
Abstract
Background
The prevalence of heart failure (HF) is rising with ageing population and constitutes a major health problem globally. A common complication of HF is pulmonary hypertension (PH) which negatively impacts survival. A pathophysiological association between HF and PH with tumorigenic processes has been suggested. We aimed to identify the plasma levels of, and the association between tumour-related proteins and hemodynamic improvements in patients with HF and PH due to left heart disease (LHD) before and 1-year after heart transplantation (HT).
Methods
Forty-eight tumour-related proteins were measured with proximity extension assay in plasma from 20 controls and 26 HF patients before and 1-year after HT. Patients’ hemodynamics were measured with right heart catheterization.
Results
Out of 48 proteins, specifically, plasma levels of endocan and brother of CDO (BOC) were elevated in end-stage HF patients compared to controls (p < 0.001), but decreased after HT (p < 0.01), towards controls’ levels. The decrease of endocan levels after HT correlated with improved mean pulmonary arterial pressure (rs = 0.80, p < 0.0001), pulmonary arterial wedge pressure (rs = 0.63, p = 0.0012), and pulmonary vascular resistance (rs = 0.70, p < 0.001). The decrease and normalization of BOC after HT correlated with decreased mean right atrial pressure (rs = 0.61 p = 0.0015) and NT-proBNP (rs = 0.57, p = 0.0022), as well as increased cardiac index (rs = − 0.51, p = 0.0086) and left-ventricular stroke work index (rs = − 0.57, p = 0.0039).
Conclusion
Our results suggest that (i) plasma endocan in HF may reflect the state of pulmonary vascular congestion and PH-LHD, whereas (ii) plasma BOC may reflect the cardiac function and the hemodynamic overload in HF. The exact role of these proteins and their clinical applicability as biomarkers in HF and PH-LHD ought to be investigated in larger cohorts.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Heart failure (HF) is a clinical syndrome with a prevalence of 1–2% of the adult population in developed countries [1]. Despite improved treatment modalities in the last 2 decades, the 5-year survival of HF patients with reduced ejection fraction remains poor [2]. A common complication in HF, irrespective of ejection fraction, is pulmonary hypertension (PH), with negative impact on survival and exercise capacity [3]. PH may arise as a consequence of left heart disease (LHD), through congestion and backward transmission of elevated left-sided filling pressures. A sustained congestion may cause endothelial dysfunction and excessive vasoconstriction with subsequent vascular remodeling [4].
The chronic progression of HF involves an array of different pathophysiological mechanisms [5]. Proteomic biomarkers are emerging as a new tool for diagnosis and prognosis, and may reflect this pathophysiological progression [5, 6]. In the fields of HF [7] and especially PH, biomarker research is of particular interest, as outlined in a recent state-of-the-art review of PH pathology and pathobiology [8]. To date, however, the clinical use of biomarkers in HF and PH remains mainly limited to natriuretic peptides and its precursors, which collectively reflect one pathophysiological pathway [9, 10].
Although tumorigenic processes and HF are two distinct entities, recent studies reported that HF could prime the onset of cancer by mechanisms involved in pathophysiology of HF, such as aberrant neuro-hormonal axis and growth hormonal overexpression with impact on proliferation [11, 12]. Additionally, the Warburg effect, originally ascribed to cancer cells undergoing higher glycolytic activity through fermenting glucose to lactate during normoxic conditions, has also been postulated to be involved in the pathobiology of HF and pulmonary arterial hypertension (PAH). For instance, in hypertrophic cardiomyopathy, metabolic dysfunction and energy deficit are functional in cardiac dysfunction and ventricular remodeling [13]. Moreover, the development of pulmonary vascular remodeling during PAH progression, which pathobiologically may imitate that observed in PH-LHD [14], involves cellular acquisition of tumorigenic traits including deranged cellular energetics, sustained proliferative signaling, and reduced susceptibility to apoptosis [15].
Intriguingly, several studies have appraised tumour-related proteins in the context of either PAH [16, 17] or HF [18]. In a murine model, deletion of the pro-apoptotic transcription factor P53 exacerbated hypoxia-induced PAH [16]. A subsequent study showed that treatment with Nutilin-3a, a cis-imidazoline analog that stabilizes the pro-apoptotic transcription factor p53 and increases the pro-senescent p21 expression, reversed PAH in mice and induced cell growth arrest and senescence in cultured human pulmonary arterial smooth muscle cells [17]. Mucin-16 or CA125, a marker of ovarian cancer, is, furthermore, elevated in HF patients with severe fluid overload and may be of prognostic value [18]. To our knowledge, there is, however, a paucity in studies with focus on tumour markers in HF and PH-LHD as well as the consequence of heart transplantation (HT).
In search of potential biomarkers reflecting alterative pathophysiological pathways in HF and related PH, such as inflammatory response, cellular proliferation, and endothelial dysfunction, we aimed to identify the levels of tumour-related proteins with associated hemodynamic improvements in HF and PH-LHD, before and after HT. Identifying such proteins may aid in generating hypothesis for clinical research and the incorporation of a multi-marker testing panel of different pathophysiological mechanisms. Moreover, a biomarker-guided phenotyping of HF and related PH may potentially optimize the clinical management and prompt the development of new therapies [6], especially in the stagnant supply of donor hearts enabling HT [19].
Materials and methods
Study population
The present study was based on 29 patients with end-stage HF with or without preoperative PH evaluated before and 1-year after HT at Skåne’s University Hospital in Lund, Sweden, as well as 20 cardiopulmonary healthy controls (≥ 18 years) with no history of ischemic heart disease, atrial fibrillation, stroke, or diabetes mellitus. Although two of the controls reported a previous thyroid illness, all were included as none of the controls exhibited cardiovascular-related comorbidities. Patients with PH after HT (n = 1) and with missing postoperative hemodynamic data (n = 2) were excluded. Left-ventricular dysfunction was diagnosed according to the routine clinical investigation with echocardiography and/or magnetic resonance imaging [1]. Informed written consent was acquired from all participants. The population has previously been characterized, including patients’ plasma creatinine and NT-proBNP, shown in Table 1 [20, 21]. Briefly, 50% of the controls were male, had a median age of 41 years and a median body surface area (BSA) of (1.92 m2, n = 19).
