
Abstract
Over the past decade, an abundance of 16S rRNA gene surveys have provided microbiologists with data regarding the prokaryotes present in a coral-associated microbial community. Functional gene studies that provide information regarding what those microbes might do are fewer, particularly for non-tropical corals. Using the GeoChip 5.0S microarray, we present a functional gene study of microbiomes from five species of cold-water corals collected from depths of 296–1567 m. These species included two octocorals, Acanthogorgia aspera and Acanthogorgia spissa, and three stony corals: Desmophyllum dianthus, Desmophyllum pertusum (formerly Lophelia pertusa), and Enallopsammia profunda. A total of 24,281 gene sequences (representing different microbial taxa) encoding for 383 functional gene families and representing 9 metabolic gene categories were identified. Gene categories included metabolism of carbon, nitrogen, phosphorus, and sulfur, as well as virulence, organic remediation, metal homeostasis, secondary metabolism and phylogeny. We found that microbiomes from Acanthogorgia spp. were the most functionally distinct but also least diverse compared against those from stony corals. Desmophyllum spp. microbiomes were more similar to each other than to E. profunda. Of 383 total gene families detected in this study, less than 20% were significantly different among these deep-water coral species. Similarly, out of 59 metabolic sub-categories for which we were able to make a direct comparison to microbiomes of tropical corals, only 7 were notably different: anaerobic ammonium oxidation (anammox), chitin degradation, and dimethylsulfoniopropionate (DMSP) degradation, all of which had higher representations in deep-water corals; and chromium homeostasis/resistance, copper homeostasis/resistance, antibiotic resistance, and methanogenesis, all of which had higher representation in tropical corals. This implies a broad-scale convergence of the microbial functional genes present within the coral holobiont, independent of coral species, depth, symbiont status, and morphology.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cold-water corals provide 3-dimensional structure in the deep ocean, creating mounds or ‘gardens’ that are rapidly populated by numerous other invertebrates and fish species (Buhl-Mortensen and Mortensen 2004, 2005; Stone 2006; Ross and Quattrini 2007; Sulak et al. 2007; Cordes et al. 2008). These deep-sea corals provide a foundation for oceanic biodiversity hot-spots and serve many of the same habitat functions as shallow-water coral species on tropical reefs (Roberts and Hirshfield 2004).
Due to the cost and difficulties of sampling corals in the deep ocean, we know relatively less about their microbiomes compared to tropical corals (Kellogg 2019). There is some evidence for bacterial community conservation in deep-sea octocorals at the host genus- rather than species-level (Lawler et al. 2016; Kellogg and Pratte 2021). A recent comparison of the prokaryotic microbiomes of deep-water scleractinian corals Desmophyllum dianthus and Desmophyllum pertusum (formerly Lophelia pertusa; Addamo et al. 2016) showed species specificity (Kellogg and Pratte 2021). However, it remains an open question whether functional redundancy underlies taxonomic diversity in these microbiomes.
The functional potential of coral-associated microbial communities, particularly deep-sea corals, can be incredibly difficult to study. In addition to the logistical challenges of deep-sea coral collections, there are also issues of large amounts of coral versus microbial DNA in genomic extractions, and a substantial amount of mucus that can complicate downstream analyses (Wegley et al. 2007; Pratte and Kellogg 2021). The metabolic roles of deep-sea coral-associated microbes have been inferred from stable isotope tracer experiments (Middelburg et al. 2015) and the known capabilities of certain taxonomic groups (Kellogg et al. 2016; Lawler et al. 2016) to include nitrogen fixation, nitrogen cycling, and inorganic carbon fixation. Application of predictive functional profiling to 16S rRNA gene sequences from D. pertusum indicated potential of the microbiome to engage in chemoautotrophy, nutrient cycling, and antibiotic production (Kellogg et al. 2017). While metagenomic data from deep-sea corals are beginning to become available (e.g., Vohsen et al. 2020), studies have not yet been published describing the functional capacity of deep-sea coral microbiomes, likely hindered by insufficient matches to reference databases (Wegley et al. 2007).
One solution to overcome this bottleneck is the use of functional gene microarrays (He et al. 2007). For example, the GeoChip microarray has been repeatedly updated to include greater numbers and diversities of environmental microbial (bacterial, archaeal, and microeukaryotic) functional gene sequences. There have been two studies that used versions of this microarray to characterize the microbiomes of tropical scleractinian coral species (Kimes et al. 2010; Zhang et al. 2015). The GeoChip 5.0S contains 57,498 oligonucleotide probes that target major metabolic categories such as carbon, sulfur, and nitrogen cycling, as well as sub-categories including antibiotic resistance, and contaminant degradation (Wang et al. 2014; Shi et al. 2019). The aim of this study was to use a functional gene microarray to assess the metabolic potential of the microbial communities associated with five species of deep-sea corals, including both octocorals and scleractinians.
Materials and methods
Sample collection
Deep-sea coral samples were collected for microarray analysis during two research cruises supporting the DeepSEARCH project (Table 1). Samples from 2018 were collected using the submersible Alvin and support vessel RV Atlantis (expedition AT41) and the samples from 2019 were collected using the remotely operated vehicle (ROV) Jason II from the NOAA ship Ronald H. Brown (expedition RB1903). Sub-samples of branching coral colonies, or in the case of D. dianthus the entire cup coral, were collected using the respective vehicle’s manipulator arm and placed into individual, thermally insulated containers that had been pre-cleaned (washed with freshwater, interiors wiped with 100% ethanol to remove any biofilms or particulates from prior collections), filled with freshwater, and sent down sealed. During collection, opening the container released the freshwater, which was then displaced with local seawater from the collection site. The containers were resealed at depth to prevent microbial contamination from other sample collections during the dive or passing through different water masses during vehicle ascent. Upon vehicle recovery, sample containers were brought into a cold room or laboratory, and samples were removed using sterile forceps. All coral samples were lightly rinsed with sterile 1 × phosphate-buffered saline (PBS) to remove any loosely associated surface microbes. Stony coral samples (D. dianthus, D. pertusum, Enallopsammia profunda) were placed into sterile aluminum weigh boats and a flame-sterilized hammer was used to break open the skeletons to expose polyp tissue. Octocoral samples (Acanthogorgia aspera, Acanthogorgia spissa) were trimmed with sterile shears to only sample the branches and not the central skeleton nor the area that contacted the manipulator. All samples were individually wrapped in pre-sterilized aluminum foil, inserted into sterile Whirlpak bags, and flash frozen in liquid nitrogen. After the cruises, the samples were transferred to a − 80 °C freezer until processed.
