genome-wide screen identified MMR gene transcripts associated with the number of café-au-lait macules (CALM) and identified MMR non-pathogenic single nucleotide variants associated with CALM count [44]. Since CALM in NF1 also develop from biallelic NF1 inactivation, this study supports the above hypothesis. In a previous study, epigenetic MMR gene events were associated with neurofibroma burden: MSH2 promoter methylation was observed in NF1 cases (n = 79) compared to controls (n = 39). Additionally, a significantly increased methylation of two MSH2 CpG dinucleotides was seen NF1 patients with higher neurofibroma count [40]. Germline epimutations of MSH2 that have been described in HNPCC support the significance of this so-far unique finding in NF1 [45].
There is no doubt that the NF1 gene is a target of reduced MMR activity (Wang 2003). A study investigating the spectrum of somatic pathogenic variants in Schwann cells from 38 neurofibromas suggested slightly reduced DNA repair efficiency as mechanism (Maertens 2006). The authors described that the spectrum of somatic non-pathogenic variants differs between microdeletion and non-microdeletion NF1 patients, and they suggested that the high frequency of somatic frameshifts can result from reduced DNA repair efficiency due to aging. Interestingly, the authors detected MSH2 variant re4987188 (G322D) to be associated with a higher neurofibroma burden and reduced MMR activity in other experiments.
Concerning MSI that would normally arise from a reduced MMR capacity, data from studies of NF1 associated neurofibroma are conflicting. Two studies detected MSI in neurofibromas, whereas others did not (for reference see Table 1). As mentioned above, due to a proposed milder effect of MMR on neurofibroma manifestation, MSI may not be detected in neurofibromas although non-pathogenic MMR gene variants influence severity of tumour manifestation. To underline this, e.g., MSH2 rs2303428 TC + CC genotype was shown to predict prognosis and adjuvant chemotherapy benefit in non-cardia gastric patients but was not associated with MSI suggesting lower penetrance and probably also MSI-independent mechanisms [46].
From a different point of view, in 2007, Garza and co-workers proposed that genomic instability influences tumour progression in NF1 and disease severity, because they discovered a mild mutator phenotype in tumours of a mouse model targeting Nf1 and p53 in cis. Although in this model TP53 haploinsufficiency is the basic cause for limited repair capacity of lesions, genomic instability seems to play a role in NF1 associated benign and malignant lesions.
Material and methods
A comprehensive literature and database search was undertaken to identify those studies reporting single-nucleotide non-pathogenic variants of MMR genes that potentially affected somatic NF1 mutation frequency and therefore possessed the ability to modify neurofibroma load in patients.
Results and discussion
Non-pathogenic MMR gene variants and neurofibroma load in NF1
We will report only those studies that investigated single-nucleotide non-pathogenic variants of MMR genes that potentially affect somatic NF1 mutation frequency and therefore possess the ability to modify neurofibroma load in patients. To date, only three studies investigated the association of specific non-pathogenic variants and load of neurofibroma (Table 2). In 2009, our group hypothesized that MMR genes may be associated with a NF1 phenotype of a high versus a low neurofibroma burden and demonstrated that the extent of methylation of two CpG dinucleotides of MSH2 promotor in leukocytes correlated with an increased number of neurofibromas [40]. In that study, we also fully sequenced MMR genes facilitating re-analysis of single nucleotide variants of MSH2, MSH6, MLH1, and PMS2. Against the background of novel studies, we compiled our data and compared non-pathogenic variants between NF1 patients with a high (n = 38) and those with a low neurofibroma burden (n = 41) which we had previously defined as major and minor disease manifestation. Overall, we detected 10 non-pathogenic variants in MSH2, 1 non-pathogenic variant in MLH1, and 10 non-pathogenic variants in PMS2, and, as expected, did not identify constitutional pathogenic MMR gene variants (see supplementary material) which is supported by other studies [53, 54, 43].