The study was conducted in accordance with the declarations of Helsinki and Istanbul and approved by the ethical board in Lund, Sweden (diary numbers: 2010/114; 2010/442; 2011/368; 2011/777; 2014/92 and 2015/270).
Protein analysis
Venous blood samples were collected from the venous introducer of the patients’ internal jugular veins during right heart catheterization (RHC) and from peripheral veins in controls, stored at − 80 °C in Lund Cardio Pulmonary Register (LCPR), a cohort of Region Skåne’s biobank. As per protocol, neither the patients nor controls were fasting during blood sample collection. Plasma aliquots, retrieved from LCPR, were analysed with proximity extension assay (PEA). PEA is based on the use of oligonucleotide-linked antibodies and qPCR for protein detection and quantification (Proseek Multiplex Cardiovascular II, III and Oncology II kits, Olink, Proteomics, Uppsala, Sweden) [22]. Proteins analysed were N-terminal pro b-type natriuretic peptide (NT-proBNP), 5′-nucleotidase, protein AMBP (AMBP), aminopeptidase N (AP-N), bleomycin hydrolase (BLM-H), brother of cell adhesion molecule-related/down-regulated by oncogenes (CDO) or (BOC), carbonic anhydrase 9 (CA9), cathepsin Z, p21 or cyclin-dependent kinase inhibitor 1A (CDKN1A), carcinoembryonic antigen-related cell adhesion molecule (CEACAM) 1 and 5, contactin-1, cornulin, carboxypeptidase A1 (CPA1), carboxypeptidase B (CPB1), carboxypeptidase E (CPE), cystatin B, endothelial cell-specific molecule 1 or endocan, epithelial cell adhesion molecule (Ep-CAM), furin, gastrotropin, glyoxalase I or lactoylglutathione lyase, kallikrein 6, 8, 11, 13 and 14, Ly6/PLAUR domain-containing protein 3 (LYPD3) or C4.4A, mesothelin, methionine aminopeptidase 2 (MetAP2), melanoma-derived growth regulatory protein or melanoma inhibitory activity (MIA), midkine, mucin-16 or CA125, podocalyxin, prostasin, PVRL4 or nectin-4, S100A11, S100A4, secretory carrier-associated membrane protein 3 (SCAMP3), secretoglobin family 3A member 2 (SCGB3A2), tyrosine-protein phosphatase non-receptor-type substrate 1 (SHPS-1), sortilin, T-cell leukemia/lymphoma protein 1A (TCL1A), trefoil factor 3 (TFF3), protein-glutamine gamma-glutamyltransferase 2 (TGM2), WAP four-disulfide core domain protein 2 (WFDC2), vimentin, V-set and immunoglobulin domain-containing protein 2 (VSIG2), and Xaa-Pro aminopeptidase 2 (XPNPEP2). NT-proBNP and all 48 proteins are expressed arbitrarily in linear normalized protein expression scale. PEA’s analytical quality in assessing proteins is rigorously validated regarding sensitivity, dynamic range, specificity, precision, and scalability. Panel and protein-specific validation documents can be found on www.olink.com/downloads.
Right heart catheterization
As a part of the clinical evaluation for HT, the patients’ hemodynamic profiles were characterized by cardiologists before and during the routine 1-year follow-up after HT by RHC, using a Swan-Ganz catheter (Baxter Health Care Corp, Santa Ana, CA, USA) inserted through the right internal jugular vein. Recorded parameters were systolic pulmonary arterial pressure (sPAP), diastolic PAP (dPAP), mean PAP (mPAP), mean right atrial pressure (MRAP), pulmonary arterial wedge pressure (PAWP), mixed venous oxygen saturation (SvO2), and arterial oxygen blood saturation (SaO2). Mean arterial pressure (MAP) was measured non-invasively and thermodilution was used to estimate cardiac output (CO).
Hemodynamic definitions
The other hemodynamic parameters were calculated with the following formulas: cardiac index (CI) = CO/BSA, stroke volume (SV) = CO/heart rate, stroke volume index (SVI) = SV/BSA, transpulmonary pressure gradient, (TPG) = mPAP − PAWP, pulmonary vascular resistance (PVR) = TPG/CO, PVR index (PVRI) = TPG/CI, diastolic pulmonary pressure gradient (DPG) = DPAP − PAWP, right-ventricular stroke work index, (RVSWI) = (mPAP − MRAP) × SVI, left-ventricular stroke work index (LVSWI) = (MAP − PAWP) × SVI, pulmonary arterial compliance, (PAC) = SV/(sPAP − dPAP), and arteriovenous oxygen difference (a − vO2diff) = (SaO2 − SvO2) × plasma hemoglobin × 1.34.
PH-LHD was defined by a resting mPAP ≥ 25 mmHg, PAWP > 15 mmHg and sub-classified into isolated post-capillary PH (DPG < 7 mmHg and/or PVR ≤ 3 WU) or combined post-capillary and pre-capillary PH (DPG ≥ 7 and/or PVR > 3 WU), according to current guidelines [10]. HT were performed at Skåne’s University Hospital in Lund, Sweden, according to the International Society for Heart and Lung Transplantation guidelines [23, 24].
Hemodynamic improvement
Hemodynamic data of patients before and 1-year after HT have previously been described [20, 21], with an additional subgroup description of patients with HF without PH (Table 2 and supplementary Table 1, respectively).
Statistics
Continuous data are presented as median (interquartile range). Distribution assumptions of normality were determined visually, using histrograms. As the data were non-Gaussian distributed, Wilcoxon signed-rank test and Mann–Whitney U test were used as appropriate. Correlation analysis of changes [∆, (Post-HT) − (Pre-HT)] was expressed by Spearman’s rank correlation coefficient (rs). The two-stage step-up procedure of false discovery rate (FDR) was used to adjust for mass significance [25] and p values less than attained thresholds were considered statistically significant. Q values were set at 0.01 for t tests and 0.1 for correlations. Statistical analyses were performed using Prism version 8.01 for Windows, GraphPad Software, La Jolla California USA, www.graphpad.com).
