
Abstract
Perovskite oxides are an extremely versatile class of materials in which functionality can, besides other routes, also be engineered via the deliberate introduction of defects. In this focused review, we will specifically look at mechanistic details of ferroelectric and magnetic functionality introduced, altered, or reinforced by point defects. An ever-growing number of related studies start to provide a basis for the mechanistic understanding of different engineering routes to be exploited in future studies. Nevertheless, this review highlights that the effect of defects is not always easily predicted, given the delicate balance of lattice, charge, spin, and orbital degrees of freedom inherent to the perovskite structure. Systematic studies across various chemistries are thus still very much needed to obtain a more complete basis for defect-engineering ferroelectric and magnetic functionality in perovskite oxides.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
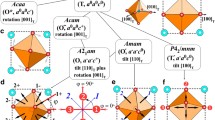
Oxides with the ABO3 perovskite crystal structure (see Fig. 1a) are a chemically extremely diverse class of materials as many elements of the periodic table can be accommodated on the A and B sites [1]. The class of materials is named after the parent compound CaTiO3, other prototypical material examples being SrTiO3, BaTiO3, or PbTiO3. In all perovskites, the B site is octahedrally coordinated with O anions, while the A site is in dodecahedral coordination (see Fig. 1a). The perovskite structure is generally stable when the ionic radii fulfill the criteria of the Goldschmidt tolerance factor

a Perovskite oxide structure, highlighting the octahedrally coordinated B-site and the A-site in dodecahedral coordination, b perovskite structure with octahedral rotations and c perovskite structure with B-site off-centering
which is based on a balance of the anion (\(r_{{\text{O}}}\)) and cation (\(r_{{\text{A}}}\) and \(r_{{\text{B}}}\)) radii to fit both the unit-cell edge (numerator) and diagonal (denominator) [2]. Generally, compositions with a smaller t (i.e., a small A site compared to the B site) will be prone to undergo antiferrodistortive octahedral rotations (see Fig. 1b), while in those with t near unity, the B site may move away from the center of the coordination octahedron (see Fig. 1c). B-site off-centering may additionally also be induced by the chemistry, for example in PbTiO3 due to the presence of lone pairs on the A site [3].
This off-center distortion is of technological relevance as the relative displacement of anions and cations induces a local electrical dipole. If coupled parallel with neighboring dipoles, this leads to polar materials than can be ferroelectric if the dipole direction is switchable by an applied electric field [4, 5]. An antiparallel coupling, at least between some neighboring dipoles, on the other hand, will lead to antiferroelectricity [6]. Ferroelectricity and antiferroelectricity are two functional properties of great relevance. While a ferroelectric can be locally poled to store digital information as a function of the polarization direction (FE-RAM) [4], antiferroelectrics are highly relevant for energy storage [7].
Perovskite oxides may also present magnetic properties, most commonly when the B site has a partially filled d-electron shell. A-site magnetism usually derives from rare-earth elements and, due to the weaker exchange interactions, is limited to very low temperatures. Depending on the exchange interaction between the magnetic sites, the material may assume antiferromagnetic, ferromagnetic, or ferrimagnetic spin configurations [8,9,10]. Apart from (anti)ferroelectricity and magnetism, perovskite oxides may also exhibit functional properties such as superconductivity [11], magnetoresistance [12], or (photo)catalytic activity [13], which we will, however, not focus on here.
Looking at the enormous compositional flexibility, it is surprising how few materials exhibit the above-mentioned functional properties. These properties also do not often co-exist [14], magnetoelectric multiferroicity (e.g., coexisting ferroelectricity and magnetism) naturally occurring at room temperature for just a few compositions such as the well-studied BiFeO3 [15].
Engineering perovskite oxides to induce or reinforce functional properties has therefore been an active field of research. The most established engineering approach is coherent epitaxial thin-film growth during which the perovskite material is strained to adopt the lattice parameters of the substrate [16]. This deformation changes the unit-cell geometry, symmetry, and interatomic interactions and may lead to emerging ferroelectricity in nominally non-polar materials [17]. Another approach to engineering properties is by alloying on either of the two cation sites. As such, adding Ba to SrTiO3 is known to promote ferroelectricity [18]. Magnetic properties may also be engineered in this way by aliovalent substitution. The LaMnO3–SrMnO3 solid solution is a prominent example where the two end members are antiferromagnets, while changes in the Mn oxidation state due to the aliovalent A site substitution result in ferromagnetism for intermediate compositions [19].
Another, less-explored route to engineer functional properties is by deliberately inducing point defects or point-defect clusters. Point defects can be missing atoms (vacancies), additional atoms (interstitials), or replaced atoms (substitutions), while clusters refer to aggregates of these individual defects (see Fig. 2a). The stability (i.e., formation energy) and therefore the relative abundance of each type of defect depends on the environment (i.e., elemental chemical potentials) [20,21,22]. While point defects are referred to as zero-dimensional (0D) defects, defects may also occur in higher dimensions such as 1D line defects (e.g., dislocations) or 2D planar defects (e.g., grain boundaries, surfaces, or interfaces), which we will, however, not focus on here.