DNA extraction
Qiagen’s DNeasy PowerBiofilm kit was used to extract duplicate samples from each coral colony that were combined at the end to ensure sufficient DNA concentrations for application to the microarrays. Coral pieces were weighed and placed directly into the PowerBiofilm bead tubes, which were held in a cold block during the weighing step. Approximately 0.2 g per sample were used for each octocoral per extraction tube. To compensate for the weight of the skeleton, the stony coral samples were 0.2–0.65 g per extraction tube to make sure sufficient tissue was included in each extraction. The manufacturer’s protocol (Handbook edition 03/2018) using a vacuum manifold was followed except that our bead-beating step substituted a Fastprep (MP Biomedicals) at setting 5 (approximately 3100 rpm). The two extractions per coral sample were combined at the end for a total of 200 µL of DNA solution per coral. Two 50-reaction kits from the same lot were used, such that each sample used one tube from each kit. Kit blanks (extractions with no sample added) were processed from each kit and then combined in the same fashion as the samples. DNA (~ 180 µL per sample or kit blank) was shipped on dry ice to Glomics Inc. (Norman, OK) for application to GeoChip 5.0S microarrays.
GeoChip 5.0S processing
For each sample, 400 ng of DNA was used directly for labeling. The kit blank did not have a detectable DNA concentration but was labeled using the same process as the samples. Fluorescence-labeling, hybridization of the microarrays, and scanning were carried out as previously described (Lu et al. 2012). Glomics conducted a data normalization, and only probe spots that were above a designated limit (a signal-to-noise ratio of ≥ 2; minimum signal intensity of 200 in at least one sample) were retained (Wu et al. 2006; He et al. 2007).
Data processing
The GeoChip 5.0S is organized into metabolic categories (e.g., carbon cycling) containing 4–118 gene families per category, with each gene family containing hundreds to thousands of oligonucleotide probes representing taxonomically different gene variants. GeoChip analysis allows estimation of both the richness of genes per gene family or metabolic category and the fluorescent intensity per probe, with intensity proportionate to gene abundance in the sample.
The raw data table with all zeroed intensities provided by Glomics was exported for further analysis. Genes (probes) that were only detected in one coral sample were removed from the data table. The quality-controlled table was imported into R version 4.0.5 (R Core Team 2018). The two Acanthogorgia spp. were pooled for statistical analyses. All statistical analyses were conducted using the R package ‘vegan’ (Oksanen et al. 2016), and separate tests were done for all genes and then for each of the nine metabolic categories (carbon cycling, nitrogen, sulfur, phosphorus, organic remediation, metal homeostasis, virulence, secondary metabolism, and phylogeny). Alpha diversity refers to the diversity within a particular coral microbiome, in terms of the number (richness) of different functional genes present in that sample or within a particular metabolic category for that sample. Shannon diversity indices (Shannon 1948) and the number of observed genes were calculated. Significant differences in functional gene alpha diversity between coral species microbiomes were evaluated with an ANOVA, and significant tests were followed up with a Tukey’s HSD (honestly significant difference) test. Beta-diversity analyses were based upon a square-root transformed Bray–Curtis dissimilarity matrix and plotted on a non-metric multi-dimensional scaling (NMDS) plot. Significant differences in microbial community structure were evaluated using PERMANOVA (adonis) using 999 permutations, and significant tests were followed up with pairwise adonis with a Bonferroni correction. Significant differences between dispersion were tested using betadisper.
For analysis of individual gene families, the total intensity and total number of genes (from different taxa) were summed for each gene family. For example, sqr is one gene family associated with sulfur cycling, and 58 different sqr genes were detected from 58 different taxa in sample L1; the sum of all their intensities was 381,669.616. The intensity of the signal detected for each gene is a direct correlation to the abundance of the gene, and assessing the total intensity associated with each gene family provided a proxy for the abundance of each gene family. Assessing the total number of genes associated with each gene family provided information regarding the number of taxa carrying this gene. For genes included on the array for the purposes of phylogenetic assignment, all taxa were clustered at the genus level (rather than gene family). Significant differences in total intensity and total number of gene families were evaluated with an ANOVA and significant tests were followed up with a Tukey’s HSD (honestly significant difference) test.
Finally, data from previously published studies were obtained for direct comparisons to the results of our study. First, 16S rRNA amplicon data generated from technical replicates of some of the same coral colonies during a previous study were re-analyzed (Kellogg and Pratte 2021). These included one A. spissa, two D. dianthus, and three D. pertussum samples (indicated by asterisks in Table 1). The 16S rRNA amplicon data for these were downloaded from the NCBI Sequence Read Archive (SRA) from BioProject number PRJNA699458 and analyzed as described in Kellogg and Pratte (2021). Second, a literature search was conducted to identify any studies that utilized the GeoChip 5.0 and reported the number of genes detected for each of the gene categories. These studies included tropical corals (Zhang et al. 2015), artificial forest soil (Li et al. 2020), rice crop soil (Das et al. 2019), sea water (McFarlin et al. 2017), sugarcane soil (Navarrete et al. 2017), and mountain soil (Shen et al. 2016). The number of genes detected for each metabolic category was then compared across all studies.
Results
Data statistics and quality control
A total of 24,281 genes were detected after Glomics’ standard quality control processing, ranging from 41 (secondary metabolism) to 10,636 (carbon cycling; Table 2), 21,278 of which were bacterial in origin, 578 were archaeal, 2,155 were eukaryotic (1,892 of which were fungi), and 270 were unclassified. The number of genes detected per coral sample ranged from 15,019 to 21,510, and the number of gene families detected per coral sample ranged from 305 to 320. Although the extraction blank returned positive spots, the extraction blank clustered separately from all coral samples in an nMDS plot (Online Resource 1) and did not resemble any of the coral communities. Therefore, the coral data were deemed valid, and the extraction blank was removed from all subsequent analyses. The microarray data analyzed during the current study are available from the USGS data release, https://doi.org/10.5066/P9RPE8YX.