Study set-up
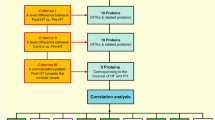
To identify plasma proteins reflecting the reversal of HF in response to HT, three criteria were set; (i) a significant plasma-level difference pre-HT vs. post-HT, (ii) a significant plasma-level difference in controls vs. pre-HT, and (iii) a plasma-level change of post-HT towards controls’ levels, resembling that of NT-proBNP, (FDR, Q = 0.01). Next, proteins reflecting a pattern consistent with the reversal of HF in response to HT were correlated with NT-proBNP and improved hemodynamic parameters of heart and pulmonary circulation, i.e., mPAP, MRAP, PAWP, PVR, PAC, CI, and LVSWI (FDR , Q = 0.1). Proteins correlating to several parameters were of particular interest; and a subgroup analysis between PH-LHD and HF without PH was performed for these proteins thereafter. The study set-up is summarized in Fig. 1.

Study set-up and biomarker selection. C control, HF heart failure, HT heart transplantation, PH pulmonary hypertension, Post-HT 1-year after HT, BOC brother of CDO, CEACAM1 carcinoembryonic antigen-related cell adhesion molecule 1, CPE carboxypeptidase E
Results
Plasma endocan, BOC, CPE, and kallikrein 11 in end-stage heart failure patients
The levels of tumour-related proteins in controls and patients at baseline and after HT are presented in Table 3 and supplementary Table 2 (FDR, Q = 0.01). In end-stage HF patients, plasma levels of endocan (Fig. 2a), BOC (Fig. 3a), CPE and kallikrein 11 were elevated compared to the controls (p < 0.01). After HT and reversal of HF and pre-existing PH, these levels decreased vs. pre-HT (p < 0.01), towards the controls’ levels, with normalization of BOC and CPE levels.

Plasma levels and correlations of endocan with hemodynamic changes following heart transplantation. Level changes (Δ) were calculated using values post-HT–pre-HT and outliers were defined with Tukey’s fence. HT heart transplantation, AU arbitrary units, rs Spearman’s correlation coefficient, mPAP mean pulmonary arterial pressure, PAWP pulmonary arterial wedge pressure, PVR pulmonary vascular resistance, WU wood units. *p < 0.01; **p < 0.001; ***p < 0.0001

Plasma levels and correlations of BOC with changes in hemodynamics following heart transplantation. Level changes (Δ) were calculated using values post-HT–pre-HT and outliers were defined with Tukey’s fence. HT heart transplantation, AU arbitrary units, rs Spearman’s correlation coefficient, BOC brother of CDO, NT-proBNP N-terminal pro b-type natriuretic peptide, MRAP mean right atrial pressure, CI cardiac index, LVSWI left-ventricular stroke work index. **p < 0.001; ***p < 0.0001
Plasma ∆endocan, ∆BOC, ∆CPE, and ∆kallikrein 11 correlate with hemodynamic changes following heart transplantation
Correlations of changes (∆) between proteins’ levels with ∆NT-proBNP and ∆hemodynamic parameters following HT are presented in Table 4. ∆endocan correlated with ∆mPAP, ∆PAWP, and PVR (Fig. 2b–d; p < 0.01). ∆BOC correlated with ∆NT-proBNP, ∆MRAP, ∆CI, and ∆LVSWI (Fig. 3b–e; p < 0.01). ∆CPE correlated with ∆mPAP, ∆PVR and ∆CI (p < 0.01). ∆kallikrein 11 correlated with ∆NT-proBNP and ∆MRAP (p < 0.01).
Other tumour-related proteins in end-stage heart failure patients
In end-stage HF patients, plasma levels of AP-N, CEACAM1, CPA1, CPB1, furin, kallikrein 14, mucin-16, and vimentin were elevated compared to controls (p < 0.01). After HT and reversal of HF and pre-existing PH, these levels decreased vs. pre-HT (p < 0.01), towards the controls’ levels. Conversely, in HF patients, plasma cornulin levels were low compared to controls (p < 0.0001), but increased after HT vs. pre-HT (p < 0.01), towards the controls’ levels (p < 0.001). ∆CEACAM1 correlated with CI, whereas ∆mucin-16 correlated with ∆NT-proBNP (p < 0.01). ∆AP-N, ∆CPA1, ∆CPB1, ∆furin, ∆kallikrein 14, ∆vimentin, and ∆cornulin did not correlate with changes in hemodynamics or NT-proBNP (Table 4).
Plasma endocan in PH-LHD patients following heart transplantation
A subgroup analysis of the plasma levels pre-HT and post-HT between HF without PH (n = 7) and PH-LHD patients (n = 19) was performed for BOC, CEACAM1, CPE, endocan, kallikrein 11, and mucin-16 (supplementary Table 3). Plasma endocan levels pre-HT were higher in PH-LHD compared to HF without PH group (p < 0.001). No differences were found in the other proteins.
Discussion
Developing a multi-marker panel reflecting different pathophysiological mechanisms underlying HF may be the future approach for individualized phenotyping and management of HF [9], and potentially PH-LHD. In the present study, we found that in end-stage HF patients, elevated endocan, BOC, CPE, and kallikrein 11 levels decreased after HT towards controls’ levels. Moreover, level changes of these proteins correlated with improved hemodynamics after HT. Our results suggest that endocan, BOC, CPE, and kallikrein 11 may reflect different pathophysiologic mechanisms and be potential biomarkers in HF and PH-LHD.
Endocan is a dermatan sulfate proteoglycan expressed by vascular endothelial cells, cardiomyocytes, and pulmonary capillaries. By virtue of its ability to interact with bioactive proteins, endocan regulates a wide range of biological processes including proliferation, neovascularization, and cellular adhesion [26]. Endocan has been implicated in vascular diseases, endothelium-dependent pathologies, and inflammatory processes including sepsis [27] and systemic sclerosis [28]. Also, endocan has been proposed as an indicator of endothelium activation [27] and dysfunction in septic patients [29]. Elevated circulating endocan levels has been reported in various conditions including hypertension and atherosclerosis [30] as well as in malignant lymphoma, renal cell carcinoma [26], and lung cancer [31].