Those non-pathogenic variants are now reviewed that were detected in NF1 patients and were analysed with respect to neurofibroma burden and/or were found in other studies which helps to underline significance. Since meanwhile a plenty of data on non-pathogenic MMR variants are available, we only focussed on those that can be evaluated as a risk factor or as modifier.
Non-pathogenic MSH2 variants and neurofibroma manifestation
There is strong evidence that the common non-pathogenic missense variant re4987188 (G322D) acts as a modifier. The residue is highly conserved between species, and an equivalent alteration was investigated in yeast [55]. It was shown to act damaging due to disruption of splicing enhancers by bioinformatic algorithms and to moderately decrease MMR efficiency as well as to produce a dominant mutator effect in yeast [22, 56, 57]. In older studies, allele frequencies did not give rise to assume that this non-pathogenic variant could be pathogenic, and interpretations varied from deleterious to benign [58,59,60,61,62]. Re4987188 was also in silico proposed to be non-pathogenic (Thompson et al., 2013). However, in our and another study, it was associated with a higher neurofibroma burden [54]. Maertens and co-workers additionally showed an effect on reduction of MMR activity. In more recent studies, re4987188 was also associated with increased risks to vestibular schwannoma (odds ratio (OR) 1.67), recurrent colon cancer, lung cancer (OR 1.29), and breast cancer (OR 10.61) clearly attributed to the A allele in most studies [16, 63,64,65]. From current data, it can therefore be concluded that the A allele, being detected only in NF1 patients with a high tumour burden, serves as a disease modifier towards an increased neurofibroma load. MSH2 re4987188 therefore needs to be evaluated in larger studies since it could be extremely useful for clinical risk assessment in NF1.
According to our analysis, two other non-pathogenic variants are of potential interest: MSH2 rs771126636 and rs63750810 are both associated with a high neurofibroma burden (see supplementary material). Both variants have not been referenced in literature so far. Genotype C/A of MSH2 missense variant rs771126636 is a still undescribed genotype in NF1 patients with a high tumour load. MSH2 rs63750810 allele G is very rare and solely occurred in a patient with a high tumour load in our study. Since both non-pathogenic variants have not yet described elsewhere, it is very difficult to assess their relevance. Given their increased frequency in the cohort with a high neurofibroma load, larger studies as well as segregation and experimental studies might provide additional useful insights.
Other non-pathogenic MSH2 variants such as rs17224360, rs2303426, rs61756467, and rs2303428 are also of potential interest, although associations are not as strong. For example, MSH2 rs2303426 was found to be significantly associated with a higher risk for gallbladder cancer (OR 1.83) but neither differences between allele frequency or heterozygosity concerning risk of tumour burden in NF1 were detected in our study nor specific risk associations evoked from several larger cancer studies [66]. The same applies to intronic rs3732183 which may influence cis-acting regulatory elements and enhance MSH2 expression: It was described to be a prognostic marker for patients with colorectal cancer (GG genotype predicted better survival), significantly associated with a better response to oxaliplatin-based chemotherapy of patients with advanced colorectal adenocarcinoma (OR for A/G + G/G genotype of 5.3). Furthermore, G/G genotype was associated with a lower risk of relapse in oral squamous cell carcinoma patients, but no specific effect was seen in our NF1 study [67,68,69]. Interestingly, data on MSH2 rs3732183 remain conflicting as shown in a meta-analysis that demonstrated that this non-pathogenic variant had no major influence on overall cancer risk, but when stratified by cancer types the risk for non-Hodgkin’s lymphomas was increased which may indicate different roles in different cancer types [70].
Another non-pathogenic variant (rs2303428) of an acceptor splice site in intron 12 within a polypyrimidine tract was detected in our NF1 cohort but did neither demonstrate differences in heterozygosity or genotype concerning neurofibroma load. Nevertheless, it was associated with partial exon skipping 13 in splicing assays and with modification of prognosis of cancer or therapy side effects as well as benefit of chemotherapy of multiple other tumour diseases (reviewed in online resources) [71]. Those characteristics convincingly favour the hypothesis of a tumour-type dependent effect of non-pathogenic MMR variants.
