
Abstract
Palau suffered massive mortality of reef corals during the 1998 mass bleaching, and understanding recovery from that catastrophic loss is critical to management for future impacts. Many reef species have shown significant genetic structure at small scales while apparently absent at large scales, a pattern often referred to as chaotic genetic patchiness. Here we use hierarchical sampling of population structure scored from a panel of microsatellite markers for the coral Acropora hyacinthus across the islands of Yap, Ngulu and Palau to evaluate hypotheses about the mechanisms of previously described chaotic genetic structure. As with previous studies, we find no isolation-by-distance within or between the three islands and high genetic structure between sites separated by as little as ~ 10 km on Palau. Using kinship among individual colonies, however, we find higher mean pairwise relatedness coefficients among individuals within sampling sites. Comparing population structure among hierarchical sampling scales, we show that the pattern of chaotic genetic patchiness reported previously appears to derive from genetic patches of local kin groups at small spatial scales. Genetic distinction of Palau from neighboring islands and high kinship among individuals within these kinship neighborhoods implies that the coral reefs of Palau apparently recovered through a mosaic of rare thermally tolerant colonies that survived the 1998 mass bleaching and are now spreading and recolonizing reefs as local kin groups. This pattern of recovery on Palau gives us a better understanding for effective coral reef conservation strategies in which protecting these rare survivors wherever they occur, rather than specific areas of reef habitat, is critical to increase coral reef resilience.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Scale plays a key role in defining ecological and evolutionary patterns (Levin 1992). Thus, the choice of scale at which to study population genetics, the patterns of population structure, their boundaries and connectivity, is critical (Hellberg 1995, 2009; Zvuloni et al. 2008). Two key components of spatial scale are (1) the “grain”: the minimum spatial resolution of the data or the measure of the smallest difference that can be detected, and (2) the “extent”: the scope of the study area. Observing a pattern at a grain that is too small can lead to the illusion of chaos or “noise” (Hewitt et al. 2010); observing a pattern at an extent not large enough may give the impression of homogeneity (Edmunds and Bruno 1996). Once a pattern is detected, it is then possible to look for the processes responsible for driving the observed variations and examine the scale at which such processes act.
Dispersal is one of the drivers of both population genetics and demography (Slatkin 1987; Cowen et al. 2007; Weersing and Toonen 2009; Lowe and Allendorf 2010), playing a role in the colonization of new spaces, as well as the persistence and recovery of populations. In marine systems, where most organisms are characterized by a bipartite life cycle with great potential for dispersal during the pelagic larval phase, it is widely expected that large scale processes play a fundamental role in population structure (Palumbi 1994; Kinlan et al. 2005; Liggins et al. 2013). High gene flow over large geographic areas will result in lower genetic differences among spatially close populations than those that are further apart (Wright 1943; Rousset 1997). However, there have been an increasing number of studies where these expected patterns either break down at small scale or do not appear at all and observations of unexpectedly high population structure over short distances are commonly identified as patterns of chaotic genetic patchiness (Johnson and Black 1982; Toonen and Grosberg 2011; Eldon et al. 2016). However, the definition of “small scale” is relative to realized dispersal, which remains a black box for most marine species (Buston and D’Aloia 2013). Thus, the scale at which gene flow will homogenize neutral genetic variation remains unknown for most species (Kinlan and Gaines 2003; Kinlan et al. 2005; Cowen et al. 2007; Selkoe and Toonen 2011). The observation of chaotic genetic patchiness could therefore be due to overestimating dispersal of many marine species (extent is too large).
A large proportion of marine metazoans have a biphasic life cycle in which the sedentary adults produce tiny pelagic larvae that disperse in the plankton before becoming competent to settle and metamorphose into the adult body form (Thorson 1950; Rieger 1994). Pelagic larval development is particularly prevalent in the tropics with a relative decrease in the frequency of pelagic larval development at higher latitudes, a trend known as Thorson’s rule (Jablonski and Lutz 1983). The discrepancy between the microscopic larvae and the scale at which we expect those larvae to interact with the environment hints at a more complex layer of processes that influence population structure in marine systems (Pineda et al. 2007; Cowen and Sponaugle 2009; Pringle et al. 2009). Studying patterns across multiple spatial scales, using spatially explicit data, could help identify medium- and small-scale processes that influence genetic structure (White et al. 2010; Selkoe et al. 2016).