a Types of point defects: anion and cation vacancies, interstitial (anion), substitution (cation) as well as point defects clusters (depicted are Schottky and Frenkel pairs as well as a substitution-anion vacancy pair). Electronic structure changes compared to b the stoichiometric structure for donor and acceptor defects when c the charge is delocalized, d localized, or e traps states exist
A defect in an ionic or mixed covalent-ionic material will lead to electron excess or deficiency when neutral atoms are removed. In the most typical situation, this leads to delocalized free electrons or holes in the conduction and valence band, respectively (see Fig. 2b). In addition, as the bonding around a defect is altered, states will shift to different energies. In a semiconductor or insulator, these states may appear in the band gap and become preferential sites for excess charge localization (see Fig. 2c). These localized occupied defect states, when located close to either the conduction or valence band edge are referred to as shallow donor and shallow acceptor states, respectively, as thermal excitation from these states to the conduction or valence band, respectively, leads to free carriers. Depending on the crystal chemistry, these states can also be unoccupied or can become unoccupied as a defect assumes a different charge state by electron exchange with a reservoir [20,21,22]. In such situations, the state can accommodate free electrons or holes and is referred to as a trap state (see Fig. 2d).
1 Ferroelectricity induced by defects
1.1 Defect pairs
Defects in ferroelectrics were long considered to be avoided as they are related to degradation phenomena such as leakage, fatigue, and aging [23,24,25,26,27,28]. More recently, it was recognized that the defect chemistry can be used to tune ferroelectric properties. Most studies considered defect pairs such as \(\left[ {{\text{Fe}}_{{{\text{Ti}}}}^{^{\prime}} {\text{V}}_{{\text{O}}}^{ \cdot \cdot } } \right]^{ \cdot }\) in perovskite titanates, using Kröger-Vink notation [29]. These defect pairs involving cation substitution and oxygen vacancies are known to bind, whereas those involving cation substitution and cation vacancies (e.g., \(\left[ {{\text{Nb}}_{{{\text{Ti}}}}^{ \cdot } {\text{V}}_{{{\text{Pb}}}}^{{^{\prime\prime}}} } \right]^{^{\prime}}\) will dissociate [30]. These defect pairs carry a defect dipole between the positively and the negatively charged defect entity (see Fig. 3a) that preferentially aligns with the ferroelectric polarization as shown for the example of a \(\left[ {{\text{Fe}}_{{{\text{Ti}}}}^{^{\prime}} {\text{V}}_{{\text{O}}}^{ \cdot \cdot } } \right]^{ \cdot }\) in PbTiO3 in Fig. 3b. Due to their dipolar field, two interacting dipoles will align parallel, when axially aligned but antiparallel when laterally aligned. In a ferroelectric, the axial dipole–dipole interactions dominate over the lateral one, favoring an overall parallel dipole alignment. A defect dipole embedded in such a dipole lattice will also most favorably be aligned parallel with the ferroelectric dipoles. As such it was demonstrated that defects pairs reorient to align with the ferroelectric polarization direction in materials such as PbTiO3 [30,31,32], BaTiO3 [33], KNbO3 [33], and BiFeO3 [27] upon cooling as schematically shown in Fig. 3c. The defect–dipole reorientation must involve migration, usually of substitutional cation defects or oxygen vacancies. The latter was shown to be energetically significantly more facile (sub-eV migration energies) than cation migration (multi-eV barriers) [34,35,36,37]. It should nevertheless be noted that, in defect clusters, the presence of oxygen vacancies can significantly affect migration pathways due to A-site-oxygen bonding optimization leading to a reduction in cation migration energies [34].

a Schematic of a defect-pair dipole formed between a substitutional Fe atom on a Ti site with a relative charge of − 1 and an oxygen vacancy with a relative charge of + 2 in PbTiO3, b Energetics of different alignments of the defect dipole (dashed white arrow) with the lattice polarization \(\vec{P}\) reprinted with permission from Ref. [30], Copyright 2013 by the American Physical Society, c Schematic 90° domain structure (black arrows) in which defect dipoles (white arrows) align at low temperatures
Once formed, a state with ordered defect dipoles can pin a certain ferroelectric polarization direction. This effect may require fairly high defect concentrations, as in perovskite manganites the polarizing effect of both \(\left[ {{\text{V}}_{Sr}^{^{\prime\prime}} {\text{V}}_{O}^{ \cdot \cdot } } \right]^{{\text{x}}}\) and \(\left[ {{\text{Fe}}_{Mn}^{^{\prime}} {\text{V}}_{O}^{ \cdot \cdot } } \right]^{{\text{X}}}\) dipoles was shown to be limited to within a radius of around 6 Å around the defect [37, 38]. Nevertheless, multiscale simulations revealed a longer-range effect [39], which could be due to periodic boundary condition effects in density functional theory or due to the different chemistry (manganite vs. titanate). Since many defect pairs not only carry an electrical dipole but also an elastic dipole, the ordering of the dipoles with external strain, applied for example via heteroepitaxial growth on a substrate with a different lattice parameter, can be used to impose a preferred defect–dipole orientation via electromechanical coupling [39]. This effect can increase the ferroelectric critical temperature, for example in BaTiO3 from the stoichiometric value of 123 °C [40] up to 800 °C [41].