Secondary to HF, malfunctioning and hemodynamically stressed cardiomyocytes result in cytokine release of tumour necrosis factor-α (TNF-α) and interleukin-1β, eliciting a sterile inflammation in the heart. As a result, cardiomyocyte apoptosis and hypertrophy, myofibroblast differentiation, and endothelial dysfunction ensue, leading to reduced myocardial perfusion, ventricular remodeling and subsequent progression of HF [32, 33]. Endothelial dysfunction, defined as an imbalance in the production of vasoactive substances, i.e., increased endothelin-1 expression and reduced nitric-oxide availability, plays a central role in the pathophysiology of HF [33], PH-LHD [34], and PAH [8] (Fig. 4a). Apart from being linked to cardiovascular risk factors, endothelial dysfunction predicts adverse clinical events, and its grade is analogous to the functional capacity and the severity of HF [33]. A recent study of chronic HF patients showed that apart from plasma endocan being elevated compared with healthy controls and patients with coronary artery disease, it emerged as an independent prognosticator of HF-related hospitalization and mortality [35]. In a rat model of PAH, endocan levels were elevated in the serum and lungs, and knockdown of endocan reversed monocrotaline-induced pulmonary vascular remodeling and reduced right-ventricular pressure. A subsequent in vitro experiment on rat pulmonary microvascular endothelial cells displayed the important interplay between TNF-α and endocan, as TNF-α upregulation induced endocan expression, whereas endocan inhibition prevented TNF-α-induced vascular permeability [36]. In the present study, we found that endocan levels were elevated in end-stage HF patients with or without PH-LHD compared to controls. After HT and reversal of HF and concomitant PH, endocan levels decreased towards controls’ levels. The following subgroup analysis revealed that plasma endocan is higher in patients with HF and PH-LHD compared to HF patients without PH, potentially suggesting endocan to be more PH-specific. Moreover, Δendocan correlated with ΔmPAP, ΔPAWP, and ΔPVR, reflecting the state of PH, passive pulmonary congestion as well as pulmonary vascular tone following HT, respectively. Taken together, elevated plasma endocan, may, theoretically have a role in endothelial dysfunction in HF patients with PH, as reflected by the correlation with PVR (Fig. 4b). It is also possible that endocan may be linked to, or involved in pulmonary vasoconstriction and pulmonary vascular remodeling, as endothelial dysfunction is a well-known trigger of these processes in PH-LHD [3]. Thus, it is encouraging to investigate the role of endocan, its interactions with TNF-α as well as its clinical implications as a biomarker of pulmonary congestion and potentially endothelial dysfunction in HF and PH-LHD.

Endothelial dysfunction in the progression of heart failure and pulmonary hypertension; and possible roles of plasma endocan and BOC. a Mechanisms involved in the progression of heart failure (HF) and pulmonary hypertension due to left heart disease (PH-LHD). In HF, along with cardiomyocyte hypertrophy and apoptosis, endothelial dysfunction leads to reduced myocardial perfusion and progression of HF. In PH-LHD, endothelial dysfunction may trigger excessive vasoconstriction and vascular remodeling. b Hypothetical mechanism of elevated plasma brother of CDO (BOC) in response to HF, i.e., whether activation of the Hedgehog (Hh) signaling augments the progression of HF, as well as possible (patho-)physiological and clinical roles of both BOC and endocan. PVR pulmonary vascular resistance
The hedgehog (Hh) signaling pathway is crucial in embryogenesis, organ development, as well as in adult tissue repair and homeostasis [37, 38]. Aberrant Hh signaling has emerged as an important pathway in human cancer, including basal cell carcinoma and medulloblastoma. BOC is a transmembrane co-receptor which through unknown molecular mechanisms enhances Hh pathway activity and facilitates Hh ligand binding to its receptor, Patched 1, which elicits responses in a dose-dependent manner [38]. In an adult murine model, Hh signaling was shown to be critical in the maintenance of coronary arteries, and ablation of Hh signaling resulted in coronary vasculature dropout, cardiomyocyte apoptosis, ventricular failure, and death. In an ensuing experiment, reduction of endogenous Hh signaling after myocardial infarction aggravated heart function and increased infarction size [39]. In another adult murine model, intramyocardial gene therapy with Shh—a Hh-specific ligand—after acute and chronic myocardial ischemia, resulted in preserved left-ventricular function by augmenting neovascularisation, as well as reducing fibrosis and cardiomyocyte apoptosis [40]. In the present study, plasma BOC levels were elevated in HF patients irrespective of PH compared to controls. These levels decreased and normalized upon the reversal of HF and PH after HT. The decrease of BOC following HT correlated with decreased NT-proBNP and MRAP as well as increased CI and LVSWI, reflecting decreased cardiac overload and improved cardiac function after HT. All in all, whether plasma BOC elevation is an endogenous response to counteract HF progression through enhancing Hh signaling reception and thereby augmenting cardiac tissue repair in chronic HF remains to be investigated (Fig. 4a, b).
CPE or enkephalin convertase is a member of metallocarboxypeptidase gene family and is most abundantly found in endocrine tissues, but also in heart and lungs [41]. CPE is involved in the biosynthesis of numerous prohormones and neurotransmitters. CPE is overly expressed in a variety of cancers including neuroendocrine tumours and small-cell lung carcinoma, and its abundance therein promotes neuropeptide biosynthesis, resulting in autocrine tumour growth [42]. In the fields of cardiovascular physiology and pathobiology, it has been proposed that CPE may be involved in atrial natriuretic peptide synthesis in rat hearts [43]. Moreover, a series of studies of Chinese patients found that specific CPE gene polymorphisms may be linked to increased severity of coronary atherosclerosis [44,45,46]. In the present study, plasma CPE levels were elevated before HT compared to controls, which thereafter decreased after HT towards controls’ levels. Plasma-level changes in CPE correlated with changes in mPAP, PVR, and CI. Whether these associations infer causality between CPE, HF, and pulmonary vascular disease remains to be further elaborated.