To conclude, there is evidence that both methylation and specific non-pathogenic variants of MSH2 serve as a disease modifier for neurofibroma load in NF1.
Non-pathogenic MLH1 and PMS2 variants and neurofibroma manifestation
In MLH1, frequent non-pathogenic missense variant rs1799977 (I219Val) was shown to be associated with increased risk of sporadic colorectal cancer but unrelated to MSI: genotypes A/G and G/Gs were associated with odds ratios from 1.89 to 3.05 indicating that G allele carriers were at greater risk [72]. Those carriers had a decreased probability of vascular invasion, distant metastasis, re-occurrence, and improved outcome. The authors interpreted that this non-pathogenic variant only modulates but not abrogates MMR activity. In our study, this non-pathogenic variant was associated with a discrete difference and no specific genotypes or alleles in dependence to tumour load were seen. Maertens and co-workers also identified s1799977 in their NF1 study (allele frequency of controls (74/184, 40%)) and of NF1 patients (6/18; 33%) with an insignificant difference, but only in patients with a severe phenotype and without microdeletions [54]. Thus, although more than 40 studies analysed rs1799977 among others in cancer, only the two above mentioned studies exist so far in NF1 patients. From these few data, an influence on neurofibroma development might be inferred, but is still inconclusive.
In PMS2, a few non-pathogenic variants were described to be associated with a decreased risk. A allele of rs3735295, which we detected in our NF1 cohort, was shown to associate with a reduced risk for papillary thyroid carcinoma in Belarusian children after exposure to fallout from the Chernobyl power plant accident [73]. Unfortunately, although we observed differences in heterozygosity, we did not detect a specific genotype concerning neurofibroma burden. Whereas only rs112796669 showed predominant A allele in patients with a high tumour burden, two other variants of PMS2, rs12532895 and rs1805326, were linked to a low tumour burden in our study (see online resources). Rs12532895 has already been detected in a cohort of colon cancer patients [74]. Thus, non-pathogenic variants in PMS2 were more often associated with a very mild NF1 phenotype. Since co-segregation studies of these rare non-pathogenic variants are missing, their association with neurofibroma burden is not confirmed and further experiments are needed. Testing larger cohorts as well as genome wide association studies of well characterized, sex, age-, and variant type-matched groups seem to be necessary to prove further associations of non-pathogenic MMR sequence variants and the extent of tumour burden in NF1.
Conclusions
Independent of constitutional pathogenic variants (mutations), specific non-pathogenic MMR gene variants are associated with decreased MMR capacity. Reduced MMR capacity can lead to accumulation of somatic NF1 sequence alterations and promote manifestation of NF1 associated cutaneous neurofibromas.
Already in childhood, NF1 patients typically develop neurofibromas within a range from single to thousands which cannot be predicted by the pathogenic germline NF1 variant instead of microdeletion cases. Some studies suggested that non-pathogenic MMR gene variants modify the number of neurofibromas in NF1 as they similarly act as a modifier in other diseases. From the data reviewed here, there is strong evidence that specific non-pathogenic single nucleotide variants of MMR genes act in the proposed way. MSH2 re4987188 is one of best analysed so far and is associated with a high burden of neurofibromas. Other promising non-pathogenic variants being associated with a high or low number of neurofibromas have been detected, but data sets are limited.
In summary, non-pathogenic germline MSH2 variants and MSH2 promotor methylation was shown to be associated with the extent of neurofibroma manifestation in NF1 patients (without microdeletions) indicating an influence on MMR activity. Accessible sequencing data should be used to establish more of those associations facilitating a far-sighted mentoring of our neurofibromatosis patients in future.
Availability of data and material
Additional data on MMR gene analysis is provided by Anja Harder.