1.2 Defect clusters
Not only defect pairs but also clusters of more than two defects may result in an electric dipole. As such, it was shown, based on a comparison of density functional theory calculations and spectroscopic experiments, that in SrTiO3 the occurrence of a \({\text{V}}_{{{\text{Sr}}}} - {\text{V}}_{{\text{O}}} - {\text{V}}_{{\text{O}}}\) trivacancy cluster is associated with the emergence of ferroelectricity [42]. In this case, many different relative arrangements of the various individual defects are possible, each having a different net polarization direction.
1.3 Single defects
While defect pairs and defect clusters require a high level of control of the synthesis conditions to form, super- or substoichiometry of a single element, leading to the formation of point defects associated with that element, promise to be easier to control. Indeed, also single point defects can promote ferroelectric properties. As such, cation antisite defects (substitution of Ti on the Sr site or Sr on the Ti site) lead to polar displacements in SrTiO3, TiSr more strongly so [36, 43, 44]. The mechanism behind the emerging polarization is a bonding optimization of the cation occupying a too-large site, resulting in off-centering within the coordination polyhedron (see Fig. 4). In this case, clustering of the antisites with oxygen vacancies should be avoided as a reduction in polarization and an increase in switching barrier result [36]. A similar effect was reported in the A-site double perovskite EuBaTi2O6, where oxygen vacancies lead to a Ti off-centering, in addition to ferromagnetic Eu2+–Ti3+–Eu2+ exchange, resulting in a ferroelectric–ferromagnetic multiferroic material [45].

a TiSr antisite defect off-centered along [001] and [110] as well as b the energy profile associated with migration between these positions, showing that the [100] direction is most favorable. Reprinted figure with permission from Ref. [36] Copyright 2017 by the American Physical Society
Perovskite oxides with a Jahn–Teller distortion were also shown to lead to polar structures around defects. Stoichiometric LaMnO3 exhibits a cooperative Jahn–Teller distortion with alternating long and short Mn–O bonds within a {001} plane [46, 47]. Relaxations around an oxygen vacancy alter these bond lengths and therefore orbital energies, leading to an asymmetric accommodation of the excess electrons at Mn in next-nearest neighbor positions to the defect, resulting in a net polarization around the defect [48].
The cation order in perovskite oxide superlattices offers an additional degree of freedom in obtaining a net polarization due to oxygen vacancies. It was shown that a stoichiometric (LaFeO3)2/(SrFeO3) superlattice (i.e., a ferrite perovskite with a LaO–LaO–SrO A-site layer sequence) adopts an antipolar Fe displacement [49]. This antipolar pattern results from hole localization in the FeO2 layer surrounded by two LaO layers (i.e., the formal Fe oxidation state in these layers is Fe4+ whereas the other Fe ions are Fe3+). This hole localization also favors oxygen vacancy formation in the FeO2 layer between two LaO layers, as the two excess electrons most favorably reduce the Fe4+ ions to Fe3+. This reduction, in turn, counteracts the antipolar displacement and favors a polar one in the presence of oxygen vacancies [49].
Just like defect-pair dipoles can be controlled by epitaxial strain [41], also single defects may have anisotropic elastic dipoles [50] than can interact with a strain field leading for example to oxygen vacancy ordering [51]. Such ordering is crucial as only a structure with ordered oxygen vacancies can lead to long-range polar and ferroelectric properties.
2 Magnetism induced or altered by defects
Defect-induced or controlled magnetism is a rich and established field of research, a full review of which would go beyond the goals of the present article, and we will here focus on examples that highlight the mechanisms by which magnetism can be affected by defects.
Both oxygen and Ti deficiency were predicted to result in ferromagnetism in nominally nonmagnetic SrTiO3 [52], a result later confirmed by experiment in BaTiO3 [53]. We want to note, however, the extreme sensitivity of both experimental synthesis and theoretical methods in obtaining non-magnetic or magnetic phases [54, 55]. The mechanism underlying this emergence of magnetism in presence of oxygen vacancies is unpaired excess electrons on the reduced Ti3+ sites. Since the concentration of magnetic Ti3+ sites is linked with the oxygen vacancy concentration, highly deficient materials are required for long-range magnetic order.
