
Abstract
Purpose
Ventriculomegaly in infants with congenital myotonic dystrophy (CDM) is common, and the neurosurgical determination of shunting is complex. The natural history of CDM-associated ventriculomegaly from prenatal to natal to postnatal stages is poorly known. The relationship between macrocephaly and ventriculomegaly, incidence of shunt necessity, and early mortality outcomes lack pooled data analysis. This study aims to review clinical features and pathophysiology of CDM, with emphasis on ventriculomegaly progression, ventriculomegaly association with macrocephaly, and incidence of shunting.
Methods
This is a literature review with pooled data analysis and case report.
Results
One hundred four CDM patients were reviewed in 13 articles that mentioned CDM with ventriculomegaly and/or head circumference. Data was very limited: only 7 patients had data on the presence or absence of prenatal ventriculomegaly, 97 on ventriculomegaly at birth, and 32 on whether or not the ventricles enlarged post-natally. Three patients of 7 (43 %) had pre-natally diagnosed ventriculomegaly, 43 of 97 (44 %) had ventriculomegaly at birth, and only 5 of 32 (16 %) had progressive enlargement of ventricles post-natally. Only 5 of 104 patients had a documented shunt placement: 1 for obstructive, 1 for a post-hemorrhagic communicating, 2 for a communicating hydrocephalus without hemorrhage, and 1 with unknown indication. Of 13 macrocephalic patients with data about ventricular size, 12 had ventriculomegaly.
Conclusions
Ventriculomegaly occurs regularly with CDM but most often does not require CSF diversion. Decisions regarding neurosurgical intervention will necessarily be based on limited information, but shunting should only occur once dynamic data confirms hydrocephalus.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Myotonia is the abnormally slow relaxation of muscles after voluntary contraction or electrical stimulation. Originally called Thomsen’s disease, myotonia was extensively discussed in the late nineteenth century medical literature and Erb’s monograph in 1886 is probably the most cited source [1–3]. Deleage in 1890 was the first to comment on a small subset of patients with Thomsen’s disease who also developed muscle atrophy. Steinert subsequently specified that the atrophy in these patients primarily involved the face, forearms, and peroneal regions, lending his name eponymously to the disease coming to be recognized as myotonica atrophica [4, 5]. Curschmann’s work in 1912 brought greater clarity to this diagnosis, including a description of other symptoms central to the disease, including cataracts, baldness, testicular atrophy, and weight loss. These findings were grouped together as “dystrophic symptoms,” giving rise to its current name, myotonic dystrophy or dystrophia myotonica (DM) [6].
Possible cases of DM have been captured by the historical record as far back as the reign of Pharaoh Akhenaton (approx. 1380–1362 bce) of Egypt. Physical features of this disease are found in the many sculpted likenesses of this pharaoh; they consistently show an elongated face, ptotic eyes, a half-open mouth, gynecomastia, a prominent abdomen, and distal limb atrophy. Statues of other members of his royal family suggest a genetic disease with autosomal dominant inheritance [7]. Claude Monet (1840–1926), diagnosed with cataracts as a sexagenarian, may also have suffered from this disease, as a late relative has been diagnosed with DM by DNA analysis [8]. A population-based risk assessment identified increased risks of sleep apnea, hypothyroidism, cardiac conduction disorders, and intellectual disability as compared to the surrounding population; however, CDM carries multisystemic phenotypes with varying severity. Thus, there is no single constellation of findings in CDM patients which complicates clinical detection and standardization of care [9].
Genetic analysis has defined two types of DM. DM1 (DM1, OMIM 160900) results from a trinucleotide CTG repeat in the 3′ untranslated region of 19q13.3; DM2—not clinically discernible in adults from DM1—results from a tetranucleotide CCTG repeat in intron 1 of 3q21.3. DM2 is not further discussed in this report, as it never manifests congenitally. DM1 is further segregated into four distinct types based on age at presentation: (1) adult onset, (2) childhood onset or juvenile, (3) congenital, and (4) late-onset oligosymptomatic. While ventriculomegaly can occur with childhood- and adult-onset DM1, it is the infant with congenital myotonic dystrophy (CDM) that presents a unique therapeutic challenge to the pediatric neurosurgeon and will be the focus of this report.