References
Strand M, Prolla TA, Liskay RM, Petes TD (1993) Destabilization of tracts of simple repetitive DNA in yeast by mutations affecting DNA mismatch repair. Nature 365:274–276. https://doi.org/10.1038/365274a0
Buermeyer AB, Deschenes SM, Baker SM, Liskay RM (1999) Mammalian DNA mismatch repair Annu Rev Genet 33:533–564. https://doi.org/10.1146/annurev.genet.33.1.533
Toledano H et al (2020) Paediatric systemic lupus erythematosus as a manifestation of constitutional mismatch repair deficiency. J Med Genet 57:505–508. https://doi.org/10.1136/jmedgenet-2019-106303
Altieri F, Grillo C, Maceroni M, Chichiarelli S (2008) DNA damage and repair: from molecular mechanisms to health implications. Antioxid Redox Signal 10:891–937. https://doi.org/10.1089/ars.2007.1830
Kebudi R et al (2020) Position paper: challenges and specific strategies for constitutional mismatch repair deficiency syndrome in low-resource settings. Pediatr Blood Cancer 67:e28309. https://doi.org/10.1002/pbc.28309
Lynch HT, Snyder CL, Shaw TG, Heinen CD, Hitchins MP (2015) Milestones of Lynch syndrome: 1895-2015 Nat Rev Cancer 15:181–194. https://doi.org/10.1038/nrc3878
Wimmer K, Etzler J (2008) Constitutional mismatch repair-deficiency syndrome: have we so far seen only the tip of an iceberg?. Hum Genet 124:105–122. https://doi.org/10.1007/s00439-008-0542-4
Plaschke J et al (2004) Lower incidence of colorectal cancer and later age of disease onset in 27 families with pathogenic MSH6 germline mutations compared with families with MLH1 or MSH2 mutations: the German Hereditary Nonpolyposis Colorectal Cancer. Consortium J Clin Oncol 22:4486–4494. https://doi.org/10.1200/JCO.2004.02.033
Carethers JM, Koi M, Tseng-Rogenski SS (2015) EMAST is a form of microsatellite instability that is initiated by inflammation and modulates colorectal cancer progression genes (Basel) 6:185–205. https://doi.org/10.3390/genes6020185
Duraturo F, Liccardo R, De Rosa M, Izzo P (2019) Genetics, diagnosis and treatment of Lynch syndrome: old lessons and current challenges. Oncol Lett 17:3048–3054. https://doi.org/10.3892/ol.2019.9945
Boland CR et al (1998) A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res 58:5248–5257
Perez-Valencia JA et al (2020) Constitutional mismatch repair deficiency is the diagnosis in 0.41% of pathogenic NF1/SPRED1 variant negative children suspected of sporadic neurofibromatosis type 1. Genet Med https://doi.org/10.1038/s41436-020-0925-z
Feitsma H, Kuiper RV, Korving J, Nijman IJ, Cuppen E (2008) Zebrafish with mutations in mismatch repair genes develop neurofibromas and other tumors. Cancer Res 68:5059–5066. https://doi.org/10.1158/0008-5472.CAN-08-0019
Esteller M, Levine R, Baylin SB, Ellenson LH, Herman JG (1998) MLH1 promoter hypermethylation is associated with the microsatellite instability phenotype in sporadic endometrial carcinomas. Oncogene 17:2413–2417. https://doi.org/10.1038/sj.onc.1202178
Richards S et al (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17:405–424. https://doi.org/10.1038/gim.2015.30
Doherty JA et al (2013) DNA repair genotype and lung cancer risk in the beta-carotene and retinol efficacy trial Int J Mol Epidemiol Genet 4:11–34
Flower M et al (2019) MSH3 modifies somatic instability and disease severity in Huntington's and myotonic dystrophy type 1. Brain. https://doi.org/10.1093/brain/awz115