The above examples referred to cases where nominally d0 Ti4+ ions were reduced to potentially magnetic Ti3+ ions. Oxygen vacancies may, however, also lead to the emergence of magnetism for non-d0 ions. One such case is LaCoO3, which is normally non-magnetic due to a full occupation of the t2g states in the low-spin d6 configuration in an octahedral crystal field. Oxygen vacancies lead to both reduction of Co3+ to Co2+ and a transition to a high-spin state (see Fig. 5) [56]. The unpaired electrons in Co2+ result in a ferromagnetic state in oxygen-deficient LaCoO3. Epitaxial strain was shown to affect the ordering and concentration of oxygen vacancies and hence the Co2+ ions, modulating the stability of the long-range ferromagnetic order [57]. A defect-free strain accommodation mechanism with modulation of lattice spacing and hence crystal field was, however, reported to result in a similar stabilization of the ferromagnetic order [58].

Copyright (2014) by the American Physical Society
Oxygen-deficient LaCoO3 with polyhedra adjacent to the defect shown in blue. Arrows show the magnetic moments arising on reduced high-spin Co2+, while low-spin Co3+ remain non-magnetic. Reprinted figure with permission from Ref. [56]
Oxygen vacancies can also be used to alter an existing magnetic order. In EuTiO3, the creation of Eu and O vacancy pairs alters the geometry and results in a weakening of the antiferromagnetic Eu–Ti–Eu interactions while introducing ferromagnetic Eu–O–Eu exchange, leading to a transition from antiferromagnetism to ferromagnetism [59]. Spin-polarized Ti3+ sites were invoked for an antiferromagnetic to ferromagnetic transition in oxygen-deficient EuBaTi2O6, where the antiferromagnetic Eu2+–Eu2+ exchange in the stoichiometric material is dominated by ferromagnetic Eu2+–Ti3+–Eu2+ exchange in presence of oxygen vacancies [60]. Epitaxial strain in conjunction with oxygen vacancies can lead to further changes in the magnetic order. For example, multiferroic BiCoO3 assumes a supertetragonal phase in which Co is in square-pyramidal coordination [61]. The Co sites adopt a C type antiferromagnetic order in the bulk, which is retained in presence of oxygen vacancies. However, while tensile epitaxial strain in stoichiometric BiCoO3 results in a transition to G-type antiferromagnetic order, it yields a ferrimagnetic state in the oxygen-deficient structure due to the reduction of Co magnetic moments around the defect. Similar results were reported for antiferromagnetic LaMnO3 thin films, where oxygen deficiency leads to mixed Mn3+ and Mn4+ oxidation states, that couple ferromagnetically [62].
The alteration of exchange becomes richer in double perovskites with two different magnetic B sites, as the cation order and oxidation states of both B sites may change. La2CoMnO6 with a checkerboard B-site order is ferromagnetic due to the dominant exchange interaction between Co2+ and Mn4+ ions. Reduction via oxygen vacancy formation preferentially occurs on the Mn sites, while oxidation via Sr doping on the A-site affects the Co sites [63]. In both cases, the Curie temperature is lowered as the vacancy and Sr defects induce strong antiferromagnetic exchange or reduce the strength of ferromagnetic exchange, respectively. In La2NiMnO6, the presence of excess electrons, for example, due to oxygen vacancy formation or a polar catastrophe mechanism, can alter the checkerboard to a columnar order, introducing antiferromagnetic Mn–Mn and Ni–Ni superexchange in addition to the ferromagnetic Mn–Ni interactions [64]. In agreement with this result, extended oxygen anneal resulted in a reduced oxygen vacancy concentration and lead to stronger ferromagnetism in this material [65].
3 Conclusions and outlook
As the above examples have shown, defects can be a rich source of added or altered functionality in perovskite oxides. Defect dipoles resulting from defect pairs or defect clusters couple with the ferroelectric polarization and can locally promote a certain polarization direction. In some cases, single defects can also lead to local polarization effects. In both situations, the fact that electric defect dipoles usually go along with elastic dipoles allows their manipulation with epitaxial strain. The resulting ordered electric defect dipoles strongly promote a polar state and dramatically increase the ferroelectric Curie temperature.
On the other hand, defects lead to changes in oxidation state and exchange pathways that can induce magnetism in otherwise non-magnetic materials or alter an existing magnetic order, for example from antiferromagnetic to ferromagnetic.