Alpha diversity
Overall, microbiomes from Acanthogorgia spp. had the lowest Shannon diversity index in all metabolic categories, while either D. dianthus or E. profunda microbiomes were the most diverse, depending on metabolic gene category (Fig. 1). For each metabolic category, the number of detected genes (observed richness) were not significantly different among the coral species (Table 2). However, the Shannon diversity index was significantly different for four metabolic categories (organic remediation, metal homeostasis, carbon cycling, and phylogeny; Table 2). Pairwise tests indicated that Shannon diversity differed significantly between microbiomes of D. dianthus and Acanthogorgia spp. for each of the four metabolic categories identified as significant via ANOVA: adjusted p values were 0.003 for Phylogeny, 0.032 for Organic Remediation, 0.003 for Metal Homeostasis, and 0.032 for Carbon Cycling. Though Acanthogorgia spp. had fewer replicates (n = 2) than the other coral species (n = 3), and thus lower statistical power, Acanthogorgia spp. microbiomes had the highest number of pairwise significant differences, particularly for the metal homeostasis category (adjusted p values ranging from 0.003 to 0.006, different from all the other coral species). Only one significant pairwise comparison did not include Acanthogorgia spp. and was between the microeukaryotic phylogenetic genes associated with D. dianthus and D. pertusum (adjusted p value = 0.049). While the global ADONIS tests were all significant (Table 2), none of the pairwise comparisons were significant, due to lack of statistical power from low sample sizes.

Shannon diversity indices calculated using all genes for microbiomes of Acanthogorgia spp., Desmophyllum dianthus, Enallopsammia profunda, and Desmophyllum pertusum
Beta diversity
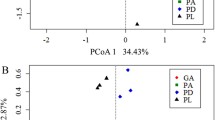
As mentioned above, beta diversity using a Bray–Curtis distance matrix was calculated based on functional gene (probe) intensities as a proxy for abundance. Clustering according to coral genus was evident, with the two octocorals A. aspera (A2) and A. spissa (A1) and the two Desmophyllum spp. (designated by D and L; Table 1) clustering together and distinct from the Enallopsammia (Fig. 2). Microbial community structure (beta diversity) was significantly different for all metabolic categories (Table 2). This means that each coral species harbored a microbial community that differed from the rest in either the presence/absence of functional genes or in the relative proportion of these genes. However, due to the lack of statistical power combined with a Bonferroni correction, no significant differences could be detected in the pairwise ADONIS results.

Non-metric multi-dimensional scaling plots (NMDS) based upon a square-root transformed Bray–Curtis dissimilarity matrix calculated using all genes for microbiomes of Acanthogorgia spp. (A1-A2), Desmophyllum dianthus (D1-D3), Desmophyllum pertusum (L1-L3), and Enallopsammia profunda (E1-E3). GeoChip IDs as listed in Table 1
Gene family comparisons–Richness
The diversity (total number of genes) and total intensity (proportional to abundance) were evaluated for each gene family. Gene annotations are given in Online Resource 2. Excluding differences in the phylogeny category, of the 383 gene families detected, only 7 differed in gene richness among the coral species (Table 3, Fig. 3, Online Resource 3). Relative to Desmophyllum spp., E. profunda and Acanthogorgia spp. each had an additional cpne gene (5-oxovalerate dehydrogenase) from a Vibrio. Acanthogorgia spp. had one additional accD gene for carbon fixation from Chloroflexus. Desmophyllum diathus had additional diversity in carbon fixation genes via two archaeal genes from Thermoproteus and Vulcanisaeta (C CoA hydratase DiC4HB) and a bacterial icd gene from Hydrogenobacter. Enallopsammia profunda had an additional mct gene from Chloroflexus. Both Desmophyllum spp. had an additional archaeal succinyl-CoA reductase gene from Thermoproteus compared to the other corals, and E. profunda had one additional nitrogen cycling gene for hydrazine synthase, critical for anammox reactions, from Planctomycetaceae. When considering the total number of genes within a gene family, microbiome comparisons between Enallopsammia profunda vs. Desmophyllum dianthus and Acanthogorgia spp. vs. Desmophyllum dianthus both had the highest number of significant gene families (10 each), while microbiomes from the two Desmophyllum spp. had the fewest significantly different gene families (3; Table 4).

All significantly different genes according to the total number of each gene family for microbiomes of Acanthogorgia spp. (A1-A2), Desmophyllum dianthus (D1-D3), Desmophyllum pertusum (L1-L3), and Enallopsammia profunda (E1-E3); (genes that were not significant are not shown). GeoChip IDs as listed in Table 1. Significant differences between total number of gene families were evaluated with an Analysis of Variance (ANOVA) and significant tests (p ≤ 0.01) were followed up with a Tukey’s HSD (honestly significant difference) test
Gene family comparisons–Abundance
Unlike richness (total number of different genes), the total intensity of a gene family drove the microbiome differences between corals (Fig. 4). Significantly different metabolic categories by total intensity include carbon cycling, metal homeostasis, organic remediation, and phylogeny (Fig. 4, Table 2, Online Resource 3). Excluding differences in the phylogeny category, of the 383 gene families detected, 60 differed in total intensity (a direct correlation to abundance) among the coral species (Table 3). Carbon fixation and degradation showed variability across the coral microbiomes, but the pmoa gene (methane oxidation) was significantly higher in D. pertusum’s microbiome compared to those of all other corals. Within the metal homeostasis category, the mer gene (mercury detoxification) was abundant across all coral microbiomes, the arxa gene (arsenic detoxification) was significantly higher in the E. profunda microbiome than in the other corals, and silicon transport was significantly higher in Acanthogorgia spp. microbiomes compared to those of stony corals. For nitrogen cycling, the amoa gene (nitrification) and the narg gene (denitrification) were highly abundant across all coral microbiomes, but E. profunda’s microbial community had significantly higher abundance of denitrification genes nirs (compared to microbiomes of all other corals) and nosz (compared to microbiomes of D. dianthus and Acanthogorgia spp.). For sulfur cycling, both significantly different genes (sir and aps apra) were more abundant in E. profunda microbiomes than those of all other corals. Organic remediation also showed a lot of variability across coral microbiomes. Acanthogorgia spp. microbiomes had significantly higher abundance of the adpb gene (organophosphorus degradation) and the nahb gene (degradation of polycyclic aromatics) relative to the stony corals. E. profunda microbiomes had significantly higher abundance relative to the other coral species in several organic contaminant degradation genes, including the fungal enzyme aromatic peroxygenase. Overall, E. profunda vs. Acanthogorgia spp. microbiomes had the most significantly different gene families when taking the total intensity into account (51), while D. dianthus vs. D. pertusum microbiomes had the fewest significantly different gene families (13; Table 4).