Lo YL et al (2011) Polymorphisms of MLH1 and MSH2 genes and the risk of lung cancer among never smokers. Lung Cancer 72:280–286. https://doi.org/10.1016/j.lungcan.2010.10.009
Morales F, Vasquez M, Santamaria C, Cuenca P, Corrales E, Monckton DG (2016) A polymorphism in the MSH3 mismatch repair gene is associated with the levels of somatic instability of the expanded CTG repeat in the blood DNA of myotonic dystrophy type 1 patients. DNA Repair (Amst) 40:57–66. https://doi.org/10.1016/j.dnarep.2016.01.001
Nogueira GAS et al (2018) Polymorphisms in DNA mismatch repair pathway genes predict toxicity and response to cisplatin chemoradiation in head and neck squamous cell carcinoma patients. Oncotarget 9:29538–29547. https://doi.org/10.18632/oncotarget.25268
Pinto RM et al (2013) Mismatch repair genes Mlh1 and Mlh3 modify CAG instability in Huntington’s disease mice: genome-wide and candidate approaches. PLoS Genet 9:e1003930. https://doi.org/10.1371/journal.pgen.1003930
Drotschmann K, Clark AB, Kunkel TA (1999) Mutator phenotypes of common polymorphisms and missense mutations in MSH2. Curr Biol 9:907–910. https://doi.org/10.1016/s0960-9822(99)80396-0
Francioli LC et al (2015) Genome-wide patterns and properties of de novo mutations in humans. Nat Genet 47:822–826. https://doi.org/10.1038/ng.3292
Kong A et al (2012) Rate of de novo mutations and the importance of father’s age to disease risk. Nature 488:471–475. https://doi.org/10.1038/nature11396
Hodgkinson A, Eyre-Walker A (2011) Variation in the mutation rate across mammalian genomes. Nat Rev Genet 12:756–766. https://doi.org/10.1038/nrg3098
Rodenhiser DI, Andrews JD, Mancini DN, Jung JH, Singh SM (1997) Homonucleotide tracts, short repeats and CpG/CpNpG motifs are frequent sites for heterogeneous mutations in the neurofibromatosis type 1 (NF1) tumour-suppressor gene. Mutat Res 373:185–195. https://doi.org/10.1016/s0027-5107(96)00171-6
Shibata D, Peinado MA, Ionov Y, Malkhosyan S, Perucho M (1994) Genomic instability in repeated sequences is an early somatic event in colorectal tumorigenesis that persists after transformation. Nat Genet 6:273–281. https://doi.org/10.1038/ng0394-273
Chao EC, Lipkin SM (2006) Molecular models for the tissue specificity of DNA mismatch repair-deficient carcinogenesis. Nucleic Acids Res 34:840–852. https://doi.org/10.1093/nar/gkj489
Jouhilahti EM, Peltonen S, Heape AM, Peltonen J (2011) The pathoetiology of neurofibromatosis 1. Am J Pathol 178:1932–1939. https://doi.org/10.1016/j.ajpath.2010.12.056
Philpott C, Tovell H, Frayling IM, Cooper DN, Upadhyaya M (2017) The NF1 somatic mutational landscape in sporadic human cancers. Hum Genomics 11:13. https://doi.org/10.1186/s40246-017-0109-3
Seton-Rogers S (2016) Oncogenes: one of these things is not like the others. Nat Rev Cancer 16:5. https://doi.org/10.1038/nrc.2015.10
Upadhyaya M, Spurlock G, Monem B, Thomas N, Friedrich RE, Kluwe L, Mautner V (2008) Germline and somatic NF1 gene mutations in plexiform neurofibromas. Hum Mutat 29:E103–E111. https://doi.org/10.1002/humu.20793
Suerink M et al (2019) Constitutional mismatch repair deficiency as a differential diagnosis of neurofibromatosis type 1: consensus guidelines for testing a child without malignancy. J Med Genet 56:53–62. https://doi.org/10.1136/jmedgenet-2018-105664
Easton DF, Ponder MA, Huson SM, Ponder BAJ (1993) An analysis of variation in expression of neurofibromatosis type 1: evidence for modifying genes. Am J Hum Genet 53:305–313