Methods
IRB approval was obtained. A Google Scholar search was conducted using the search phrase “congenital myotonic dystrophy.” These papers were reviewed carefully along with their reference lists in order to enlarge the literature base. Articles reporting on CDM patients with ventriculomegaly or macrocephaly or both were included in this analysis. Studies presenting CDM patients but not commenting explicitly on either ventriculomegaly and/or macrocephaly were excluded from the analysis. Articles meeting these criteria were queried for presence of pre-natal ventriculomegaly, presence of ventriculomegaly at birth, worsening or development of ventriculomegaly after birth, head circumference (HC) at birth, abnormally increased HC velocity after birth, infant demise before discharge from the neonatal intensive care unit (NICU), and placement of and indication for a shunt. This allowed for an analysis of the natural progression of CDM-associated ventriculomegaly from prenatal to early childhood. A chart review was performed to present data on the patient included in the case report. For the case report, informed consent was obtained from the patient’s mother.
Genetics and pathophysiology
DM1 belongs to a class of genetic abnormalities called repeat-associated disorders which arise from lengthy repeated sequences of nucleotides in untranslated regions of DNA (rather than from altered protein structures secondary to mutations in translated DNA). The DM1 gene codes for myotonic dystrophy protein kinase (DMPK), a 75–80 kDa protein kinase found in highest concentrations in the heart and skeletal muscle and, to a lesser extent, in smooth muscle and other non-muscle tissues [1]. DMPK plays many roles; the more clearly defined roles include determination of cell shape and coupling of excitation to the contraction of skeletal muscles by maintaining calcium homeostasis [10]. The mutation giving rise to DM1 lies in the 3′ intron region of the 19q13.3 chromosome and consists of variable numbers of CTG trinucleotide repeats. Normal individuals have between 3 and 35 repeats, a pre-mutation state is defined as 36–50 repeats, and DM1 phenotypes are seen in individuals with > 50 CTG repeats. DM1 displays increased severity (potentiation) and earlier onset (anticipation) as the number of CTG repeats present in the germline is amplified; in DM1, these processes almost always perpetuate generationally via maternal transmission [11]. Within a kinship, the phenotypic severity of anticipation as well as potentiation is well correlated with increasing CTG repeats. This relationship is less pronounced when comparing non-related individuals. CTG repeat length varies not only within germlines across generations but also somatically in tissues of the same patient, rendering genetic testing difficult. Germline repeat length instability and the process leading to somatic mosaicism are unclear [12].
The pathophysiology behind the DM1 phenotype is very complicated and has therefore been hard to define. Several abnormal intracellular processes have been identified to date, including the following: transcriptome-wide changes in gene expression in muscle biopsies, dysregulated microRNA interference with normal RNA-dependent processes, and presence of homopolymeric polyglutamate peptides due to repeat-associated non-ATG translation of the DMPK antisense transcript [13]. Previously defined pathophysiology involves the accumulation of large amounts of mutant messenger RNA (mRNA) in the cell nucleus [14–16]. These repeat-CUG mRNAs form large nuclear inclusions which are unable to pass out of the cell nucleus. These inclusions in turn disrupt the function of two proteins important in managing the process of mRNA splicing into their proteomically relevant isoforms. The first is muscleblind binding protein 1 (MBNL-1) which becomes sequestered in the mRNA inclusions, reducing its nuclear concentration. MBNL-1 in normal concentrations drives mRNA splicing toward greater concentrations of adult isoforms; conversely, a shortage of active MBNL-1 will elevate mRNA isoforms destined to generate fetal types of proteins. The second disrupted protein is CUG-binding protein 1 (CUG-BP1) which is up-regulated by hyperphosphorylation secondary to the nuclear mRNA inclusions. Up-regulated CUG-BP1 actively increases mRNA splicing, resulting in fetal isoforms of its dependent proteins [10]. The imbalance in MBNL-1 and CUG-BP1 has been found to have wide proteomic and cellular biological effects. The transport of chloride channels to the myofiber membrane appears to be compromised, causing an impairment of chloride conductance which some believe to be the etiology of the myotonic aspect of this disease. Insulin receptors, sarcoplasmic reticulum chloride channels, cardiac troponin transcripts, and MBNL-1 itself all appear to be negatively affected in DM1. In the brain, splicing abnormalities have been found in the N-methyl-C aspartate receptor 1, the microtubule-associated tau protein (from which arise the tau protein abnormalities found histopathologically), the amyloid precursor protein, and the SLITRK family of proteins (found predominantly in neural tissues which appear to have a neurite-modulating function) [10, 11]. The fetal isoform shift of tissue proteins has been found in muscle biopsy analysis in DM1 patients; in CDM patients, serial muscle biopsies have demonstrated a gradual normalization of the fetal isoform abnormality in parallel to its clinical course.