All significantly different genes according to the total intensity of each gene family for microbiomes of Acanthogorgia spp. (A1-A2), Desmophyllum dianthus (D1-D3), Desmophyllum pertusum (L1-L3), and Enallopsammia profunda (E1-E3); (genes that were not significant are not shown). GeoChip IDs as listed in Table 1. Note scale differences for each panel. Significant differences between total intensity of gene families were evaluated with an Analysis of Variance (ANOVA) and significant tests (p ≤ 0.01) were followed up with a Tukey’s HSD (honestly significant difference) test
Gene family comparisons–Phylogeny
Within the phylogeny category, GeoChip 5.0S is equipped with 472 probes to detect up to 219 different genera of protists (microeukaryotes), 60 of which were detected in this study. The phylogeny category had 8 significant differences when considering numbers of each gene family (genera; Fig. 3) compared to 9 significant differences when considering the total intensity of each gene family (genera; Fig. 4). These included common marine protists, such as heterotrophic algae, green algae, brown algae, diatoms, choanoflagellates, and coccolithophorids. Various pathogenic protists were detected, including the genera Perkinsus, Giardia, Leishmania, Bonamia, Trypanosoma, Blastocystis, Rhynchopus, Entamoeba, and Pythium. Finally, ten Parabasalids were detected, four of which were significantly different. Only the phylogeny category (protists) was different between microbiomes of the closely related D. dianthus and D. pertusum, indicating that while bacterial metabolic categories remain similar among the two coral species, they each harbor a unique protist community.
Comparisons to previously published studies
16S rRNA gene surveys generated from technical replicates of some of the coral colonies analyzed here, and that were previously published in Kellogg and Pratte (2021), reflected diversity patterns of functional genes; Acanthogorgia spp. microbiomes had the lowest taxonomic diversity while D. dianthus had the highest. However, it must be noted that 16S rRNA data were only available for 6 of the 11 samples presented here, thus our conclusions are tentative.
Finally, a literature search was conducted to identify any studies that utilized the same version of this microarray and reported the number of genes detected for each of the gene categories (Online Resource 4). Of particular interest, one of these studies represented four species of tropical corals (Zhang et al. 2015). More carbon, nitrogen, and phosphorus cycling genes were detected in deep-water and tropical corals than seawater or several types of soil (Online Resource 4). The number of genes detected for both tropical and deep-water coral microbiomes were remarkably similar for the vast majority of metabolic categories when compared to the other ecosystems (Online Resource 4). However, seven metabolic sub-categories had a calculated percent difference outside the 99% upper confidence interval limit (> 12.3%) when comparing between tropical and deep coral microbiomes (Online Resource 4). Deep-water coral microbiomes had higher representation of anaerobic ammonium oxidation (anammox; 23.53%), chitin degradation (14.26%), and dimethylsulfoniopropionate (DMSP) degradation (44.57%). Tropical coral microbiomes had higher representation of antibiotic resistance (63.18%), chromium homeostasis/resistance (23.81%), copper homeostasis/resistance (16.67%), and methanogenesis (25.00%).
Discussion
Our study provides the first comparison of microbial functional genes across five species of cold-water corals from the deep ocean, including both octocoral and scleractinian representatives. On a broad scale, all five deep-water coral species’ functional microbiomes were highly similar to one another, although some significant differences could be detected. Of 383 total microbial gene families detected in this study, less than 20% were significantly different among these deep-water coral species, in spite of their very different life-history traits. Of the 7 gene families that were significantly different by total number of genes per coral species (Table 3, Fig. 3, Online Resource 3), none were different by more than one gene, implying that the total diversity of any one gene family was not critical in differentiating the coral microbiomes. Further, most of the significant differences detected among deep-water coral microbiomes were associated with the intensity of gene families rather than the taxonomic diversity within any given gene family (Table 4, Fig. 4). Differences in gene intensity rather than taxonomic diversity within a gene family imply that the overall abundance of a gene family is more important in a deep-sea coral microbiome, rather than the number of the different microbes that carry the gene or their identity. This result supports the idea of specific symbionts being dominant contributors to certain functions in addition to the many low abundance microbes that typically make up a distinct coral microbiome. If the corals were selecting for function alone, we might expect to see more taxonomic variability within a gene family across the different coral species. The functional complexity of the microbiota of Acanthogorgia spp. is less diverse compared to the scleractinian samples, contributing to these octocorals being most distinct from other deep-water coral species examined.
Prior 16S rRNA gene taxonomic studies of deep-sea coral microbiomes have shown that the host is the dominant driver of microbiome structure (Kellogg 2019), and we observe a similar effect based on functional genes in the clustering of coral species within a genus (i.e., the Desmophyllum spp. and Acanthogorgia spp.) and the difference between more divergent lineages of stony corals like Desmophyllum spp. and E. profunda (Fig. 2). This clustering could be indicative of phylosymbiosis, an eco-evolutionary pattern in which host-associated microbiome relationships mirror the phylogeny of their hosts (O’Brien et al. 2020). Phylosymbiosis (based on 16S rRNA genes rather than functional genes) has been proposed for Mediterranean octocorals (van de Water et al. 2018) and tropical scleractinian corals (Pollock et al. 2018). Using a number of statistical tests, a phylosymbiotic signal was confirmed across several Australian marine invertebrate groups including scleractinian corals, octocorals, and sponges (O’Brien et al. 2020).
Carbon cycling
Carbon fixation genes showed variability across the coral species both in terms of significantly different numbers of genes (Fig. 3) and abundance (Fig. 4). Five of the 7 genes that were significantly different by number were related to carbon fixation, with D. dianthus having more diversity in the C CoA hydratase DiC4HB gene (dicarboxylate/4-hydroxybutyrate cycle) and isocitrate dehydrogenase gene (tricarboxylic acid cycle) compared to the other coral species, while Acanthogorgia spp. and E. profunda had more diversity in the 3-hydroxypropionate bicycle. Of particular note, the intensity of pmoa genes for methane oxidation in Desmophyllum spp. microbiomes was higher compared to the other coral species, with the highest abundance in D. pertusum (Fig. 4). Previous functional predictions based on 16S rRNA gene data from D. pertusum showed high values for carbon fixation and methane metabolism (Kellogg et al. 2017). There has been recent evidence that deep-sea octocorals may derive nutritional benefits from seeps via their microbiomes (Osman et al. 2023) and one of the D. pertusum samples (L3; Table 1) was collected from a seep site; however, the relative abundance of the pmoa gene was similar between the seep and non-seep samples.