Defects thus hold great promise to engineer both properties in perovskite oxides, in particular inducing ferroelectricity in already (anti)ferromagnetic materials or magnetism in ferroelectric materials to obtain magnetoelectric multiferroics. Despite this promise, a rational design of these properties is still hampered by the lack of a general understanding and predictability of defect-induced functionality. This primarily stems from the fact that defects induce complicated alterations in both the ionic and electronic structures. Since the physics of stoichiometric perovskite oxides is already dominated by an intricate interplay of lattice, charge, spin, and orbital degrees of freedom, the effect of defects is not always easily foreseen. This is further complicated in double perovskites and thin films/superlattices, where order/disorder phenomena and altered electrostatic boundary conditions, respectively, affect charge localization and defect stability. As a result, excess charge localization, which is at the origin of both defect dipoles and changes in magnetism, is not readily predicted without resorting to involved experiments such as scanning transmission microscopy, electron energy loss spectroscopy, or computational investigations at a high level of theory. Nevertheless, it seems hopeful that the ever-growing number of such studies will eventually lead to sufficient data for a mechanistic understanding of defect-induced ferroelectric and magnetic functionality and hence their rational engineering across the wide compositional range offered by perovskite oxides.
Data availability
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
References
M.A. Peña, J.L.G. Fierro, Chemical structures and performance of perovskite oxides. Chem. Rev. 101, 1981–2018 (2001). https://doi.org/10.1021/cr980129f
V.M. Goldschmidt, Die Gesetze der Krystallochemie. Naturwissenschaften 14, 477–485 (1926). https://doi.org/10.1007/bf01507527
R.E. Cohen, Origin of ferroelectricity in perovskite oxides. Nature 358, 136–138 (1992). https://doi.org/10.1038/358136a0
J.F. Scott, Applications of modern ferroelectrics. Science 315(954), 959 (2007). https://doi.org/10.1126/science.1129564
J.F. Scott, A review of ferroelectric switching. Ferroelectrics 503, 117–132 (2016). https://doi.org/10.1080/00150193.2016.1236611
C. Kittel, Theory of antiferroelectric crystals. Phys Rev. 82, 729–732 (1951). https://doi.org/10.1103/physrev.82.729
Z. Liu, T. Lu, J. Ye, G. Wang, X. Dong, R. Withers, Y. Liu, Antiferroelectrics for energy storage applications: a review. Adv. Mater. Technolo. 3, 1800111 (2018). https://doi.org/10.1002/admt.201800111
J.B. Goodenough, Theory of the role of covalence in the perovskite-type manganites [La, M(II)]MnO3. Phys. Rev. 100, 564–573 (1955). https://doi.org/10.1103/physrev.100.564
J.B. Goodenough, An interpretation of the magnetic properties of the perovskite-type mixed crystals La1−xSrxCoO3−λ. J. Phys. Chem. Solids. 6, 287–297 (1958). https://doi.org/10.1016/0022-3697(58)90107-0
J. Kanamori, Superexchange interaction and symmetry properties of electron orbitals. J. Phys. Chem. Solids 10, 87–98 (1959). https://doi.org/10.1016/0022-3697(59)90061-7
J.G. Bednorz, K.A. Müller, Perovskite-type oxides-the new approach to high-Tc superconductivity. Rev. Mod. Phys. 60, 585–600 (1988). https://doi.org/10.1103/revmodphys.60.585
Y. Tokura, Y. Tomioka, H. Kuwahara, A. Asamitsu, Y. Moritomo, M. Kasai, Origins of colossal magnetoresistance in perovskite-type manganese oxides (invited). J. Appl. Phys. 79, 5288 (1996). https://doi.org/10.1063/1.361353
J. Hwang, R.R. Rao, L. Giordano, Y. Katayama, Y. Yu, Y. Shao-Horn, Perovskites in catalysis and electrocatalysis. Science 358, 751–756 (2017). https://doi.org/10.1126/science.aam7092
N.A. Hill, Why are there so few magnetic ferroelectrics? J Phys Chem B. 104, 6694–6709 (2000). https://doi.org/10.1021/jp000114x