Clinical congenital myotonic dystrophy
Musculoskeletal
CDM patients can expect one hospital admission per year for a DM1-associated complication [17]. Infants with CDM have obvious facial weakness with a tented upper lip, mild ptosis, and bi-temporal wasting which gives their face a long thin appearance. Myotonia, despite lending its name to the disease, is not present congenitally, although 100 % of surviving patients will develop this problem by the time that they are older than 11 years [18]. Diffuse hypotonia and hyporeflexia are present, and global weakness is often apparent because of a lack of anti-gravity movement. The sensory exam is usually normal, but the withdrawal responses and facial grimace will be muted due to the motor effects of the disease; in some cases, only an alteration in vital signs will indicate discomfort to noxious stimulus. Many CDM patients will go on to experience orthopedic problems like joint contractures and pathologic fractures, and scoliosis will affect 30 % of these patients with 10 % of these requiring operative intervention [19].
Cardiopulmonary
The CDM neonate will almost certainly have breathing and feeding difficulties significant enough to require assistance. The respiratory problems are mechanical: muscle weakness, thin ribs, and a raised right hemi-diaphragm (arising from global immaturity of the diaphragm with greater resistance from the liver) all conspire to impede adequate ventilation [20]. Pulmonary hypoplasia can sometimes complicate this mechanical picture. Cardiac abnormalities are common: CDM carries a relative risk of cardiac conduction anomalies 60 times that of the population; 80 % of patients will have some type of ECG abnormality [9, 21]. Even asymptomatic DM1 patients are at risk for lethal cardiac abnormalities. All CDM children require annual Holter monitoring [21].
Gastrointestinal
Feeding is similarly hindered by profound muscular weakness of the oropharynx, leaving CDM patients at high risk for aspiration. Literature from the early 1990s indicated that CDM infants have a poor prognosis if they are ventilated for more than 30 days, and this information has been widely repeated even as recently as 2004 in a neuromuscular workforce report from Europe [22]. More recent data has, however, shown that many infants receiving prolonged ventilation can wean successfully and have meaningful lives. Once out of the NICU, respiratory infections and gastrointestinal (GI) dysmotility account for the majority of morbidity in CDM [23].
Central nervous system
One of the major histopathological characteristics of DM1 is the presence of abnormal aggregates of Tau proteins, leading to CDM categorization as a tauopathy. Because there is very little long-term data, let-alone data with adequate patient categorization, little more is known beyond the knowledge that CDM patients surviving infancy develop significant cognitive and behavioral problems. Common clinical issues in these patients are as follows: compromised executive function, avoidant behavior, cognitive impairment, apathy, fatigue, and over-sleeping. These cognitive-behavioral problems lead to significant difficulty in maintaining employment and participating in social interactions. Cognitive-behavioral problems are progressive in the majority of patients, though severity and progression are highly variable. A recent workshop on DM1 central nervous system (CNS) research issues stated that “it is unclear if CNS dysfunction in DM1 is neurodevelopmental, neurofunctional and/or neurodegenerative even though several clinical parameters such as stamina, sleepiness, pain and systemic disease show degenerative progression” [24]. Communication difficulty is cited as the most impacting symptom of the disease, and there is some suggestion of similarity to autistic features [25]. There is some debate about IQ levels. Data places children with CDM in the 43rd to 74th percentiles, and special education programs are required by most patients [10]. However, others have drawn attention to the fact that these patients have delayed motor response due to muscle impairment, reduced facial expressivity, and a lack of initiative that may falsely lower scores on IQ tests. Many studies using the Wechsler Adult Intelligence Scale have found a normal range of IQ in DM1 patients [26]. More advanced imaging techniques offer the hope of greater clarity on the nature of CNS effects in DM1 patients. While repeat length has not been found to correlate tightly with degree of CNS impairment, Minnerop et al. have found a significantly higher fractional anisotropy in DTI in DM1 patients with higher scores on validated measures of fatigue and depression [27]. Future research integrating sensitive and specific neuropsychological testing with functional neuroimaging holds promise for greater understanding of DM1 cognitive effects.
Neuroradiology and neuropathology
Neuroradiologically, patients with DM1 have been found to have many abnormalities, but none is specific to the disease. Ventriculomegaly is a common finding, often with maintained extra-axial CSF spaces and a well-delineated sulcal/gyral pattern. White matter abnormalities are virtually universal, though their distribution is far from stereotyped. These abnormalities are seen as high signal on T2-weighted images and are more commonly found in the periventricular, temporal, and parietal regions. Many other findings are common: mild atrophy of the frontal cortex, hypoplasia of the corpus callosum, brainstem hypoplasia, neuronal migration abnormalities, and cerebellar abnormalities [28–34].