Nitrogen cycling
Nitrogen cycling in deep-sea corals has been hypothesized based on the presence of bacterial genera known to be capable of the various steps (Kellogg et al. 2016; Kellogg 2019) and detection of fixed nitrogen incorporation (Middelburg et al. 2015), but previously there were no direct links between the microbial taxonomy present and specific functional capabilities. Results of the microarray analysis revealed archaeal and bacterial genes for ammonification, fungal nitrogen cycling genes, and numerous bacterial genes for nitrogen fixation, nitrate reduction, nitrite reduction, and denitrification in these five species of deep-sea corals. The genes detected in these deep-sea corals include those associated with many of the bacterial genera previously hypothesized to play roles in nitrogen cycling: e.g., Bradyrhizobium, Methylobacterium, Campylobacter, Acidovorax, Propionibacterium (Kellogg et al. 2016; Kellogg 2019). For nitrogen cycling, nitrification (amoa gene) and denitrification (narg gene) were highly abundant across all corals, but E. profunda was significantly higher in abundance of denitrification genes nirs (compared to all other corals) and nosz (compared to D. dianthus and Acanthogorgia spp.). Compared to the other coral species, E. profunda had one additional nitrogen cycling gene for hydrazine synthase, critical for anammox reactions, from Planctomycetaceae.
Microeukaryotic phylogeny
While many of the coral-associated protists detected seem logical, such as the marine protists, some of them require more critical contemplation. Pathogenic genera such as Giardia, Trypanosoma, and Leishmania are terrestrial/mammal associated, and Parabasalids are commonly associated with the hindguts of termites–unlikely associates of deep-sea corals. There are three plausible explanations for these unlikely protists. (1) Although the microarray data were “zeroed” in order to remove noise, some noise signals were still detected, (2) non-specific binding of probes occurred, or (3) analogues of these protists were present in the deep-water coral samples, either as food, pathogens, or commensals (e.g., as in Vohsen et al. 2020). While it is likely that a combination of these explanations is at play and these results must be interpreted with caution, certain patterns emerge from these data that provoke thought. For example, different genera of Parabasalids were quite specific to coral species; for example, the genus Foaina was far more abundant in D. dianthus, while the genus Devescovina was more abundant in E. profunda. The implications of these findings are unclear. It is likely that many of these protists have never been reported in association with coral reefs simply because no one is looking for them (del Campo et al. 2020). Given the diversity of protists detected in the study, we suggest that protists may play critical roles in coral health and disease, and that future studies consider examining protists in studies of the coral microbiome.
Comparisons to tropical corals
The number of gene families detected in this study (between 305 and 320 per deep-sea coral sample) were within the same range as Zhang et al. (2015), which detected 288–348 gene families per tropical coral sample, with an average of only 8.3% (99% CI[4,12.3]) difference between the two. Unexpectedly, given the environmental differences and absence of an obligate photosynthetic algal symbiont, out of 59 metabolic sub-categories for which we were able to make a direct comparison to tropical coral microbiomes, only 7 were notably different based on number of gene families observed (above the 95% confidence interval). This is in contrast to GeoChip microarray studies that have shown substantial functional gene differences (i) inside and outside an oil spill plume (Lu et al. 2012), (ii) between soil ecotones (Shen et al. 2016), and (iii) in different marine water masses (McFarlin et al. 2017). This implies a broad-scale convergence of the functional genes present within the coral holobiont, independent of coral species, depth, symbiont status, and morphology (e.g., branching colonies versus solitary polyp cup corals). A similar broad-scale convergence has also been reported for sponges (Fan et al. 2012; Bayer et al. 2014). However, these conclusions are drawn from the presence/absence data as the available data did not enable a comparison of intensity across studies. It remains to be determined whether the relative abundance of these functional genes, rather than their presence, absence, or taxonomic diversity, differentiates tropical from cold-water coral microbiomes.
Dependence on heterotrophic feeding is a likely explanation for the higher chitin degradation capacity detected in this study compared to values in tropical coral microbiomes (Online Resource 4). Chitin is a source for carbon and nitrogen and is present in the exoskeletons of zooplankton consumed by these corals. Moreover, a recent study has confirmed the presence of various types of chitinase enzymes in shallow octocoral microbiomes by both culture-based and functional metagenomic methods (Raimundo et al. 2021). Based on their findings, Raimundo et al. hypothesized a chitin-degrading role for Gammaproteobacterial coral symbionts in the family Endozoicomonaceae/Hahellaceae as well as Chloroflexi. Gammaproteobacteria were the dominant class in these deep-sea corals, and Endozoicomonas made up a substantial part of the microbiome for two of the D. pertusum samples and both Acanthogorgia spp. based on 16S rRNA gene surveys (Kellogg and Pratte 2021). The phylum Chloroflexi was also present as 2 out of 16 most abundant classes detected for these corals (Kellogg and Pratte 2021). Another possible role for chitinase-producing bacteria in the coral holobiont is to protect the host from chitin-containing fungal pathogens (Douglas et al. 2007).