Kallikrein 11 is a member of soluble serine proteases and is regulated in a steroid hormone-dependent manner. It is highly expressed in human prostatic and tracheal tissues, but also present in lungs and serum [47]. Despite several studies addressing its potential diagnostic or prognostic properties in prostate, ovarian [47], and lung cancer [48], the precise physiological function of kallikrein 11 remains largely unknown. In assessing the enzymatic function and its physiological substrates, unlike some kallikreins, kallikrein 11 is incapable of cleaving kininogen [49], an activator of the kallikrein–kinin system that has been implicated in left-ventricular dysfunction [50]. Instead, it cleaves and degrades insulin-like growth factor-binding protein 3 (IGFBP3) [51]. As a carrier protein, IGFBP3 extends the half-life of IGFs, and upon the cleavage of the IGF–IGFBP3 complex, IGFs are released to bind and activate IGF-1 receptor signaling [52]. In dilated cardiomyopathy, elevated IGFBP3 and IGF-1 mRNA tissue expressions have been reported in comparison to controls [53]. Moreover, IGF-1 signaling has been implicated in cardiac ageing and dysfunction [54]. Another study reported that elevated plasma IGF-1 in HF patients appeared to be associated with angiotensin-converting enzyme inhibitor (ACEi) treatment and increased risk of cardiovascular mortality [55]. Herein, kallikrein 11 plasma levels were elevated in advanced HF patients in comparison to controls. These levels decreased after HT matching controls’ levels. Furthermore, a decrease in plasma kallikrein 11 correlated with a decrease in NT-proBNP and MRAP, reflecting an alleviated cardiac hemodynamic overload. Hypothetically, elevated circulating kallikrein 11 in HF may have a role in promoting cardiac ageing and accelerating ventricular dysfunction through increasing the bioactivity of IGF-1. Hence, the role of plasma kallikrein 11 in HF warrants further investigation.
Moreover, plasma CEACAM1 and mucin-16 levels were elevated in HF patients, with these levels decreasing after HT towards controls’ levels. The decrease of CEACAM1 and mucin-16 correlated with improved CI and NT-proBNP, respectively, supporting the previously reported association between mucin-16 and volume overload [18]. Intriguingly, a study showed that CEACAM1 upregulation after hypoxic cardiomyocyte injury promoted unfavorable cardiac remodeling by inducing apoptosis [56]. Whether elevated CEACAM1 levels play a role in the chronic progression of HF remains to be investigated.
Strengths and limitations
Although concordant with the size of other studies, the relatively small population and the lack of validation cohorts constitute limitations. Despite the inability to provide absolute protein concentrations, tissue-specific expression and differentiation of protein isoforms, PEA, compared to conventional multiplex immunoassays, warrant high specificity and sensitivity [22], which is crucial in the process of identifying biomarker candidates for future clinical utility. Thus, the use of PEA and the invasive hemodynamic measurements constitutes major strengths in our study. Noteworthy is, however, that the present study is hypothesis generating and our results do not necessarily imply causality. Hence, our results do not allow for definite mechanistical conclusions. Factors including comorbidities, age and sex disparities, diurnal variations, and medication intake may have affected the proteins’ levels. Although it is well established that β-blockers improve left-ventricular function [57] and ACEi increase CO and attenuate left-ventricular wall stress [58], their postoperative withdrawal effects remain unknown. Analogously, antihypertensive and HF-specific medications including ACEi, angiotensin II receptor blockers, and calcium channel antagonists attenuate vascular inflammation and/or endothelial dysfunction [59], specifically valsartan and amlodipine, which may affect plasma endocan levels [27]. Conversely, first- and second-generation β-blockers and diuretics have not been shown to affect inflammation. Moreover, the role of diuretics in endothelial function remains unknown [59]. Although the effects of immunosuppressants on plasma protein levels have not been investigated in the present study, calcineurin and mTOR inhibitors are associated with endothelial dysfunction and increased risk of cardiovascular morbidity [60], whereas mycophenolate mofetil reduces immune-mediated vascular injury and possibly exert positive effects on endothelial function [61]. Furthermore, given the large number of statistical tests conducted, false-positive results may be present, even though FDR was used to accommodate for mass significance. Larger studies are necessary to confirm and validate our findings.
Conclusions
In the present study, we identified the tumour-related proteins endocan, BOC, kallikrein 11, CPE, CEACAM1, and mucin-16 in end-stage HF patients before and after HT. Specifically, the decrease of high plasma levels of endocan in HF patients after HT was associated with improved mPAP, PAWP, and PVR. Moreover, the decrease after HT of elevated BOC levels was associated with decreased MRAP and NT-proBNP, as well as increased CI and LVSWI. Our results suggest that endocan may be a potential biomarker reflecting the state of PH, pulmonary congestion, and potentially endothelial dysfunction in HF and PH-LHD. Additionally, plasma BOC may be a biomarker candidate, potentially reflecting the hemodynamic overload and heart function in HF, irrespective of concomitant PH. The exact roles of endocan and BOC as well as their potential clinical applicability in HF and PH-LHD remain to be elaborated in future studies.