Beyond these radio-anatomical abnormalities, recent exploration of radio-functional parameters has shown that DM1 is a diffuse white matter disease. Perfusion scans of DM1 patients show variable areas of decreased perfusion, but the left hemisphere appears significantly more often affected [35]. MR spectroscopy studies demonstrate abnormally low levels of N-acetylaspartate, creatine, and choline and more pronounced in the frontal white matter. Finally, DTI images have demonstrated both lower fractional anisotropy values and elevated levels of mean diffusivity [36]. Several radiofunctionality studies have found significant correlations between degree of cognitive and behavioral dysfunction and the degree of brain abnormality on imaging [35–37].
The neuropathology of this disease is not well understood, a result of the paucity of subjects who allow for autopsy and the relatively rare occurrence of CDM. Some authors have found intact cytoarchitecture [38]. On the whole, however, neuropathological inquiry has found a wide range of abnormalities including disturbed neuronal migration with neurons present in the subcortical white matter, abnormal cortical layering, polygyria, and leptomeningeal neuroglional heterotopia. Other abnormalities such as periventricular leukomalacia, ventriculomegaly, olivary dysplasia, small tegmentum, defects of the septum pellucidum, corpus callosum hypoplasia, and hypoxic-ischemic encephalopathy and basal ganglia abnormalities all have been reported [30, 39–42]. Patterns of radiological and histopathological in DM1 are highly variable indicating a highly complex pathophysiology and inconsistent findings.
Genetic testing and mortality
Generally speaking, data on CDM severity, progression, morbidity, and mortality are limited due to the low incidence of disease. The infant with CDM will often be born to a mother who is unaware of her diagnosis due to the subtlety of her symptoms; the infant will be the “clinically evident” index case in DM1 families approximately 50 % of the time [10]. Extremely rarely, the CDM infant will result from paternal transmission [11]. Because repeat size only weakly correlates with anticipation and potentiation outside of kinship lines, genetic counseling is difficult and many women decline genetic testing. When surveyed, about half of a group of 25 women with DM1 said that having the disease did not affect their child-bearing decisions [43]. CDM mortality from disease process is unclear, as many of the studies included infants receiving comfort measures only. Reardon et al. have created a life table using 115 CDM patients, indicating that there is a 50 % mortality rate by the mid-1930s [44]. Schara comments that collecting comprehensive data from patients and family with eventual creation of an international registry would further the understanding and potential standard of care for CMD patients [45].
Results
Thirteen articles were identified which both presented details of CDM patients and mentioned ventriculomegaly or macrocephaly or both [7, 30, 40, 46–55]. A total of 104 CDM patients were reviewed in these articles, but conclusions are severely limited by a lack of data.
Early mortality
Twenty-three of these 102 patients passed away before discharge from the NICU. None of these 23 patients received a shunt.
Ventriculomegaly and shunt necessity
Out of these 102 patients, only 7 patients had data on the presence or absence of prenatal ventriculomegaly, 97 had similar data on ventriculomegaly at birth, and 32 had data on whether or not the ventricles enlarged post-natally. Three patients of 7 (43 %) had pre-natally diagnosed ventriculomegaly, 43 of 97 (44 %) had ventriculomegaly at birth, and only 5 of 32 (16 %) had progressive enlargement of ventricles post-natally. Out of all 104 patients, only 5 had a documented shunt placement, 1 for radiographically determined obstructive hydrocephalus with ventriculomegaly, 1 for communicating hydrocephalus secondary to intraventricular hemorrhage (IVH) with progressive ventriculomegaly, 1 for communicating hydrocephalus with macrocephaly but no hemorrhage, and 1 for communicating hydrocephalus with progressive ventriculomegaly without hemorrhage, and in 1 patient, the indication was unknown. No further information was available on the patient with obstructive hydrocephalus [7]. The patient with post-IVH hydrocephalus had progressive dilatation of the ventricles as did one of the non-hemorrhagic communicating hydrocephalus shunt patients [47, 48, 55]. The second non-hemorrhagic communicating hydrocephalus patient had a birth HC of 37 cm (98th percentile) [49]. Data on ventricular size or HC post-shunt placement was not available.