Acropora hyacinthus is a widely distributed table coral that can be found commonly on shallow reefs of Palau between 3 and 10 m depth but is rare or absent on patch reefs, fringing reefs and within the lagoon (Bruno et al. 2001). A. hyacinthus reaches maturity around four to 5 years of age (Wallace 1985), which corresponds to approximately a 15–20 cm diameter colony (Guest et al. 2005; Baria et al. 2012). Although A. hyacinthus can reproduce asexually through fragmentation, previous studies show that relatively few clones are found in the field (Ayre and Hughes 2000; Márquez et al. 2002; Cros et al. 2016, 2017). A. hyacinthus is a hermaphroditic broadcast spawner with a larval pelagic duration of ~ 90 days under laboratory conditions (Márquez et al. 2002). It is both historically and currently one of the dominant coral species growing on the barrier reef of Palau (Golbuu et al. 2007; Victor et al. 2009). However, A. hyacinthus suffered nearly complete mortality from the 1998 El Niño bleaching event in Palau, virtually disappearing from the entire atoll with estimates of mortality approaching 100% (Bruno et al. 2001). In contrast, Yap and Ngulu populations were spared similar losses, and Palauan reefs recovered quickly with the population of A. hyacinthus rebounding by 2005, presumably due to larval dispersal from Yap and Ngulu (Golbuu et al. 2007). Subsequent genetic surveys found Palau was highly differentiated from sites on Yap and Ngulu which did not support the recolonization hypothesis (Cros et al. 2016). Further study revealed that virtually every sampling location around Palau was significantly differentiated from the others (Cros et al. 2017).
Previously, we studied population genetic structure of the coral Acropora hyacinthus at a regional scale, between the Micronesian islands of Yap, Ngulu and Palau as well as at an island scale, at 25 sites around Palau (Cros et al. 2016, 2017). Comparing population genetic structure (Fst & kinship) among sites, we found similar genetic structure among sites around Palau (island scale) as at regional scales among islands, with virtually every sampling location significantly differentiated from the others. There was no geographic patterning to the magnitude of genetic structure nor isolation-by-distance (IBD), giving the impression of chaotic genetic patchiness. Additionally, we found higher pairwise kinship values within sites than between sites and concluded that self-seeding likely played a role in structuring the populations of A. hyacinthus around Palau.
In this study, we combine the datasets (1418 colonies) from Cros et al. (2016, 2017) with new data (593 colonies) to determine whether the patterns of chaotic genetic patchiness we observed were the result of sampling and analyzing genetic structure at the wrong spatial extent and grain. To test this hypothesis, we vary the extent of the observations with an explicit hierarchical sampling design at four different spatial scales from a few centimeters to 500 km, and the grain with two different measures of genetic variation: a fixation index (F’st) measuring population differentiation at coarse grain and a kinship coefficient estimating genetic differences between each pair of individuals and representing population structure at a fine grain. Based on the previous work, we hypothesized that, using a smaller grain and extent, we would recover patterns of IBD at the appropriate scales because self-seeding impacts the genetic structure of A. hyacinthus at large scales, thereby explaining the previously observed patterns of chaotic genetic patchiness.
Methods
We combined the dataset previously published in Cros et al. (2016, 2017) as well as new data collected from a subset of six sites in which we exhaustively sampled all individuals of Acropora hyacinthus observed within a 2 × 100 m belt transect (Table 1). Individuals were confirmed to be the correct species (see below), and only Acropora hyacinthus were included in these analyses. Combining these new fine-scale samples with previous data creates an explicit hierarchical sampling design among sites and allows us to re-analyze the previous data to address whether grain or extent impact our interpretation of the results of previous studies, and the central question of what role scale plays in interpreting patterns of genetic structure in natural populations.