References
Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JGF, Coats AJS, Falk V, González-Juanatey JR, Harjola VP, Jankowska EA, Jessup M, Linde C, Nihoyannopoulos P, Parissis JT, Pieske B, Riley JP, Rosano GMC, Ruilope LM, Ruschitzka F, Rutten FH, van der Meer P, ESC Scientific Document Group (2016) 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: the Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC)Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J 37:2129–2200
Heggermont WA, Papageorgiou A-P, Heymans S, van Bilsen M (2016) Metabolic support for the heart: complementary therapy for heart failure? Eur J Heart Fail 18:1420–1429
Vachiéry J-L, Adir Y, Barberà JA, Champion H, Coghlan JG, Cottin V, De Marco T, Galiè N, Ghio S, Gibbs JSR, Martinez F, Semigran M, Simonneau G, Wells A, Seeger W (2013) Pulmonary hypertension due to left heart diseases. J Am Coll Cardiol 62:D100–D108
Fang JC, DeMarco T, Givertz MM, Borlaug BA, Lewis GD, Rame JE, Gomberg-Maitland M, Murali S, Frantz RP, McGlothlin D, Horn EM, Benza RL (2012) World Health Organization Pulmonary Hypertension Group 2: pulmonary hypertension due to left heart disease in the adult—a summary statement from the Pulmonary Hypertension Council of the International Society for Heart and Lung Transplantation. J Heart Lung Transplant 31:913–933
Braunwald E (2008) Biomarkers in heart failure. N Engl J Med 358:2148–2159
Ibrahim NE, Januzzi JL (2017) Beyond natriuretic peptides for diagnosis and management of heart failure. Clin Chem 63:211–222
Magnussen C, Blankenberg S (2018) Biomarkers for heart failure: small molecules with high clinical relevance. J Intern Med 283:530–543
Humbert M, Guignabert C, Bonnet S, Dorfmüller P, Klinger JR, Nicolls MR, Olschewski AJ, Pullamsetti SS, Schermuly RT, Stenmark KR, Rabinovitch M (2019) Pathology and pathobiology of pulmonary hypertension: state of the art and research perspectives. Eur Respir J 53:1801887
Ibrahim NE, Januzzi JL Jr (2018) Established and emerging roles of biomarkers in heart failure. Circ Res 123:614–629
Galiè N, Humbert M, Vachiery J-L, Gibbs S, Lang I, Torbicki A, Simonneau G, Peacock A, Vonk Noordegraaf A, Beghetti M, Ghofrani A, Gomez Sanchez MA, Hansmann G, Klepetko W, Lancellotti P, Matucci M, McDonagh T, Pierard LA, Trindade PT, Zompatori M, Hoeper M, ESC Scientific Document Group (2015) 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: the Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J 37:67–119
Sakamoto M, Hasegawa T, Asakura M, Kanzaki H, Takahama H, Amaki M, Mochizuki N, Anzai T, Hamasaki T, Kitakaze M (2017) Does the pathophysiology of heart failure prime the incidence of cancer? Hypertens Res 40:831–836
Hasin T, Gerber Y, McNallan SM, Weston SA, Kushwaha SS, Nelson TJ, Cerhan JR, Roger VL (2013) Patients with heart failure have an increased risk of incident cancer. J Am Coll Cardiol 62:881–886
Chen Z, Liu M, Li L, Chen L (2018) Involvement of the Warburg effect in non-tumor diseases processes. J Cell Physiol 233:2839–2849
Du Bois D, Du Bois EF (1916) Clinical calorimetry: tenth paper a formula to estimate the approximate surface area if height and weight be known. Arch Intern Med 17:863–871
Boucherat O, Vitry G, Trinh I, Paulin R, Provencher S, Bonnet S (2017) The cancer theory of pulmonary arterial hypertension. Pulm Circ 7(2):285–299
Mizuno S, Bogaard HJ, Kraskauskas D, Alhussaini A, Gomez-Arroyo J, Voelkel NF, Ishizaki T (2011) p53 Gene deficiency promotes hypoxia-induced pulmonary hypertension and vascular remodeling in mice. Am J Physiol Lung Cell Mol Physiol 300:L753–L761
Mouraret N, Marcos E, Abid S, Gary-Bobo G, Saker M, Houssaini A, Dubois-Rande JL, Boyer L, Boczkowski J, Derumeaux G, Amsellem V, Adnot S (2013) Activation of lung p53 by Nutlin-3a prevents and reverses experimental pulmonary hypertension. Circulation 127:1664–1676
Li KHC, Gong M, Li G, Baranchuk A, Liu T, Wong MCS, Jesuthasan A, Lai RWC, Lai JCL, Lee APW, Bayes-Genis A, de la Espriella R, Sanchis J, Wu WKK, Tse G, Nunez J (2018) Cancer antigen-125 and outcomes in acute heart failure: a systematic review and meta-analysis. Heart Asia 10(2):e011044
Lund LH, Edwards LB, Kucheryavaya AY, Dipchand AI, Benden C, Christie JD, Dobbels F, Kirk R, Rahmel AO, Yusen RD, Stehlik J (2013) The registry of the international society for heart and lung transplantation: thirtieth official adult heart transplant report—2013; focus theme: age. J Heart Lung Transplant 32:951–964
Ahmed S, Ahmed A, Säleby J, Bouzina H, Lundgren J, Rådegran G (2020) Elevated plasma tyrosine kinases VEGF-D and HER4 in heart failure patients decrease after heart transplantation in association with improved haemodynamics. Heart Vessels 35:786–799
Ahmed A, Ahmed S, Arvidsson M, Bouzina H, Lundgren J, Rådegran G (2020) Prolargin and matrix metalloproteinase-2 in heart failure after heart transplantation and their association with haemodynamics. ESC Heart Failure 7:223–234
Assarsson E, Lundberg M, Holmquist G, Björkesten J, Bucht Thorsen S, Ekman D, Eriksson A, Rennel Dickens E, Ohlsson S, Edfeldt G, Andersson AC, Lindstedt P, Stenvang J, Gullberg M, Fredriksson S (2014) Homogenous 96-Plex PEA immunoassay exhibiting high sensitivity, specificity, and excellent scalability. PLoS ONE 9:e95192
Mehra MR, Kobashigawa J, Starling R, Russell S, Uber PA, Parameshwar J, Mohacsi P, Augustine S, Aaronson K, Barr M (2006) Listing criteria for heart transplantation: international society for heart and lung transplantation guidelines for the care of cardiac transplant candidates—2006. J Heart Lung Transplant 25:1024–1042