Macrocephaly
HC data was similarly limited; only 17 of 104 patients had HC data. The average HC percentile was 88.3, and 15 patients had an HC at or above the 80th percentile (Fig. 1) [30, 46, 49, 50]. Out of these 17 patients, 16 had data on presence or absence of ventriculomegaly with 13 of these patients having ventriculomegaly. Two of these 13 patients had progressive dilatation of the ventricles. Only one of these two patients was shunted. The other patient’s progression stopped after day of life (DOL) 40.

Scatter plot of head circumference percentiles in the 17 patients with available data
Case study
ZC was born at 36-week estimated gestational age and weighed 3670 g. The mother is a 31-year-old G1P0 woman with a known diagnosis of DM. Family history was also significant for several maternal uncles and a maternal grandfather, all diagnosed with severe DM. ZC’s genetic testing found a trinucleotide repeat number of between 1700 and 1850. Delivery was by Cesarean section, and pregnancy was complicated by lack of pre-natal care, polyhydramnios, and breech presentation. Apgars were 1, 4, and 7 at 1, 5, and 10 min, respectively, and the patient was intubated at 2.5 min of life and required cardiac compression for 1.5 min soon after delivery. Early support included dopamine for blood pressure support. The patient was intubated at 2.5 min of life. An initial echocardiogram showed mild left ventricular hypertrophy with decreased systolic function and left wall motion hyperkinesia. X-rays revealed fractures of the left tibia, fibula, and humerus. An EEG was performed on DOL 5 for suspicion of subclinical seizures; the EEG was within normal limits for a sleeping patient. A gastrostomy tube was placed on DOL 28, and a tracheostomy was placed at 4 months of age with ventilator support lasting until late into the fifth month of life.
At birth, HC was 35.0 cm (66th percentile) and remained so for the first month of life. Head ultrasound on DOL 1 was unremarkable; a repeat at 1.5 months showed new, mild, ventriculomegaly of the lateral, third, and fourth ventricles without evidence of intraventricular or parenchymal hemorrhage. A subsequent head ultrasound at 3 months demonstrated significant interval enlargement of the lateral, third, and fourth ventricles compared to the previous ultrasound, also without evidence of hemorrhage (Fig. 2). During this time, the patient’s HC had increased, from 35.0 cm (27th percentile) at birth to 38.0 cm at 7 weeks and 42.3 cm (95th percentile) at 3 months at which point, the neurosurgery service was consulted.

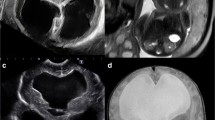
a Coronal ultrasound on DOL 1 was unremarkable. b Repeated scan at 3 months showed significantly enlarged bilateral lateral, third, and fourth ventricles without evidence of intraventricular hemorrhage
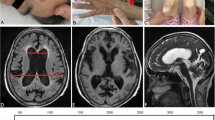
An MRI brain demonstrated moderate to marked diffuse ventriculomegaly without a focal obstructing lesion, as well as diffuse white matter disease throughout both cerebral hemispheres with a vertical orientation of the hippocampi. Sagittal view shows downward bowing of the floor of the third ventricle; however, there was a patent aqueduct with motion artifact indicating CSF flow and a moderately dilated fourth ventricle without posterior fossa cyst and mild hypoplasia of the vermis. The Evan’s ratio was 0.5. Extra-axial cerebrospinal fluid (CSF) spaces were not compressed. There was a normal sulcal/gyral pattern (Fig. 3). Because the patient’s HC seemed to stabilize at about the 90th percentile and extra-axial CSF spaces were preserved, neurosurgical intervention was deferred. However, at 3.5 months, the patient’s HC reached 45 cm (>99.9th percentile) and a ventriculoperitoneal shunt with a programmable valve set at 150 mm H2O was placed. Intraoperative assessment revealed an intracranial pressure of 21 cm H2O with an end tidal CO2 of 35 and with the patient apneic. In the 2 months following shunt placement, HC has stabilized close to the 50th percentile most currently at 42.5 cm. In addition, occupational therapy noted a clear and consistent increase in the level of activity after shunting.