Most striking between deep-sea and tropical coral microbes were the differences in antibiotic resistance genes and metal homeostasis genes. Tropical coral microbiomes were enriched in antibiotic resistance genes, with nearly twice as many antibiotic resistance genes reported compared to deep-water corals (Zhang et al. 2015). Collins et al. (2021) also found a low number of antibiotic resistance genes in microbiomes of deep-water fishes, suggesting that this trend is common across all deep-water macro-organisms. Further, a survey of antibiotic resistance genes in water depths below 100 m in the Black Sea found that some were undetectable, and others detected at a much lower abundance than in coastal waters (Sabatino et al. 2020). Although this pattern should be confirmed in additional datasets, it may indicate that the deep sea is relatively protected from the anthropogenic distribution of antibiotics compared to more shallow-water environments, or that chemical antagonism between microbial taxa is more common in tropical coral microbiomes. Metal homeostasis genes were significantly less diverse for Acanthogorgia spp. microbiomes compared to all other coral species, possibly suggesting that stony corals undergo more metals stress. Despite the clear differences in diversity of metal homeostasis genes in microbiomes of soft and hard corals, the intensities of only a few gene families were significantly different with no clear patterns. When compared to deep-water coral microbiomes, those of tropical corals were enriched in copper and chromium detoxification genes, suggesting a higher copper and chromium exposure (potentially from anti-fouling paint of recreational vessels or run-off, e.g., Pait et al. 2009; Burgin and Hardiman 2011). Recent studies in freshwater systems and at marine shallow-water hydrothermal vents have found linkages between higher heavy metal concentrations and increased abundance of antibiotic resistance genes (Komijani et al. 2021; Arcadi et al. 2022).
Coral reefs are one of the most prolific producers of dimethylsulfoniopropionate (DMSP) and its byproduct dimethylsulphide (DMS), an important negative greenhouse gas that promotes cloud formation and precipitation (Broadbent and Jones 2004). Previously, it was thought that the production of DMSP was only through photosynthetic marine algae, particularly the coral endosymbiont zooxanthellae (Broadbent et al. 2002; Van Alstyne et al. 2006). Production of DMSP by azooxanthellate deep-water corals below the photic zone therefore was not considered important. However, there is now evidence that coral animals themselves (independent from the zooxanthellae) possess the capacity to produce DMSP (Raina et al. 2013), as do many marine bacteria (Curson et al. 2017), reopening the possibility for DMSP production in deep-water coral habitats. Indeed, Burdett et al. (2014) detected intracellular DMSP levels in the deep-water coral D. pertusum equivalent to that of tropical zooxanthellate scleractinian corals, which they hypothesize is possible through food uptake. Our study suggests that D. pertusum is not the only deep-water coral that contains DMSP, as DMSP-degradation genes were also detected in microbiomes from D. dianthus, E. profunda, and even the octocorals Acanthogorgia spp. It should be noted that Geochip 5.0 only includes the DMSP-degradation gene dmdA (the demethylation pathway producing acrylate) and not the alternative cleavage pathway that converts DMSP to DMS. However, the deep-water coral microbiomes in this study had nearly twice the diversity of dmdA genes compared to tropical corals, suggesting a previously unknown significant source of DMSP and DMS.
There was a surprising amount of functional similarity between deep-sea and shallow-water coral microbiomes which may indicate a broad-scale convergence of the functional genes present within the coral holobiont, independent of coral species, depth, symbiont status, and morphology. The small number of significant differences between microbiomes of cold-water and tropical corals point to differences in nutrition strategies (heterotrophy vs autotrophy), differing environmental levels of pollution exposure, and an unexpected abundance of DMSP-degradation genes occurring in deep-sea corals. We found the use of a microarray with a broad range of microbial functional and taxonomic genes to be a solution to the currently unresolved problem of coral metagenomes being dominated by host coral sequences, particularly for uncovering microeukaryote diversity. Our findings suggest there remains a great deal to discover about the roles and abilities of coral-associated microbes, and the importance of functional redundancy versus taxonomic diversity.
Data availability
The microarray data and collection metadata generated and analyzed in this study can be found in the USGS data release https://doi.org/10.5066/P9RPE8YX.
References
Addamo AM, Vertino A, Stolarski J, García-Jiménez R, Taviani M, Machordom A (2016) Merging scleractinian genera: the overwhelming genetic similarity between solitary Desmophyllum and colonial Lophelia. BMC Evol Biol 16:108
Arcadi E, Rastelli E, Tangherlini M, Rizzo C, Mancuso M, Sanfilippo M, Esposito V, Andaloro F, Romeo T (2022) Shallow-water hydrothermal vents as natural accelerators of bacterial antibiotic resistance in marine coastal areas. Microorganisms 10:479
Bayer K, Moitinho-Silva L, Brümmer F, Cannistraci CV, Ravasi T, Hentschel U (2014) GeoChip-based insights into the microbial functional gene repertoire of marine sponges (high microbial abundance, low microbial abundance) and seawater. FEMS Microbiol Ecol 90:832–843
Broadbent AD, Jones GB (2004) DMS and DMSP in mucus ropes, coral mucus, surface films and sediment pore waters from coral reefs in the Great Barrier Reef. Mar Freshwater Res 55:849–855
Broadbent AD, Jones GB, Jones RJ (2002) DMSP in corals and benthic algae from the Great Barrier Reef. Estuar Coast Shelf S 55:547–555
Buhl-Mortensen L, Mortensen PB (2004) Symbiosis in deep-water corals. Symbiosis 37:33–61
Buhl-Mortensen L, Mortensen PB (2005) Distribution and diversity of species associated with deep-sea gorgonian corals off Atlantic Canada. In: Freiwald A, Roberts JM (eds) Cold-water Corals and Ecosystems. Springer-Verlag, Berlin, pp 849–879
