
Abstract
Melanotic neuroectodermal tumor of infancy (MNTI) is a rare pigmented tumor generally occurring in the head and neck region in children 12 months of age or younger. Approximately 380 instances of this tumor are reported in the medical literature. We presented a case of MNTI that occurred in the left temporal bone of a 2-month old female infant. And, the clinical assessment, histologic diagnosis, and management is reviewed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Melanotic neuroectodermal tumor of infancy (MNTI) is a rare neoplasm of neural crest origin, which is composed of relatively primitive pigment-producing cells and usually arises in infants within the first year of life [2, 7]. No gender predilection has been described [4, 7]. The initial description of MNTI has been attributed to Krompecher in 1918, who described it as a melanocarcinoma [6]. MNTI usually arises in the head and neck region and predominantly affects the maxilla, although other sites, such as brain, epididymis, mediastinum, femur, and ovary have also been reported [5].
MNTI is generally a benign lesion, but it requires aggressive surgical treatment because it behaves in a locally aggressive fashion and has a high rate of recurrence, i.e., from 15% to 45% [3, 4]. Furthermore, although the tumor is considered benign, cases of malignant transformation manifesting as metastasis have been reported. However, its malignant potential is difficult to determine based on clinical or histologic information [7]. In cases where complete resection has not been possible, some have tried chemotherapy and radiation, but reports are too limited to establish a preferred treatment algorithm [2]. Histologically, the tumor is a nonencapsulated neoplasm of dense fibrovascular tissue composed of small nests or basophilic cells. Moreover, MNTIs are composed of two cell types that express neural, epithelial, and melanocytic markers [4, 7]. Here, the authors present the case of a 2-month-old female infant with a melanotic neuroectodermal tumor of infancy in an uncommon site—the left temporal bone.
Case report
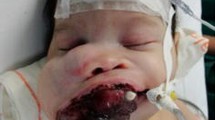
A 2-month-old female infant was admitted to the Samsung Medical Center, Seoul, in January of 2007 because of a mass in the left temporal area (Fig. 1). No other physical, clinical, or laboratory abnormalities were identified, and there were no congenital anomalies. The mass appeared to be growing rapidly. Radiographic studies revealed focal skull thickening with a soft tissue mass on the left temporal bone. The first impression of the radiologist was of Langerhans histiocytosis or neuroblastoma. A subsequent Tc-99m muramyl dipeptide bone scan study revealed increased radio uptake with a photon defect lesion in the left temporal bone, but findings were otherwise unremarkable.

Initial pre-operative brain CT
An incisional biopsy was performed under general anesthesia. Perioperatively, we found a dark pigmented bony mass with an overlying fibrotic capsule. Upon receiving a histologic diagnosis of MNTI, a second operation was required to achieve complete surgical extirpation (Fig. 2). The excised specimen obtained had a rubbery consistency and was a brownish black fibrotic mass, measuring 3 × 3 cm. Its cut surface revealed an almost entirely black pigmented area among yellowish/white fibrotic tissue. Microscopically, numerous nests of tightly packed immature cells were observed to be surrounded by proliferative, fibrous stroma containing large, melanin-laden cells, which were distributed throughout the mass. Furthermore, the tumor showed a biphasic proliferation of small rounded neuroblast-like cells and larger epithelioid cells with eosinophilic cytoplasm containing variable amounts of melanin (Fig. 3). Moreover, the small neuroblast-like cells expressed synaptophysin, and the melanin-containing epithelioid cells were highly immunoreactive for beta-hydroxy beta-methylbutyrate (HMB)-45 but rarely expressed S-100. Postoperative imaging showed surgical changes but no evidence of residual tumor. The patient followed an uneventful postoperative course and remains alive and well without evidence of disease or recurrence 12 months after initial presentation.

Post-incisional biopsy brain CT (a) and magnetic resonance imaging (b). The tumor appeared to grow faster after biopsy