G. Catalan, J.F. Scott, Physics and applications of bismuth ferrite. Adv. Mater. 21, 2463–2485 (2009). https://doi.org/10.1002/adma.200802849
D.G. Schlom, L.-Q. Chen, C.-B. Eom, K.M. Rabe, S.K. Streiffer, J.-M. Triscone, Strain tuning of ferroelectric thin films. Annu. Rev. Mater. Res. 37, 589–626 (2007). https://doi.org/10.1146/annurev.matsci.37.061206.113016
J.M. Rondinelli, N.A. Spaldin, Structure and properties of functional oxide thin films: insights from electronic-structure calculations. Adv.Mater. 23, 3363–3381 (2011). https://doi.org/10.1002/adma.201101152
V.V. Lemanov, E.P. Smirnova, P.P. Syrnikov, E.A. Tarakanov, Phase transitions and glasslike behavior in Sr1-xBaxTiO3. Phys. Rev. B. 54, 3151–3157 (1996). https://doi.org/10.1103/physrevb.54.3151
A. Urushibara, Y. Moritomo, T. Arima, A. Asamitsu, G. Kido, Y. Tokura, Insulator-metal transition and giant magnetoresistance in La1−xSrxMnO3. Phys. Rev. B. 51, 14103–14109 (1995). https://doi.org/10.1103/physrevb.51.14103
S.B. Zhang, J.E. Northrup, Chemical potential dependence of defect formation energies in GaAs: application to Ga self-diffusion. Phys. Rev. Lett. 67, 2339–2342 (1991). https://doi.org/10.1103/physrevlett.67.2339
J.E. Northrup, S.B. Zhang, Dopant and defect energetics: Si in GaAs. Phys. Rev. B. 47, 6791–6794 (1993). https://doi.org/10.1103/physrevb.47.6791
C. Freysoldt, B. Grabowski, T. Hickel, J. Neugebauer, G. Kresse, A. Janotti, C.G.V. de Walle, First-principles calculations for point defects in solids. Rev. Mod. Phys. 86, 253–305 (2014). https://doi.org/10.1103/revmodphys.86.253
U. Robels, G. Arlt, Domain wall clamping in ferroelectrics by orientation of defects. J. Appl. Phys. 73, 3454–3460 (1993). https://doi.org/10.1063/1.352948
W.L. Warren, G.E. Pike, K. Vanheusden, D. Dimos, B.A. Tuttle, J. Robertson, Defect-dipole alignment and tetragonal strain in ferroelectrics. J. Appl. Phys. 79, 9250 (1996). https://doi.org/10.1063/1.362600
X. Ren, Large electric-field-induced strain in ferroelectric crystals by point-defect-mediated reversible domain switching. Nat. Mater. 3, 91–94 (2004). https://doi.org/10.1038/nmat1051
D.C. Lupascu, Y.A. Genenko, N. Balke, Aging in ferroelectrics. J. Am. Ceram. Soc. 89, 224–229 (2006). https://doi.org/10.1111/j.1551-2916.2005.00663.x
C.M. Folkman, S.H. Baek, C.T. Nelson, H.W. Jang, T. Tybell, X.Q. Pan, C.-B. Eom, Study of defect-dipoles in an epitaxial ferroelectric thin film. Appl. Phys. Lett. 96, 052903 (2010). https://doi.org/10.1063/1.3298362
Y.A. Genenko, J. Glaum, M.J. Hoffmann, K. Albe, Mechanisms of aging and fatigue in ferroelectrics. Mater. Sci. Eng. B 192, 52–82 (2015). https://doi.org/10.1016/j.mseb.2014.10.003
F.A. Kröger, H.J. Vink, Relations between the concentrations of imperfections in crystalline solids. Solid State Phys. 3, 307–435 (1956). https://doi.org/10.1016/s0081-1947(08)60135-6
A. Chandrasekaran, D. Damjanovic, N. Setter, N. Marzari, Defect ordering and defect–domain-wall interactions in PbTiO3: A first-principles study. Phys. Rev. B 88, 214116 (2013). https://doi.org/10.1103/physrevb.88.214116
P. Erhart, P. Träskelin, K. Albe, Formation and switching of defect dipoles in acceptor-doped lead titanate: a kinetic model based on first-principles calculations. Phys. Rev. B 88, 024107 (2013). https://doi.org/10.1103/physrevb.88.024107
P. Marton, C. Elsässer, Switching of a substitutional-iron/oxygen-vacancy defect complex in ferroelectric PbTiO3 from first principles. Phys. Rev. B 83, 020106 (2011). https://doi.org/10.1103/physrevb.83.020106
A.V. Kimmel, P.M. Weaver, M.G. Cain, P.V. Sushko, Defect-mediated lattice relaxation and domain stability in ferroelectric oxides. Phys. Rev. Lett 109, 117601 (2012). https://doi.org/10.1103/physrevlett.109.117601
A. Walsh, C.R.A. Catlow, A.G.H. Smith, A.A. Sokol, S.M. Woodley, Strontium migration assisted by oxygen vacancies in SrTiO3 from classical and quantum mechanical simulations. Phys. Rev. B 83, 220301 (2011). https://doi.org/10.1103/physrevb.83.220301