Burdett HL, Carruthers M, Donohue PJC, Wicks LC, Hennige SJ, Roberts JM, Kamenos NA (2014) Effects of high temperature and C02 on intracellular DMSP in the cold-water coral Lophelia pertusa. Mar Biol 161:499–1506
Burgin S, Hardiman N (2011) The direct physical, chemical and biotic impacts on Australian coastal waters due to recreational boating. Biodivers Conserv 20:683–701
Collins FWJ, Walsh CJ, Gomez-Sala B, Guijarro-García E, Stokes D, Jakobsdóttir KB, Kristjánsson K, Burns F, Cotter PD, Rea MC, Hill C, Ross RP (2021) The microbiome of deep-sea fish reveals new microbial species and a sparsity of antibiotic resistance genes. Gut Microbes 13:1921924
Cordes EE, McGinley MP, Podowski EL, Becker EL, Lessard-Pilon S, Viada ST, Fisher CR (2008) Coral communities of the deep Gulf of Mexico. Deep Sea Res Part I Oceanogr Res Pap 55:777–787
Curson AR, Liu J, Bermejo Martínez A, Green RT, Chan Y, Carrión O, Williams BT, Zhang S-H, Yang G-P, Bulman Page PC, Zhang X-H, Todd JD (2017) Dimethylsulfoniopropionate biosynthesis in marine bacteria and identification of the key gene in this process. Nat Microbiol 2:17009
Das S, Gwon HS, Khan MI, Van Nostrand JD, Alam MA, Kim PJ (2019) Taxonomic and functional responses of soil microbial communities to slag-based fertilizer amendment in rice cropping systems. Environ Int 127:531–539
del Campo J, Bass D, Keeling PJ (2020) The eukaryome: Diversity and role of microeukaryotic organisms associated with animal hosts. Funct Ecol 34:2045–2054
Douglas NL, Mullen KM, Talmage SC, Harvell CD (2007) Exploring the role of chitinolytic enzymes in the sea fan coral, Gorgonia ventalina. Mar Biol 150:1137–1144
Fan L, Reynolds D, Liu M, Stark M, Kjelleberg S, Webster NS, Thomas T (2012) Functional equivalence and evolutionary convergence in complex communities of microbial sponge symbionts. Proc Nat Acad Sci USA 109:E1878–E1887
He Z, Gentry TJ, Schadt CW, Wu L, Liebich J, Chong SC, Huang Z, Wu W, Gu B, Jardine P, Criddle C, Zhou J (2007) GeoChip: A comprehensive microarray for investigating biogeochemical, ecological and environmental processes. ISME J 1:67–77
Kellogg CA (2019) Microbiomes of stony and soft deep-sea corals share rare core bacteria. Microbiome 7:90
Kellogg CA, Pratte ZA (2021) Unexpected diversity of Endozoicomonas in deep-sea corals. Mar Ecol Prog Ser 673:1–15
Kellogg CA, Ross SW, Brooke SD (2016) Bacterial community diversity of the deep-sea octocoral Paramuricea placomus. PeerJ 4:e2529
Kellogg CA, Goldsmith DB, Gray MA (2017) Biogeographic comparison of Lophelia-associated bacterial communities in the western Atlantic reveals conserved core microbiome. Front Microbiol 8:796
Kimes NE, Van Nostrand JD, Weil E, Zhou J, Morris PJ (2010) Microbial functional structure of Montastraea faveolata, an important Caribbean reef-building coral, differs between healthy and yellow-band diseased colonies. Environ Microbiol 12:541–556
Komijani M, Shamabadi NS, Shahin K, Eghbalpour F, Tahsili MR, Bahram M (2021) Heavy metal pollution promotes antibiotic resistance potential in the aquatic environment. Environ Poll 274:116569
Lawler SN, Kellogg CA, France SC, Clostio RW, Brooke SD, Ross SW (2016) Coral-associated bacterial diversity is conserved across two deep-sea Anthothela species. Front Microbiol 7:458
Li J, Wang G, Yan B, Liu G (2020) The responses of soil nitrogen transformation to nitrogen addition are mainly related to the changes in functional gene relative abundance in artificial Pinus tabulaeformis forests. Sci Total Environ 723:137679
Lu Z, Deng Y, Van Nostrand JD, He Z, Voordeckers J, Zhou A, Lee Y-J, Mason OU, Dubinsky EA, Chavarria KL, Tom LM, Fortney JL, Lamendella R, Jansson JK, D’haeseleer P, Hazen TC, Zhou J (2012) Microbial gene functions enriched in the Deepwater Horizon deep-sea oil plume. ISME J 6:451–460
McFarlin KM, Questel JM, Hopcroft RR, Leigh MB (2017) Bacterial community structure and functional potential in the northeastern Chukchi Sea. Cont Shelf Res 136:20–28
Middelburg JJ, Mueller CE, Veuger B, Larsson AI, Form A, van Oevelen D (2015) Discovery of symbiotic nitrogen fixation and chemoautotrophy in cold-water corals. Sci Rep 5:17962
Navarrete AA, Mellis EV, Escalas A, Lemos LN, Lavres J Jr, Quaggio JA, Zhou J, Tsai SM (2017) Zinc concentration affects the functional groups of microbial communities in sugarcane-cultivated soil. Agr Ecosyst Environ 236:187–197
O’Brien PA, Tan S, Yang C, Frade PR, Andreakis N, Smith HA, Miller DJ, Webster NS, Zhang G, Bourne DG (2020) Diverse coral reef invertebrates exhibit patterns of phylosymbiosis. ISME J 14:2211–2222
Oksanen J, Blanchet FG, Friendly M, Kindt R, Legendre P, McGlinn D, Minchin PR, O’Hara RB, Simpson GL, Solymos P, Stevens MHH, Szoecs E, Wagner H (2016) vegan: Community Ecology PackageR package version 2.4–0:https://CRAN.R-project.org/package=vegan
Osman EO, Vohsen SA, Girard F, Cruz R, Glickman O, Bullock LM, Anderson KE, Weinnig AM, Cordes EE, Fisher CR, Baums IB (2023) Capacity of deep-sea corals to obtain nutrition from cold seeps aligned with microbiome reorganization. Glob Change Biol 29:189–205
Pait AS, Jeffrey CFG, Caldow C, Whitall DR, Hartwell SI, Mason AL, Christensen JD (2009) Chemical contamination in southwest Puerto Rico: A survey of contaminants in the coral Porites astreoides. Carib J Sci 45:191–203
Pollock FJ, McMinds R, Smith S, Bourne DG, Willis BL, Medina M, Thurber RV, Zaneveld JR (2018) Coral-associated bacteria demonstrate phylosymbiosis and cophylogeny. Nat Commun 9:4921
Pratte ZA, Kellogg CA (2021) Comparison of preservation and extraction methods on five taxonomically disparate coral microbiomes. Front Mar Sci 8:684161
R Core Team (2018) R: A language and environment for statistical computing, R Foundation for Statistical Computing, Vienna, Austria
Raimundo I, Silva R, Meunier L, Valente SM, Lago-Lestón A, Keller-Costa T, Costa R (2021) Functional metagenomics reveals differential chitin degradation and utilization features across free-living and host-associated marine microbiomes. Microbiome 9:43
Raina J-B, Tapiolas DM, Forêt S, Lutz A, Abrego D, Ceh J, Seneca FO, Clode PL, Bourne DG, Willis BL, Motti CA (2013) DMSP biosynthesis by an animal and its role in coral thermal stress response. Nature 502:677–680