Mehra MR, Canter CE, Hannan MM, Semigran MJ, Uber PA, Baran DA, Danziger-Isakov L, Kirklin JK, Kirk R, Kushwaha SS, Lund LH, Potena L, Ross HJ, Taylor DO, Verschuuren EAM, Zuckermann A (2016) The 2016 international society for heart lung transplantation listing criteria for heart transplantation: a 10-year update. J Heart Lung Transplant 35:1–23
Benjamini Y, Krieger AM, Yekutieli D (2006) Adaptive linear step-up procedures that control the false discovery rate. Biometrika 93:491–507
Zhang SM, Zuo L, Zhou Q, Gui SY, Shi R, Wu Q, Wei W, Wang Y (2012) Expression and distribution of endocan in human tissues. Biotech Histochem 87:172–178
Balta S, Mikhailidis DP, Demirkol S, Ozturk C, Celik T, Iyisoy A (2015) Endocan: a novel inflammatory indicator in cardiovascular disease? Atherosclerosis 243:339–343
Bălănescu P, Lădaru A, Bălănescu E, Voiosu T, Băicuş C, Dan GA (2016) Endocan, novel potential biomarker for systemic sclerosis: results of a pilot study. J Clin Lab Anal 30:368–373
Scherpereel A, Depontieu F, Grigoriu B, Cavestri B, Tsicopoulos A, Gentina T, Jourdain M, Pugin J, Tonnel A-B, Lassalle P (2006) Endocan, a new endothelial marker in human sepsis*. Crit Care Med 34:532–537
Wang X-s, Yang W, Luo T, Wang J-m, Jing Y-y (2015) Serum endocan levels are correlated with the presence and severity of coronary artery disease in patients with hypertension. Genet Test Mol Biomark 19:124–127
Grigoriu BD, Depontieu F, Scherpereel A, Gourcerol D, Devos P, Ouatas T, Lafitte J-J, Copin M-C, Tonnel A-B, Lassalle P (2006) Endocan expression and relationship with survival in human non-small cell lung cancer. Clin Cancer Res 12:4575
Van Linthout S, Tschöpe C (2017) Inflammation—cause or consequence of heart failure or both? Curr Heart Fail Rep 14:251–265
Marti CN, Gheorghiade M, Kalogeropoulos AP, Georgiopoulou VV, Quyyumi AA, Butler J (2012) Endothelial dysfunction, arterial stiffness, and heart failure. J Am Coll Cardiol 60:1455–1469
Breitling S, Ravindran K, Goldenberg NM, Kuebler WM (2015) The pathophysiology of pulmonary hypertension in left heart disease. Am J Physiol Lung Cell Mol Physiol 309:L924–L941
Kosir G, Jug B, Novakovic M, Mijovski MB, Ksela J (2019) Endocan is an independent predictor of heart failure-related mortality and hospitalizations in patients with chronic stable heart failure. Dis Mark 2019:9134096
Zhao H, Xue Y, Guo Y, Sun Y, Liu D, Wang X (2017) Inhibition of endocan attenuates monocrotaline-induced connective tissue disease related pulmonary arterial hypertension. Int Immunopharmacol 42:115–121
Petrova R, Joyner AL (2014) Roles for Hedgehog signaling in adult organ homeostasis and repair. Development 141:3445–3457
Yao E, Chuang P-T (2015) Hedgehog signaling: from basic research to clinical applications. J Formos Med Assoc 114(7):569–576
Lavine KJ, Kovacs A, Ornitz DM (2008) Hedgehog signaling is critical for maintenance of the adult coronary vasculature in mice. J Clin Invest 118(7):2404–2414
Kusano KF, Pola R, Murayama T, Curry C, Kawamoto A, Iwakura A, Shintani S, Ii M, Asai J, Tkebuchava T, Thorne T, Takenaka H, Aikawa R, Goukassian D, von Samson P, Hamada H, Yoon Y-S, Silver M, Eaton E, Ma H, Heyd L, Kearney M, Munger W, Porter JA, Kishore R, Losordo DW (2005) Sonic hedgehog myocardial gene therapy: tissue repair through transient reconstitution of embryonic signaling. Nat Med 11(11):1197–1204
Fricker LD (1988) Carboxypeptidase E. Annu Rev Physiol 50:309–321
Cawley NX, Wetsel WC, Murthy SRK, Park JJ, Pacak K, Loh YP (2012) New roles of carboxypeptidase E in endocrine and neural function and cancer. Endocr Rev 33(2):216–253
Lynch DR, Venable JC, Snyder SH (1988) Enkephalin convertase in the heart: similar disposition to atrial natriuretic factor. Endocrinology 122(6):2683–2691
Jia E-Z, Wang J, Yang Z-J, Zhu T-B, Wang L-S, Wang H, Li C-J, Chen BO, Cao K-J, Huang J, Ma W-Z (2009) Association of the mutation for the human carboxypeptidase E gene exon 4 with the severity of coronary artery atherosclerosis. Mol Biol Rep 36(2):245–254
Wang J, Zhang Y, Yang Z-J, Zhu T-b, Wang L-s, Chen B, Cao K-j, Huang J, Ma W-z, Jia E-z (2008) Association of human carboxypeptidase E exon5 gene polymorphisms with angiographical characteristics of coronary atherosclerosis in a Chinese population. Acta Pharmacol Sin 29:736–744
Jia E-Z, Wang J, Yang Z-J, Zhu T-B, Wang L-S, Chen BO, Cao K-J, Huang J, Ma W-Z (2008) Molecular scanning of the human carboxypeptidase E gene for mutations in Chinese subjects with coronary atherosclerosis. Mol Cell Biochem 307(1–2):31–39
Diamandis EP, Okui A, Mitsui S, Luo LY, Soosaipillai A, Grass L, Nakamura T, Howarth DJ, Yamaguchi N (2002) Human kallikrein 11: a new biomarker of prostate and ovarian carcinoma. Cancer Res 62:295–300
Unal D, Tasdemir A, Oguz A, Eroglu C, Cihan YB, Turak EE, Karaman H, Soyuer S (2013) Is human kallikrein-11 in gastric cancer treated with surgery and adjuvant chemoradiotherapy associated with survival? Pathol Res Pract 209:779–783
Luo L-Y, Shan SJC, Elliott MB, Soosaipillai A, Diamandis EP (2006) Purification and characterization of human kallikrein 11, a candidate prostate and ovarian cancer biomarker, from seminal plasma. Clin Cancer Res 12:742–750
Wei C-C, Chen Y, Powell LC, Zheng J, Shi K, Bradley WE, Powell PC, Ahmad S, Ferrario CM, Dell'Italia LJ (2012) Cardiac kallikrein-kinin system is upregulated in chronic volume overload and mediates an inflammatory induced collagen loss. PLoS ONE 7:e40110
Sano A, Sangai T, Maeda H, Nakamura M, Hasebe T, Ochiai A (2007) Kallikrein 11 expressed in human breast cancer cells releases insulin-like growth factor through degradation of IGFBP-3. Int J Oncol 30:1493–1498