D.D. Cuong, B. Lee, K.M. Choi, H.-S. Ahn, S. Han, J. Lee, Oxygen vacancy clustering and electron localization in oxygen-deficient SrTiO3: LDA+U study. Phys. Rev. Lett. 98, 115503 (2007). https://doi.org/10.1103/physrevlett.98.115503
K. Klyukin, V. Alexandrov, Effect of intrinsic point defects on ferroelectric polarization behavior of SrTiO3. Phys. Rev. B 95, 035301 (2017). https://doi.org/10.1103/physrevb.95.035301
C. Ricca, D. Berkowitz, U. Aschauer, Ferroelectricity promoted by cation/anion divacancies in SrMnO3. J. Mater. Chem. C 9, 13321–13330 (2021). https://doi.org/10.1039/d1tc02317a
C. Ricca, U. Aschauer, Interplay between polarization, strain, and defect pairs in Fe-doped SrMnO3−δ. Phys. Rev. Res. 3, 033237 (2021). https://doi.org/10.1103/physrevresearch.3.033237
S. Liu, R.E. Cohen, Multiscale simulations of defect dipole–enhanced electromechanical coupling at dilute defect concentrations. Appl. Phys. Lett. 111, 082903 (2017). https://doi.org/10.1063/1.4989670
K. Sakayori, Y. Matsui, H. Abe, E. Nakamura, M. Kenmoku, T. Hara, D. Ishikawa, A. Kokubu, K. Hirota, T.I.T. Ikeda, Curie temperature of BaTiO3. Jpn. J. Appl. Phys. 34, 5443 (1995). https://doi.org/10.1143/jjap.34.5443
A.R. Damodaran, E. Breckenfeld, Z. Chen, S. Lee, L.W. Martin, Enhancement of ferroelectric curie temperature in BaTiO3 films via strain-induced defect dipole alignment. Adv. Mater. 26, 6341–6347 (2014). https://doi.org/10.1002/adma.201400254
Y.S. Kim, J. Kim, S.J. Moon, W.S. Choi, Y.J. Chang, J.-G. Yoon, J. Yu, J.-S. Chung, T.W. Noh, Localized electronic states induced by defects and possible origin of ferroelectricity in strontium titanate thin films. Appl. Phys. Lett. 94, 202906 (2009). https://doi.org/10.1063/1.3139767
M. Choi, F. Oba, I. Tanaka, Role of Ti antisitelike defects in SrTiO3. Phys Rev Lett. 103, 185502 (2009). https://doi.org/10.1103/physrevlett.103.185502
F. Yang, Q. Zhang, Z. Yang, J. Gu, Y. Liang, W. Li, W. Wang, K. Jin, L. Gu, J. Guo, Room-temperature ferroelectricity of SrTiO3 films modulated by cation concentration. Appl. Phys. Lett. 107, 082904 (2015). https://doi.org/10.1063/1.4929610
W. Li, Q. He, L. Wang, H. Zeng, J. Bowlan, L. Ling, D.A. Yarotski, W. Zhang, R. Zhao, J. Dai, J. Gu, S. Shen, H. Guo, L. Pi, H. Wang, Y. Wang, I.A. Velasco-Davalos, Y. Wu, Z. Hu, B. Chen, R.-W. Li, Y. Sun, K. Jin, Y. Zhang, H.-T. Chen, S. Ju, A. Ruediger, D. Shi, A.Y. Borisevich, H. Yang, Manipulating multiple order parameters via oxygen vacancies: the case of Eu0.5Ba0.5TiO3−δ. Phys. Rev. B 96, 115105 (2017). https://doi.org/10.1103/physrevb.96.115105
P. Norby, I.G.K. Andersen, E.K. Andersen, N.H. Andersen, The crystal structure of lanthanum manganite (III), LaMnO3, at room temperature and at 1273 K under N2. J. Solid State Chem. 119, 191–196 (1995). https://doi.org/10.1016/0022-4596(95)80028-n
J. Rodríguez-Carvajal, M. Hennion, F. Moussa, A.H. Moudden, L. Pinsard, A. Revcolevschi, Neutron-diffraction study of the Jahn–Teller transition in stoichiometric LaMnO3. Phys. Rev. B 57, R3189–R3192 (1998). https://doi.org/10.1103/physrevb.57.r3189
C. Ricca, N. Niederhauser, U. Aschauer, Local polarization in oxygen-deficient LaMnO3 induced by charge localization in the Jahn–Teller distorted structure. Phys. Rev. Res. 2, 042040 (2020). https://doi.org/10.1103/physrevresearch.2.042040
R. Mishra, Y.-M. Kim, J. Salafranca, S.K. Kim, S.H. Chang, A. Bhattacharya, D.D. Fong, S.J. Pennycook, S.T. Pantelides, A.Y. Borisevich, Oxygen-vacancy-induced polar behavior in (LaFeO3)2/(SrFeO3) superlattices. Nano. Lett. 14, 2694–2701 (2014). https://doi.org/10.1021/nl500601d
D.A. Freedman, D. Roundy, T.A. Arias, Elastic effects of vacancies in strontium titanate: Short- and long-range strain fields, elastic dipole tensors, and chemical strain. Phys. Rev. B 80, 064108 (2009). https://doi.org/10.1103/physrevb.80.064108
U. Aschauer, R. Pfenninger, S.M. Selbach, T. Grande, N.A. Spaldin, Strain-controlled oxygen vacancy formation and ordering in CaMnO3. Phys. Rev. B 88, 10423 (2013). https://doi.org/10.1103/physrevb.88.054111