Roberts S, Hirshfield M (2004) Deep-sea corals: out of sight, but no longer out of mind. Front Ecol Environ 2:123–130
Ross SW, Quattrini AM (2007) The fish fauna associated with deep coral banks off the southeastern United States. Deep Sea Res Part I Oceanogr Res Pap 54:975–1007
Sabatino R, Di Cesare A, Dzhembekova N, Fontaneto D, Eckert EM, Corno G, Moncheva S, Bertoni R, Callieri C (2020) Spatial distribution of antibiotic and heavy metal resistance genes in the Black Sea. Mar Poll Bull 160:111635
Shannon CE (1948) The mathematical theory of communication. Bell Syst Tech J 27:379–423
Shen C, Shi Y, Ni Y, Deng Y, Van Nostrand JD, He Z, Zhou J, Chu H (2016) Dramatic increases of soil microbial functional gene diversity at the treeline ecotone of Changbai Mountain. Front Microbiol 7:1184
Shi Z, Yin H, Van Nostrand JD, Voordeckers JW, Tu Q, Deng Y, Yuan M, Zhou A, Zhang P, Xiao N, Ning D, He Z, Wu L, Zhou J (2019) Functional gene array-based ultrasensitive and quantitative detection of microbial populations in complex communities. mSystems 4:00296–00219
Stone RP (2006) Coral habitat in the Aleutian Islands of Alaska: depth distribution, fine-scale species associations, and fisheries interactions. Coral Reefs 25:229–238
Sulak KJ, Brooks RA, Luke KE, Norem AD, Randall M, Quaid AJ, Yeargin GE, Miller JM, Harden WM, Caruso JH, Ross SW (2007) Demersal fishes associated with Lophelia pertusa coral and hard-substrate biotypes on the continental slope, northern Gulf of Mexico. Bull Mar Sci 81:65–92
Van Alstyne KL, Schupp P, Slattery M (2006) The distribution of dimethylsulfoniopropionate in tropical Pacific coral reef invertebrates. Coral Reefs 25:321–327
van de Water JAJM, Voolstra CR, Rottier C, Cocito S, Peirano A, Allemand D, Ferrier-Pagès C (2018) Seasonal stability in the microbiomes of temperate gorgonians and the red coral Corallium rubrum across the Mediterranean Sea. Microb Ecol 75:274–288
Vohsen SA, Anderson KE, Gade AM, Gruber-Vodicka HR, Dannenberg RP, Osman EO, Dubilier N, Fisher CR, Baums IB (2020) Deep-sea corals provide new insight into the ecology, evolution, and the role of plastids in widespread apicomplexan symbionts of anthozoans. Microbiome 8:34
Wang C, Wang X, Liu D, Wu H, Lü X, Fang Y, Cheng W, Luo W, Jiang P, Shi J, Yin H, Zhou J, Han X, Bai E (2014) Aridity threshold in controlling ecosystem nitrogen cycling in arid and semi-arid grasslands. Nat Commun 5:4799
Wegley L, Edwards R, Rodriguez-Brito B, Liu H, Rohwer F (2007) Metagenomic analysis of the microbial community associated with the coral Porites astreoides. Environ Microbiol 9:2707–2719
Wu L, Liu X, Schadt CW, Zhou J (2006) Microarray-based analysis of subnanogram quantities of microbial community DNAs by using whole-community genome amplification. Appl Environ Microbiol 72:4931–4941
Zhang Y, Ling J, Yang Q, Wen C, Yan Q, Sun H, Van Nostrand JD, Shi Z, Zhou J, Dong J (2015) The functional gene composition and metabolic potential of coral-associated microbial communities. Sci Rep 5:16191
Acknowledgements
We thank the captains and crews of the RV Atlantis and NOAA ship Ronald H. Brown and the submersible groups supporting Alvin and Jason II (WHOI). We also acknowledge the chief scientists of the Deep-SEARCH expeditions: Erik Cordes (Temple University) and Amanda Demopoulos (U.S. Geological Survey). Andrea Quattrini (Smithsonian Institution) and Catherine McFadden (Harvey Mudd College) confirmed octocoral host species identities. This project was sponsored by the National Oceanographic Partnership Program with funding from the Bureau of Ocean Energy Management (contract M17PC00009), the U. S. Geological Survey, and the National Oceanic and Atmospheric Administration Office of Ocean Exploration and Research (for ship time). Any use of trade, firm, or product names is for descriptive purposes only and does not imply endorsement by the U.S. Government.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there are no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
338_2023_2409_MOESM1_ESM.pdf
Online Resource 1. A non-metric multidimensional scaling (NMDS) plot comparing all the microbial genes present in the coral samples and the kit blank control. A1 = Acanthogorgia spissa; A2 = Acanthogorgia aspera; D1-D3 = Desmophyllum dianthus; L1-L3 = Desmophyllum pertusum (formerly Lophelia pertusa); E1-E3 = Enallopsammia profunda; Blank = kit extraction control (PDF 154 KB)
338_2023_2409_MOESM2_ESM.pdf
Online Resource 2. A table listing GeoChip 5.0S gene annotations for significantly different gene families between deep-sea corals in this study (differences based on either total number of gene families or total intensities of gene families). A complete list of all gene families on the microarray can be found in Shi et al. (2019), Table S1. NA means not applicable. (PDF 153 KB)
338_2023_2409_MOESM3_ESM.xlsx
Online Resource 3. An Excel workbook with one sheet providing data on genes that were significantly different in number between two or more corals in this study and one sheet providing data on genes that were significantly different based on intensity (abundance) between two or more corals in this study. A dash indicates there was no significant difference. (XLSX 30 KB)
338_2023_2409_MOESM4_ESM.xlsx
Online Resource 4. An Excel workbook with one sheet comparing the number of genes detected for each of the metabolic categories in this study versus previously published studies using GeoChip 5.0 (S or M versions) in other environments, including a comparison against tropical corals. A dash indicates that a comparable gene count value was not available in the specific paper. The second sheet further compares this study’s gene counts and taxonomic sources of those genes against the data from tropical corals in Zhang et al. 2015. (XLSX 31 KB)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pratte, Z.A., Stewart, F.J. & Kellogg, C.A. Functional gene composition and metabolic potential of deep-sea coral-associated microbial communities. Coral Reefs 42, 1011–1023 (2023). https://doi.org/10.1007/s00338-023-02409-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00338-023-02409-0