Kaufmann D et al (2001) Spinal neurofibromatosis without cafe-au-lait macules in two families with null mutations of the NF1 gene. Am J Hum Genet 69:1395–1400. https://doi.org/10.1086/324648
Szudek J, Joe H, Friedman JM (2002) Analysis of intrafamilial phenotypic variation in neurofibromatosis 1 (NF1). Genet Epidemiol 23:150–164. https://doi.org/10.1002/gepi.1129
Wimmer K et al (2002) A patient severely affected by spinal neurofibromas carries a recurrent splice site mutation in the NF1 gene. Eur J Hum Genet 10:334–338. https://doi.org/10.1038/sj.ejhg.5200807
Wiest V, Eisenbarth I, Schmegner C, Krone W, Assum G (2003) Somatic NF1 mutation spectra in a family with neurofibromatosis type 1: toward a theory of genetic modifiers. Hum Mutat 22:423–427. https://doi.org/10.1002/humu.10272
Gutmann DH, Winkeler E, Kabbarah O, Hedrick N, Dudley S, Goodfellow PJ, Liskay RM (2003) Mlh1 deficiency accelerates myeloid leukemogenesis in neurofibromatosis 1 (Nf1) heterozygous mice. Oncogene 22:4581–4585. https://doi.org/10.1038/sj.onc.12067681206768 [pii]
Titze S et al (2010) Differential MSH2 promoter methylation in blood cells of Neurofibromatosis type 1 (NF1) patients. Eur J Hum Genet 18:81–87. https://doi.org/10.1038/ejhg.2009.129
Gonzalez-Acosta M et al (2020) High-sensitivity microsatellite instability assessment for the detection of mismatch repair defects in normal tissue of biallelic germline mismatch repair mutation carriers. J Med Genet 57:269–273. https://doi.org/10.1136/jmedgenet-2019-106272
Shuen AY et al (2019) Functional repair assay for the diagnosis of constitutional mismatch repair deficiency from non-neoplastic tissue. J Clin Oncol 37:461–470. https://doi.org/10.1200/JCO.18.00474
Wang Q et al (2003) Neurofibromatosis type 1 gene as a mutational target in a mismatch repair-deficient cell type. Hum Genet 112:117-123. https://doi.org/10.1007/s00439-002-0858-4
Pemov A et al (2014) Genetic modifiers of neurofibromatosis type 1-associated cafe-au-lait macule count identified using multi-platform analysis. PLoS Genet 10:e1004575. https://doi.org/10.1371/journal.pgen.1004575
Chan TL et al (2006) Heritable germline epimutation of MSH2 in a family with hereditary nonpolyposis colorectal cancer. Nat Genet 38:1178–1183. https://doi.org/10.1038/ng1866
Zhao X, Dai D, Li X, Shen B, Chen X, Shu Y, Wang D (2019) A polymorphism within the mismatch repair gene predicts prognosis and adjuvant chemotherapy benefit in gastric cancer. Gastric Cancer 22:1121–1129. https://doi.org/10.1007/s10120-019-00962-8
Ottini L et al (1995) Alterations of microsatellites in neurofibromas of von Recklinghausen’s disease. Cancer Res 55:5677–5680
Serra E et al (1997) Confirmation of a double-hit model for the NF1 gene in benign neurofibromas. Am J Hum Genet 61:512–519
Luijten M, Redeker S, van Noesel MM, Troost D, Westerveld A, Hulsebos TJ (2000) Microsatellite instability and promoter methylation as possible causes of NF1 gene inactivation in neurofibromas. Eur J Hum Genet 8:939–945. https://doi.org/10.1038/sj.ejhg.5200565
Upadhyaya M et al (2004) Characterization of the somatic mutational spectrum of the neurofibromatosis type 1 (NF1) gene in neurofibromatosis patients with benign and malignant tumors. Hum Mutat 23:134–146
Stewart H et al (2008) Congenital disseminated neurofibromatosis type 1: a clinical and molecular case report. Am J Med Genet A 146A:1444–1452. https://doi.org/10.1002/ajmg.a.32305