Hoeflich A, David R, Hjortebjerg R (2018) Current IGFBP-related biomarker research in cardiovascular disease-we need more structural and functional information in clinical studies. Front Endocrinol (Lausanne) 9:388
Pucci A, Zanini C, Granata R, Ghignone R, Iavarone A, Broglio F, Sorrentino P, Bergamasco L, Rinaldi M, Ghigo E (2009) Myocardial insulin-like growth factor-1 and insulin-like growth factor binding protein-3 gene expression in failing hearts harvested from patients undergoing cardiac transplantation. J Heart Lung Transplant 28:402–405
Lee W-S, Kim J (2018) Insulin-like growth factor-1 signaling in cardiac aging. Biochim Biophys Acta Mol Basis Dis 1864:1931–1938
Chisalita SI, Dahlström U, Arnqvist HJ, Alehagen U (2011) Increased IGF1 levels in relation to heart failure and cardiovascular mortality in an elderly population: impact of ACE inhibitors. Eur J Endocrinol 165:891–898
Wang Y, Chen Y, Yan Y, Li X, Chen G, He N, Shen S, Chen G, Zhang C, Liao W, Liao Y, Bin J (2016) Loss of CEACAM1, a tumor-associated factor, attenuates post-infarction cardiac remodeling by inhibiting apoptosis. Sci Rep 6:21972
Hjalmarson Å, Goldstein S, Fagerberg B, Wedel H, Waagstein F, Kjekshus J, Wikstrand J, El Allaf D, Vítovec J, Aldershvile J, Halinen M, Dietz R, Neuhaus K-L, Jánosi A, Thorgeirsson G, Dunselman PHJM, Gullestad L, Kuch J, Herlitz J, Rickenbacher P, Ball S, Gottlieb S, Deedwania P, Group ftM-HS (2000) Effects of controlled-release metoprolol on total mortality, hospitalizations, and well-being in patients with heart failure the metoprolol CR/XL randomized intervention trial in congestive heart failure (MERIT-HF). JAMA 283:1295–1302
López-Sendón J, Swedberg K, McMurray J, Tamargo J, Maggioni AP, Dargie H, Tendera M, Waagstein F, Kjekshus J, Lechat P, Pedersen CT, Members TF (2004) Expert consensus document on angiotensin-converting enzyme inhibitors in cardiovascular disease: the Task Force on ACE inhibitors of the European Society of Cardiology. Eur Heart J 25:1454–1470
Silva IVG, de Figueiredo RC, Rios DRA (2019) Effect of different classes of antihypertensive drugs on endothelial function and inflammation. Int J Mol Sci 20(14):3458
Shing CM, Fassett RG, Brown L, Coombes JS (2012) The effects of immunosuppressants on vascular function, systemic oxidative stress and inflammation in rats. Transpl Int 25:337–346
Fréguin-Bouilland C, Godin M, Bellien J, Richard V, Remy-Jouet I, Dautreaux B, Henry J-P, Compagnon P, Thuillez C, Plissonnier D, Joannidès R (2011) Protective effect of mycophenolate mofetil on endothelial function in an aortic allograft model. Transplantation 91(1):35–41
Nyman U, Grubb A, Larsson A, Hansson LO, Flodin M, Nordin G, Lindström V, Björk J (2014) The revised Lund-Malmö GFR estimating equation outperforms MDRD and CKD-EPI across GFR, age and BMI intervals in a large Swedish population. Clin Chem Lab Med 52:815–824
Acknowledgements
Open access funding provided by Lund University. We acknowledge the support of the Hemodynamic Laboratory staff, The Section for Heart Failure and Valvular Disease, Skåne University Hospital, Lund, Sweden; and The Section for Cardiology, the Department of Clinical Sciences, Lund University, Lund, Sweden. We also acknowledge the biobank services and maintaining facility, blood sample storage, and retrieval from LCPR at Labmedicine Skåne, University and Regional Laboratories, Region Skåne, Sweden. Exceptional gratitude to Anneli Ahlqvist for her efforts in LCPR administration and blood sample management.
Funding
The work was supported by unrestricted research grants from Avtal om Läkarutbildning och Forskning (ALF) and Actelion Pharmaceuticals Sweden AB. The funding organizations played no role in the collection, analysis, or interpretation of the data, and had no right to restrict the publishing of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Mr. Salaheldin Ahmed and Mr. Abdulla Ahmed report no conflicts of interest. Mr. Bouzina reports an unrestricted research grant from The Swedish Society of Pulmonary Hypertension on behalf of GlaxoSmithKline. Dr. Lundgren reports unrestricted research grants from The Swedish Society of Pulmonary Hypertension on behalf of Actelion Pharmaceuticals Sweden AB. Dr. Rådegran reports unrestricted research grants from ALF and Actelion Pharmaceuticals Sweden AB, during the conduct of the study. Mr. Abdulla Ahmed and Mr. Salaheldin Ahmed report no personal lecture fees. Mr. Bouzina reports personal lecture fees from Actelion Pharmaceuticals Sweden AB outside the submitted work. Dr. Lundgren reports personal lecture fees from Actelion Pharmaceuticals Sweden AB and GlaxoSmithKline outside the submitted work. Dr. Rådegran reports personal lecture fees from Actelion Pharmaceuticals Sweden AB, GlaxoSmithKline, Bayer, Nordic Infucare, and Bayer Health Care outside the submitted work. Dr. Rådegran is, and has been primary-, or co-, investigator in; clinical PAH trials for GlaxoSmithKline, Actelion Pharmaceuticals Sweden AB, Pfizer, Bayer and United Therapeutics, and in clinical heart transplantation immuno-suppression trials for Novartis.The companies had no role in the data collection, analysis, and interpretation, and had no right in disapproving of the manuscript.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ahmed, S., Ahmed, A., Bouzina, H. et al. Elevated plasma endocan and BOC in heart failure patients decrease after heart transplantation in association with improved hemodynamics. Heart Vessels 35, 1614–1628 (2020). https://doi.org/10.1007/s00380-020-01656-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00380-020-01656-3