a Axial and b coronal T2 MRI demonstrating marked dilatation of the lateral and third ventricles without evidence of hemorrhage or focal mass. Diffuse T2 white matter prolongation bilaterally due to white matter myelination abnormality secondary to myotonic dystrophy. Hippocampi are vertically oriented bilaterally. Sulci and basal cisterns are well visualized. c Sagittal view shows downward bowing of the floor of the third ventricle; however, there was a patent aqueduct with motion artifact indicating CSF flow and a moderately dilated fourth ventricle without posterior fossa cyst and mild hypoplasia of the vermis. d No cytotoxic edema on the diffusion sequence
Conclusions
Understanding of CDM has advanced gradually since its first recognition as a distinct clinical entity, but the pathophysiology has yet to be fully elucidated, particularly within the CNS. Multiple organ systems are affected. CDM presents a particular treatment challenge to the pediatric neurosurgeon, as features of this disease process confound establishing a diagnosis of hydrocephalus. Profound hypotonia obscures detailed neurologic examination, particularly in infants. Ventriculomegaly and macrocephaly can each be independently associated with CDM, likely resulting from underlying structural white matter pathology as reflected on MR imaging. The etiology of associated hydrocephalus in some patients has not been well defined. Previously reported CDM patients requiring CSF diversion demonstrated progressive ventriculomegaly or HCs crossing percentile lines upward. Abnormal HC growth was ultimately the finding that prompted intervention in the presently reported case, rather than findings on radiology or neurologic exam. Until more comprehensive understanding of the neuropathology associated with CDM is achieved, decisions regarding neurosurgical intervention will necessarily be based on limited information, but shunting should only occur once dynamic data confirms hydrocephalus.
References
Adie WJ, Greenfield JG (1923) Dystrophia myotonica (myotonia atrophica). Brain 46:73–127
Thomsen DJ (1876) Tonische Krämpfe in willkürlich beweglichen Muskeln in Folge von ererbter psychischer Disposition. Arch Für Psychiatr Nervenkrankh 6:702–718
Erb WH (1886) Die Thomsen’sche Krankheit. Verlag von F.C.W. Vogel, Leipzig
Déléage F (1890) Etude clinique sur la maladie de Thomsen. Octave Dion, Paris
Steinert PDH (1909) Myopathologische Beiträge. Dtsch Z Für Nervenheilkd 37:58–104
Curschmann H (1912) Über familiäre atrophische Myotonie. Dtsch Z Für Nervenheilkd 45:161–202
Ekström A-B (2009) Congenital and childhood myotonic dystrophy type 1—the impact on central nervous system, visual and motor function. Available via GUPEA. https://gupea.ub.gu.se/handle/2077/19387
Lane R, Carey N, Orrell R, Moxley RT 3rd (1997) Claude Monet’s vision. Lancet 349:734
Johnson NE, Abbott D, Cannon-Albright L (2015) Relative risks for comorbidities associated with myotonic dystrophy: a population-based analysis. Muscle Nerve. doi:10.1002/mus.24766
Campbell C (2012) Congenital myotonic dystrophy. J Neurol Neurophysiol. doi:10.4172/2155-9562.S7-001
Di Costanzo A, de Cristofaro M, Di Iorio G, Daniele A, Bonavita S, Tedeschi G (2009) Paternally inherited case of congenital DM1: brain MRI and review of literature. Brain Dev 31:79–82. doi:10.1016/j.braindev.2008.04.008
Pearson CE, Nichol Edamura K, Cleary JD (2005) Repeat instability: mechanisms of dynamic mutations. Nat Rev Genet 6:729–742. doi:10.1038/nrg1689
Udd B, Krahe R (2012) The myotonic dystrophies: molecular, clinical, and therapeutic challenges. Lancet Neurol 11:891–905. doi:10.1016/S1474-4422(12)70204-1
Taneja KL, McCurrach M, Schalling M, Housman D, Singer RH (1995) Foci of trinucleotide repeat transcripts in nuclei of myotonic dystrophy cells and tissues. J Cell Biol 128:995–1002. doi:10.1038/jcb.128.6.995
Amack JD, Mahadevan MS (2001) The myotonic dystrophy expanded CUG repeat tract is necessary but not sufficient to disrupt C2C12 myoblast differentiation. Hum Mol Genet 10:1879–1887. doi:10.1093/hmg/10.18.1879