Spurlock G, Griffiths S, Uff J, Upadhyaya M (2007) Somatic alterations of the NF1 gene in an NF1 individual with multiple benign tumours (internal and external) and malignant tumour types. Fam Cancer 6:463-471. https://doi.org/10.1007/s10689-007-9149-5
Thomas L, Kluwe L, Chuzhanova N, Mautner V, Upadhyaya M (2010) Analysis of NF1 somatic mutations in cutaneous neurofibromas from patients with high tumor burden. Neurogenetics 11:391–400. https://doi.org/10.1007/s10048-010-0240-y
Maertens O et al (2006) Comprehensive NF1 screening on cultured Schwann cells from neurofibromas. Hum Mutat 27:1030–1040. https://doi.org/10.1002/humu.20389
Gammie AE, Erdeniz N, Beaver J, Devlin B, Nanji A, Rose MD (2007) Functional characterization of pathogenic human MSH2 missense mutations in Saccharomyces cerevisiae. Genetics 177:707–721. https://doi.org/10.1534/genetics.107.071084
Doss CGP, Sethumadhavan R (2009) Investigation on the role of nsSNPs in HNPCC genes - a bioinformatics approach. J Biomed Sci 16. https://doi.org/10.1186/1423-0127-16-42
Ellison AR, Lofing J, Bitter GA (2001) Functional analysis of human MLH1 and MSH2 missense variants and hybrid human-yeast MLH1 proteins in Saccharomyces cerevisiae. Hum Mol Genet 10:1889–1900. https://doi.org/10.1093/hmg/10.18.1889
Barnetson RA et al (2008) Classification of ambiguous mutations in DNA mismatch repair genes identified in a population-based study of colorectal cancer. Hum Mutat 29:367–374. https://doi.org/10.1002/humu.20635
Chao EC et al (2008) Accurate classification of MLH1/MSH2 missense variants with multivariate analysis of protein polymorphisms-mismatch repair (MAPP-MMR). Hum Mutat 29:852–860. https://doi.org/10.1002/humu.20735
Liu T et al (1998) MSH2 codon 322 Gly to Asp seems not to confer an increased risk for colorectal cancer susceptibility. Eur J Cancer 34:1981. https://doi.org/10.1016/s0959-8049(98)00217-2
Ollila S, Dermadi Bebek D, Greenblatt M, Nystrom M (2008) Uncertain pathogenicity of MSH2 variants N127S and G322D challenges their classification. Int J Cancer 123:720–724. https://doi.org/10.1002/ijc.23573
Raptis S et al (2007) MLH1 -93G>A promoter polymorphism and the risk of microsatellite-unstable colorectal cancer. J Natl Cancer Inst 99:463–474. https://doi.org/10.1093/jnci/djk095
Mik M, Dziki L, Malinowska K, Trzcinski R, Majsterek I, Dziki A (2017) Polymorphism of MSH2 Gly322Asp and MLH1 -93G>A in non-familial colon cancer - a case-controlled study. Arch Med Sci 13:1295–1302. https://doi.org/10.5114/aoms.2017.67024
Rajaraman P et al (2010) DNA repair gene polymorphisms and risk of adult meningioma, glioma, and acoustic neuroma. Neuro Oncol 12:37–48. https://doi.org/10.1093/neuonc/nop012
Smolarz B, Brys M, Forma E, Zadrozny M, Bienkiewicz J, Romanowicz H (2019) Data on single nucleotide polymorphism of DNA repair genes and breast cancer risk from Poland. Pathol Oncol Res 25:1311–1317. https://doi.org/10.1007/s12253-017-0370-8
Srivastava K, Srivastava A, Kumar A, Mittal B (2011) Gallbladder cancer predisposition: a multigenic approach to DNA-repair, apoptotic and inflammatory pathway genes. PLoS One 6:e16449. https://doi.org/10.1371/journal.pone.0016449
Kim JG et al (2010) IVS10+12A>G polymorphism in hMSH2 gene associated with prognosis for patients with colorectal cancer. Ann Oncol 21:525–529. https://doi.org/10.1093/annonc/mdp338