Study species identification
We confirmed species identity by running both a principal component analysis (PCA) in genodive v.2.0b27 (Meirmans 2014) and the Bayesian clustering algorithm implemented in structure ver. 2.3.4 (Pritchard et al. 2000), as advocated by Ladner and Palumbi (2012) to exclude individuals of other cryptic genetic lineages. The PCA was run on individuals using a covariance matrix with 10,000 permutations (Fig. S1, Supporting information). structure was run using an admixture model, without location as a prior, independent allele frequencies among populations and with a burn-in of 10,000 chains followed by 10,000 MCMC replications. Twenty independent runs were carried out for each K from 2 to 8. Summary of K values was plotted using clumpak (Kopelman et al. 2015, Fig S2. Supporting information). The analyses here and in the previous studies (Cros et al. 2016, 2017) only include those individuals who belong to the same taxonomic group based on these genetic tests, which we refer to as Acropora hyacinthus pending taxonomic clarification of the putative cryptic species complex (Ladner and Palumbi 2012; Sheets et al. 2018).
Sampling locations and methodology
Palau and Yap are island nations with large barrier reefs in Micronesia, separated roughly 450 km from each other. Ngulu is a coral atoll approximately 105 km from Yap and 350 km from Palau.
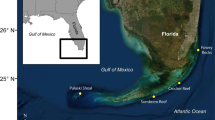
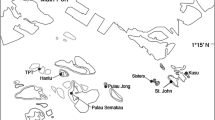
Spatial resolution for this study was defined within four spatial scales: (1) large scale (100 s of km) as the distance between the islands of Yap, Ngulu and Palau, separated by 160 to 550 km (Fig. 1a); (2) medium scale (10 s of km) as the distance between sites on the same island, separated by 5 to 150 km (Fig. 1b–d); (3) small scale (10 s of m) by distances between individuals within a site separated by 5 to 400 m (Fig. 1b); and (4) fine scale (10 s of cm) by distances between all individuals sampled within a single belt transect separated by less than a meter on average (Fig. 1b).

Location of sites where samples of Acropora hyacinthus were collected. a Location of Palau, Yap and Ngulu Atolls in Micronesia and collection sites in b Palau (25 sites and 6 transects marked in solid black), c Yap (3 sites) and d Ngulu Atoll (1 site) (adapted from Cros et al. 2016)
To study population structure at all four scales, we combined previously collected coral colonies from Yap, Ngulu Atoll and Palau with the new belt-transect samples collected for this study (Fig. 1a, Table 1). Sampling in Yap, Ngulu Atoll and Palau (Fig. 1a, Table 1) was carried out by two different laboratories in different years. In 2009–2012, Davies et al. (2015) sampled Yap and Ngulu and shared the extracted nucleic DNA, but all these samples were run from the DNA extracts so that results were not biased by laboratory protocols or scoring differences, and were directly comparable within this study (Cros et al. 2016). Briefly, at three sites on the barrier reef of Yap and a single site on the barrier reef of Ngulu (Fig. 1c, d), approximately 50 colonies (> 2 m apart) were randomly sampled using SCUBA or snorkeling. One small (~ 2 cm3) branch tip was collected, preserved in 96% ethanol and stored at 20 °C (Davies et al. 2015). Our laboratory sampled Palau in February and May 2012, at 25 sites along the outer barrier reef at a shallow depth (< 10 m) using SCUBA (Fig. 1b). Sites were selected in each of the four exposure zones around Palau, northeast (NE), northwest (NW), southeast (SE) and southwest (SW). One small branch tip per colony (< 2 cm3) was cut and preserved in salt-saturated DMSO buffer at room temperature (Gaither et al. 2011). A total of 1200 × 2 cm3 colony tips were collected by sampling haphazardly 48 colonies (> 2 m apart) of A. hyacinthus along transects of 4 × 200 m at each of these 25 sites.
At six of the 25 sites, we subsequently sampled all colonies of A. hyacinthus 5 cm or greater in diameter that could be identified reliably along a belt transect of 2 × 100 m (Fig. 1b). Each colony in the transect was photographed, measured and the collection position on the X and Y axis relative to the bottom left corner of the transect was recorded. One small branch tip per colony (< 2 cm3) was cut and preserved in salt-saturated DMSO at room temperature as above until extracted as outlined below.