Mankodi A, Logigian E, Callahan L, McClain C, White R, Henderson D et al (2000) Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat. Science 289:1769–1773. doi:10.1126/science.289.5485.1769
Campbell C, Sherlock R, Jacob P, Blayney M (2004) Congenital myotonic dystrophy: assisted ventilation duration and outcome. Pediatrics 113:811–816. doi:10.1542/peds.113.4.811
Harper PS (1975) Congenital myotonic dystrophy in Britain. I. Clinical aspects. Arch Dis Child 50:505–513. doi:10.1136/adc.50.7.505
Canavese F, Sussman MD (2009) Orthopaedic manifestations of congenital myotonic dystrophy during childhood and adolescence. J Pediatr Orthop 29:208–213. doi:10.1097/BPO.0b013e3181982bf6
Aicardi J, Conti D, Goutières F (1974) Les formes néo-natales de la dystrophie myotonique de steinert. J Neurol Sci 22:149–164
Johnson ER, Abresch RT, Carter GT, Kilmer DD, Fowler WM, Sigford BJ et al (1995) Profiles of neuromuscular diseases: myotonic dystrophy. Am J Phys Med Rehabil 74:S104–S116
Wallgren-Pettersson C, Bushby K, Mellies U, Simonds A (2004) 117th ENMC workshop: ventilatory support in congenital neuromuscular disorders—congenital myopathies, congenital muscular dystrophies, congenital myotonic dystrophy and SMA (II) 4–6 April 2003, Naarden, The Netherlands. Neuromuscul Disord 14:56–69. doi:10.1016/j.nmd.2003.09.003
Echenna B, Rideua A, Roubertica A, Sebire G, Riviera F, Lemieux B (2008) Myotonic dystrophy type I in childhood: long-term evolution in patients surviving the neonatal period. Eur J Paedriatr Neurol. doi:10.1016/j.ejpn.2007.07.014
Axford MM, Pearson CE (2013) Illuminating CNS and cognitive issues in myotonic dystrophy: workshop report. Neuromuscul Disord 23:370–374. doi:10.1016/j.nmd.2013.01.003
Johnson NE, Luebbe E, Eastwood E, Chin N, Moxley R, Heatwole C (2014) The impact of congenital and myotonic distrophy on quality of life: a qualitative study of associated symptoms. J Child Neurol, 10.117/0883073813484804
Meola G, Sansone V (2007) Cerebral involvement in myotonic dystrophies. Muscle Nerve 36:294–306. doi:10.1002/mus.20800
Minnerop M, Weber B, Schoene-Bake J-C, Roeske S, Mirbach S, Anspach C et al (2011) The brain in myotonic dystrophy 1 and 2: evidence for a predominant white matter disease. Brain J Neurol 134:3530–3546. doi:10.1093/brain/awr299
Bergoffen F, Kant J, Sladky J, McDonald-McGinn D, Zackai E, Fischbeck K (1994) Paternal transmission of congenital myotonic dystrophy. J Med Genet 31:518–20. doi:10.1136/jmg.31.7.518
Di Costanzo A, Di Salle F, Santoro L, Bonavita V, Tedeschi G (2002) Brain MRI features of congenital- and adult-form myotonic dystrophy type 1: case-control study. Neuromuscul Disord 12:476–483. doi:10.1016/S0960-8966(01)00324-8
Garcia-Alix A, Cabañas F, Morales C, Pellicer A, Echevarria J, Paisan L et al (1991) Cerebral abnormalities in congenital myotonic dystrophy. Pediatr Neurol 7:28–32. doi:10.1016/0887-8994(91)90102-Q
Hashimoto T, Tayama M, Miyazaki M, Murakawa K, Kawai H, Nishitani H et al (1995) Neuroimaging study of myotonic dystrophy. I. Magnetic resonance imaging of the brain. Brain Dev 17:24–27. doi:10.1016/0387-7604(94)00096-G
Kuo H-C, Hsiao K-M, Chen C-J, Hsieh Y-C, Huang C-C (2005) Brain magnetic resonance image changes in a family with congenital and classic myotonic dystrophy. Brain Dev 27:291–296. doi:10.1016/j.braindev.2004.09.002
Martinello F, Piazza A, Pastorello E, Angelini C, Trevisan CP (1999) Clinical and neuroimaging study of central nervous system in congenital myotonic dystrophy. J Neurol 246:186–192. doi:10.1007/s004150050332
Regev R, de Vries LS, Heckmatt JZ, Dubowitz V (1987) Cerebral ventricular dilation in congenital myotonic dystrophy. J Pediatr 111:372–376
Romeo V, Pegoraro E, Ferrati C, Squarzanti F, Sorarù G, Palmieri A et al (2010) Brain involvement in myotonic dystrophies: neuroimaging and neuropsychological comparative study in DM1 and DM2. J Neurol 257:1246–1255. doi:10.1007/s00415-010-5498-3