Park JH et al (2010) MGMT -535G>T polymorphism is associated with prognosis for patients with metastatic colorectal cancer treated with oxaliplatin-based chemotherapy. J Cancer Res Clin Oncol 136:1135–1142. https://doi.org/10.1007/s00432-010-0760-8
Senghore T, Wang WC, Chien HT, Chen YX, Young CK, Huang SF, Yeh CC (2019) Polymorphisms of Mismatch Repair Pathway Genes Predict Clinical Outcomes in Oral Squamous Cell Carcinoma Patients Receiving Adjuvant Concurrent Chemoradiotherapy Cancers (Basel) 11. https://doi.org/10.3390/cancers11050598
Wu S et al (2011) Association between the hMSH2 IVS12-6 T>C polymorphism and cancer risk: A meta-analysis. Exp Ther Med 2:1193–1198. https://doi.org/10.3892/etm.2011.336
Tournier I et al (2008) A large fraction of unclassified variants of the mismatch repair genes MLH1 and MSH2 is associated with splicing defects. Hum Mutat 29:1412–1424. https://doi.org/10.1002/humu.20796
Nejda N, Iglesias D, Moreno Azcoita M, Medina Arana V, Gonzalez-Aguilera JJ, Fernandez-Peralta AM (2009) A MLH1 polymorphism that increases cancer risk is associated with better outcome in sporadic colorectal cancer. Cancer Genet Cytogenet 193:71–77. https://doi.org/10.1016/j.cancergencyto.2009.04.011
Lonjou C et al (2017) Investigation of DNA repair-related SNPs underlying susceptibility to papillary thyroid carcinoma reveals MGMT as a novel candidate gene in Belarusian children exposed to radiation. BMC Cancer 17:328. https://doi.org/10.1186/s12885-017-3314-5
Chang YC et al (2016) Mutation analysis of 13 driver genes of colorectal cancer-related pathways in Taiwanese patients. World J Gastroenterol 22:2314–2325. https://doi.org/10.3748/wjg.v22.i7.2314
Acknowledgements
I thank Sigrid Tinschert and Hartmut Peters for original contribution of material and data to our previously published MMR study that gained the potential to be re-analysed for MMR variants in the present context. We thank Stefan Mundlos for technical support and Katrin Guse for lab processing and organizing analyses. We appreciate the help of Anna Dombrowski and Sabrina Titze for evaluating sequencing data.
Funding
Open Access funding enabled and organized by Projekt DEAL. The present review was not financially supported. The project on MMR gene analysis that was previously published and was at that time supported by a Rahel-Hirsch-Fellowship to Anja Harder from Universtitätsmedizin Charité Berlin, by a grant from the Berliner Krebsgesellschaft (HAFF200809) and by a patient’s donation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethics approval
No approval is necessary for this review.
Conflict of interest
The author declares no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Below is the link to the electronic supplementary material.
381_2021_5436_MOESM1_ESM.pdf
Supplementary file1 (PDF 729 KB) Overview of non-pathogenic germline MMR gene variants detected in Neurofbromatosis type 1 (NF1) patients of the study (A). Selected data on non-pathogenic MMR gene variants from the study that demonstrated a difference in heterozygosity or a rare genotype comparing Neurofibromatosis type 1 (NF1) groups of low and high neurofibroma load (B).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Harder, A. Do non-pathogenic variants of DNA mismatch repair genes modify neurofibroma load in neurofibromatosis type 1?. Childs Nerv Syst 38, 705–713 (2022). https://doi.org/10.1007/s00381-021-05436-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00381-021-05436-w