a Hematoxylin and eosin stained specimen showing a biphasic cell population comprising larger epithelioid cells and small rounded neuroblast-like cells. b Synaptophysin was expressed by the small round neuroblast-like cells. c Clusters of large epithelial-like cells were positive for HMB-45. d Peripheral epithelioid cells stained positively for cytokeratin
Discussion
Conventional radiographs of bony lesions usually reveal radiolucency with or without irregular margins. Typically, computed tomography (CT) scans reveal hyperdense masses, but hypodense variants have also been reported [1]. CT can accurately define lesion extent and, thus, provides a good basis for surgical planning. Melanotic neuroectodermal tumors of infancy tend to be isointense on T1-weighted images and slightly hyperintense on T2-weighted images. Furthermore, there may be an inhomogeneous increase in signal on T1-weighted images, which also may reflect intratumoral melanin, whereas T2-weighted images visualize the tumor as isointense or hypointense to grey matter. Contrast enhancement is usually quite marked [1, 8].
The differential diagnosis of MNTI is quite broad, but it must be separated from other pediatric “small round cell” neoplasms, such as neuroblastoma, Ewing’s sarcoma, peripheral neuroepithelioma, rhabdomyosarcoma, peripheral primitive neuroectodermal tumor, desmoplastic small round cell tumor, malignant melanoma, and lymphoma. However, immunohistochemistry, electron microscopy, histochemistry, and the detection of melanotransferrin mRNA transcripts now have confirmed that MNTIs are derived from neural crest cells, derived in turn from neuroectoderm [7, 8]. Microscopically, MNTIs are biphasic tumors characterized by larger polygonal epithelioid cells, which resemble melanocytes and contain variable deposits of melanin and smaller neuroblast-like round cells. Reported MNTI immunostaining results tend to vary [5, 7, 8]. The larger epithelioid cellular component expresses a number of cytokeratins, usually HMB-45, but rarely S-100, whereas the small neuroblast-like cells mainly express CD56 and synaptophysin. However, in most cases, both cell populations are positive for neuron-specific enolase. Moreover, the expression of Ki-67/CD99 in MNTI, which is uncommon, might be correlated with aggressive tumor growth. In our case, immunohistologic features were similar to previously published MNTIs [1, 7].
Complete surgical excision, with negative margins, can be performed without prior biopsy. This approach avoids unnecessary anesthetic risks and reduces lesion manipulation, which is important because it has been reported that tumors appear to grow faster at previous biopsy sites [1, 7].
A conservative approach, consisting of local excision and curettage, has been adopted for the management of MNTI. However, the extent of surgical excision is debatable because these tumors may have the potential to behave in a malignant fashion [1, 2, 5].
Malignant MNTIs develop widespread metastasis and cause death within months, and these tumors are histologic mimics of neuroblastoma rather than of MNTI. However, unfortunately, the biologic behavior of MNTIs, i.e., the likelihoods of local recurrence, malignancy, or metastasis, cannot be predicted from their clinical or histologic features [1, 7]. Flow cytometry has been examined in the context of elucidating these relations, but it was found to be unhelpful [4, 5].
Conclusions
MNTI is a rare, normally benign but locally aggressive tumor that most commonly arises from the maxilla of children less than 1 year of age. Its distinguishing feature is a biphasic cell population with melanin pigment, which allows it to be differentiated from other pediatric neoplasms. Close clinical follow-up is suggested for the first few years after presentation to identify recurrence or the rare development of metastatic disease. The present clinical report is unique as the tumor was located in the temporal bone. Melanotic neuroectodermal tumor of infancy should be included in the differential diagnosis of head and neck neoplasms in infants and young children.
References
Fowler DJ, Chisholm J, Roebuck D, Newman L, Malone M, Sebire NJ (2006) Melanotic neuroectodermal tumor of infancy: clinical, radiological, and pathological features. Fetal Pediatr Pathol 25:59–72
Gaiger de Oliveira M, Thompson LD, Chaves AC, Rados PV, da Silva Lauxen I, Filho MS (2004) Management of melanotic neuroectodermal tumor of infancy. Ann Diagn Pathol 8:207–212
Judd PL, Harrop K, Becker J (1990) Melanotic neuroectodermal tumor of infancy. Oral Surg Oral Med Oral Pathol 69:723–726
Kapadia SB, Frisman DM, Hitchcock CL, Ellis GL, Popek EJ (1993) Melanotic neuroectodermal tumor of infancy. Clinicopathological, immunohistochemical, and flow cytometric study. Am J Surg Pathol 17:566–573
Kaya S, Unal OF, Sarac S, Gedikoglu G (2000) Melanotic neuroectodermal tumor of infancy: report of two cases and review of literature. Int J Pediatr Otorhinolaryngol 52:169–172
Krompecher E (1918) Zur Histogenese und Morphologie der Adamantinome und sonstiger Kiefergeschwülste. Beitr Pathol Anat 64:165–197
Kruse-Losler B, Gaertner C, Burger H, Seper L, Joos U, Kleinheinz J (2006) Melanotic neuroectodermal tumor of infancy: systematic review of the literature and presentation of a case. Oral Surg Oral Med Oral Pathol Oral Radiol Endo 102:204–216
Matsumoto M, Sakuma J, Suzuki K, Kawakami M, Sasaki T, Kodama N (2005) Melanotic neuroectodermal tumor of infancy in the skull: case report and review of the literature. Surg Neurol 63:275–280
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Jeon, H.J., Kong, D.S. & Shin, H.J. Melanotic neuroectodermal tumor of infancy: a case report. Childs Nerv Syst 24, 1489–1492 (2008). https://doi.org/10.1007/s00381-008-0671-z
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00381-008-0671-z