Wozniak JR, Mueller BA, Bell CJ, Muetzel RL, Lim KO, Day JW (2013) Diffusion tensor imaging reveals widespread white matter abnormalities in children and adolescents with myotonic dystrophy type 1. J Neurol 260:1122–1131. doi:10.1007/s00415-012-6771-4
Wozniak JR, Mueller BA, Ward EE, Lim KO, Day JW (2011) White matter abnormalities and neurocognitive correlates in children and adolescents with myotonic dystrophy type 1: a diffusion tensor imaging study. Neuromuscul Disord 2:89–96. doi:10.1016/j.nmd.2010.11.013
Young R, Gang D (1981) Dysmaturation in infants of mothers with myotonic dystrophy. Arch Neurol 38:716–719. doi:10.1001/archneur.1981.00510110076013
Endo A, Motonaga K, Arahata K, Harada K, Yamada T, Takashima S (2000) Developmental expression of myotonic dystrophy protein kinase in brain and its relevance to clinical phenotype. Acta Neuropathol 100:513–520. doi:10.1007/s004010000216
Hageman ATM, Gabreëls FJM, Liem KD, Renkawek K, Boon JM (1993) Congenital myotonic dystrophy; a report on thirteen cases and a review of the literature. J Neurol Sci 115:95–101. doi:10.1016/0022-510X(93)90072-7
Rosman NP, Kakulas BA (1966) Mental deficiency associated with muscular dystrophy a neuropathological study. Brain 89:769–788
Sarnat HBSS (1976) Maturational arrest of fetal muscle in neonatal myotonic dystrophy: a pathologic study of four cases. Arch Neurol 33:466–474. doi:10.1001/archneur.1976.00500070008002
Faulkner C, Kingston H (1998) Knowledge, views, and experience of 25 women with myotonic dystrophy. J Med Genet 35:1020–1025. doi:10.1136/jmg.35.12.1020
Reardon W, Newcombe R, Fenton I, Sibert J, Harper PS (1993) The natural history of congenital myotonic dystrophy: mortality and long term clinical aspects. Arch Dis Child 68:177–181. doi:10.1136/adc.68.2.177
Schara U (2015) Congenital and childhood-onset myotonic dystrophy: importance of long-term data in natural history. Dev Med Child Neurol. doi:10.1111/dmcn.12960
Bell DB, Smith DW (1972) Myotonic dystrophy in the neonate. J Pediatr 81:83–86. doi:10.1016/S0022-3476(72)80378-0
Rutherford MA, Heckmatt JZ, Dubowitz V (1989) Congenital myotonic dystrophy: respiratory function at birth determines survival. Arch Dis Child 64:191–195. doi:10.1136/adc.64.2.191
Sinha R, Jayawant S (2006) Myotonic dystrophy with symptomatic hydrocephalus in early infancy: a case report. J Pediatr Neurol 4:215–217
Rettwitz-Volk W, Wikstroem M, Flodmark O (2001) Occlusive hydrocephalus in congenital myotonic dystrophy. Brain Dev 23:122–124. doi:10.1016/S0387-7604(01)00176-0
Fox GN, Gravett MG (1986) Neonatal myotonic dystrophy associated with prenatal ventriculomegaly. A case report. J Reprod Med 31:729–731
Pearse RG, Howeler CJ (1979) Neonatal form of dystrophia myotonica. Five cases in preterm babies and a review of earlier reports. Arch Dis Child 54:331–338. doi:10.1136/adc.54.5.331
Tanabe Y, Iai M, Tamai K, Fujimoto N, Sugita K (1992) Neuroradiological findings in children with congenital myotonic dystrophy. Acta Paediatr 81:613–617. doi:10.1111/j.1651-2227.1992.tb12312.x
Nazir MA, Dillon WP, McPherson EW (1984) Myotonic dystrophy in pregnancy. Prenatal, neonatal and maternal considerations. J Reprod Med 29:168–172
Webb D, Muir I, Faulkner J, Johnson G (1978) Myotonia dystrophica: obstetric complications. Am J Obstet Gynecol 132:265–270
Carroll J, Quaid K, Stone K, Jones R, Shubert F, Griffith C (2013) Two is better than one: a case of homozygous myotonic dystrophy type I. Am J Med Genet. doi:10.1002/ajmg.a.35967
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Financial disclosure
This work received no financial support.
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
No portion of this work has been published or presented in any other forum.
Rights and permissions
About this article

Cite this article
Mutchnick, I.S., Thatikunta, M.A., Gump, W.C. et al. Congenital myotonic dystrophy: ventriculomegaly and shunt considerations for the pediatric neurosurgeon. Childs Nerv Syst 32, 609–616 (2016). https://doi.org/10.1007/s00381-015-2993-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00381-015-2993-y