I.R. Shein, A.L. Ivanovskii, First principle prediction of vacancy-induced magnetism in non-magnetic perovskite SrTiO3. Phys. Lett. A 371, 155–159 (2007). https://doi.org/10.1016/j.physleta.2007.06.013
F. Yang, K. Jin, H. Lu, M. He, C. Wang, J. Wen, G. Yang, Oxygen vacancy induced magnetism in BaTiO3−δ and Nb:BaTiO3−δ thin films. Sci. China Phys. Mech. Astronomy 53, 852–855 (2010). https://doi.org/10.1007/s11433-010-0187-x
C. Ricca, I. Timrov, M. Cococcioni, N. Marzari, U. Aschauer, Self-consistent DFT+U+V study of oxygen vacancies in SrTiO3. Phys. Rev. Res. 2, 023313 (2020). https://doi.org/10.1103/physrevresearch.2.023313
A. Raeliarijaona, H. Fu, Ferromagnetism in ferroelectric BaTiO3 induced by vacancies: Sensitive dependence on charge state, origin of magnetism, and temperature range of existence. Phys. Rev. B 96, 144431 (2017). https://doi.org/10.1103/physrevb.96.144431
N. Biškup, J. Salafranca, V. Mehta, M.P. Oxley, Y. Suzuki, S.J. Pennycook, S.T. Pantelides, M. Varela, Insulating ferromagnetic LaCoO3−δ films: a phase Induced by ordering of oxygen vacancies. Phys Rev Lett. 112, 087202 (2014). https://doi.org/10.1103/physrevlett.112.087202
V.V. Mehta, N. Biskup, C. Jenkins, E. Arenholz, M. Varela, Y. Suzuki, Long-range ferromagnetic order in LaCoO3−δ epitaxial films due to the interplay of epitaxial strain and oxygen vacancy ordering. Phys. Rev. B 91, 144418 (2015). https://doi.org/10.1103/physrevb.91.144418
W.S. Choi, J.-H. Kwon, H. Jeen, J.E. Hamann-Borrero, A. Radi, S. Macke, R. Sutarto, F. He, G.A. Sawatzky, V. Hinkov, M. Kim, H.N. Lee, Strain-induced spin states in atomically ordered cobaltites. Nano Lett. 12, 4966–4970 (2012). https://doi.org/10.1021/nl302562f
D. Shin, I. Kim, S. Song, Y. Seo, J. Hwang, S. Park, M. Choi, W.S. Choi, Defect engineering of magnetic ground state in EuTiO3 epitaxial thin films. J. Am. Ceram. Soc. 104, 4606–4613 (2021). https://doi.org/10.1111/jace.17870
W. Li, R. Zhao, L. Wang, R. Tang, Y. Zhu, J.H. Lee, H. Cao, T. Cai, H. Guo, C. Wang, L. Ling, L. Pi, K. Jin, Y. Zhang, H. Wang, Y. Wang, S. Ju, H. Yang, Oxygen-vacancy-induced antiferro magnetism to ferromagnetism transformation in Eu0.5Ba0.5TiO3−δ multiferroic thin films. Sci Rep. 3, 2618 (2013). https://doi.org/10.1038/srep02618
C. Menéndez, D. Chu, C. Cazorla, Oxygen-vacancy induced magnetic phase transitions in multiferroic thin films. Npj Comput. Mater. 6, 76 (2020). https://doi.org/10.1038/s41524-020-0344-3
W. Niu, W. Liu, M. Gu, Y. Chen, X. Zhang, M. Zhang, Y. Chen, J. Wang, J. Du, F. Song, X. Pan, N. Pryds, X. Wang, P. Wang, Y. Xu, Y. Chen, R. Zhang, Direct demonstration of the emergent magnetism resulting from the multivalence Mn in a LaMnO3 epitaxial thin film system. Adv. Electron. Mater. 4, 1800055 (2018). https://doi.org/10.1002/aelm.201800055
A. Khan, S. Chatterjee, T.K. Nath, A. Taraphder, Defect-induced modulation of magnetic, electronic, and optical properties of the double-perovskite oxide La2CoMnO6. Phys. Rev. B 104, 035152 (2021). https://doi.org/10.1103/physrevb.104.035152
G.D. Luca, J. Spring, M. Kaviani, S. Jöhr, M. Campanini, A. Zakharova, C. Guillemard, J. Herrero-Martin, R. Erni, C. Piamonteze, M.D. Rossell, U. Aschauer, M. Gibert, Top-layer engineering reshapes charge transfer at polar oxide interfaces. Adv. Mater. 34, 2203071 (2022). https://doi.org/10.1002/adma.202203071
J.P. Palakkal, T. Schneider, L. Alff, Oxygen defect engineered magnetism of La2NiMnO6 thin films. AIP Adv. 12, 035116 (2022). https://doi.org/10.1063/9.0000360
Funding
Open access funding provided by University of Bern.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ricca, C., Aschauer, U. Mechanisms for point defect-induced functionality in complex perovskite oxides. Appl. Phys. A 128, 1083 (2022). https://doi.org/10.1007/s00339-022-06210-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00339-022-06210-8