DNA extraction and sequencing
A detailed description of DNA extraction and sequencing is described in (Cros et al. 2016). All the colonies from all three islands that were collected by both research groups were amplified and sequenced using the same protocol in the core laboratory at the Hawai'i Institute of Marine Biology starting with extracted genomic DNA for this study over the course of a couple years. Briefly, genomic DNA was amplified at eighteen microsatellite loci (Table S1, Supporting information) with colony identification (ID) tags (Table S2, Supporting information) and pooled by sites. We used a barcoded ligated Illumina adaptor (Illumina Inc., Hayward, CA, USA) for each colony from the same collection site to generate a library with a unique ID per individual per site. Individually barcoded amplicons were sequenced on an Illumina MiSeq at the Hawai'i Institute of Marine Biology, and each sample could be identified bioinformatically to an individual and site based on the unique barcode ID.
Data processing
We used the bioinformatics pipeline in Cros et al. (2016) to process all raw sequences from previous studies as well as the new data collected here. In brief, we demultiplexed the sequences by site, merged, separated according to primer and colony and trimmed for low quality sequences. We collapsed reads into unique sequences and used depth to apply filters developed in python (https://github.com/annickcros/Ahyacinthus-filters.git) for PCR and sequencing artifacts. Simple tandem repeats (STR) were separated from flanking regions with emboss: etandem and used to generate genotypes. Data were transformed in genodive v. 2.0b27 (Meirmans 2014) file format using formatting as described in Cros et al. (2016). The final analysis was carried out on two different datasets. We used 11 loci with less than 15% missing data (Table 2) to calculate measures of population differences. We used 9 of these loci to calculate kinship by eliminating two that had greater than 10% missing data (Table 2). The final number of colonies per site analyzed for each locus varied between 37 and 48 and the final number of colonies per transects for each locus varied between 61 and 178 (Table 2). We used genodive v.2.27 to confirm none of the loci included here showed evidence of null alleles. In total we analyzed 1418 colonies among sites and an additional 593 colonies from within transects.
Analysis
Descriptive analysis
We used genodive v.2.27 to test for clones within our samples, and to calculate descriptive statistics including the number of alleles, the effective number of alleles, and indices of genetic diversity at each of the 35 sites, as well as observed (HO), expected (HE) and corrected heterozygosity (H’T), inbreeding coefficient (GIS) and Nei’s fixation index GST (Table 2).
Measure of genetic structure at four scales
To understand the importance of scale in interpreting the population structure observed at each of the four different scales (large, medium small and fine), we used both an individual- and population-level approach. We used F-statistics as our measures of population structure (measured in genodive v.2.0b27, Meirmans 2014). F-statistics were measured at the large and medium scale where populations were defined as those individuals collected at sites identified in Fig. 2.

Comparison of mean pairwise F’ST values by islands at different scales and sites. Bars represent 95% confidence intervals. Islands are represented by colors. Pink: Palau (between sites within Palau). Olive: Yap (between sites within Yap). Green: YN (sites between Yap to Ngulu). Blue and Turquoise (overlapping) PN (sites between Palau and Ngulu) and PY (sites between Palau and Yap). Purple represents a reference point of the mean kinship of all the pairwise comparisons between all the colonies on the three islands: PYN (Palau-Yap-Ngulu)
We used kinship coefficients (Loiselle et al. 1995, measured in genodive v.2.0b27, Meirmans 2014) as our measure of genetic differences between individuals. The kinship index calculated in genodive uses the allele frequencies within the entire chosen dataset, making the distances between pairs of individuals dependent on all other individuals in the dataset and effectively corrects for allele frequency differences in calculating the relative probability of identity by descent of the alleles within the two compared individuals. The difference in sample size in each island can bias the calculation of kinship if we pooled all samples into a single combined dataset. Therefore, we created several datasets to avoid bias in calculation of kinship coefficients. These coefficients were calculated among: (1) all pairs of individuals within each island (Ngulu, Yap, Palau), (2) each pair of islands (Palau-Yap, Palau-Ngulu, Yap-Ngulu), and as a reference, (3) all individuals from all 3 islands (Palau-Yap-Ngulu).
Large and medium scales: Population structure and isolation-by-distance
To understand population structure at the large and medium scales, we created a matrix of pairwise standardized F’st (Hedrick 2005; Meirmans 2006) among sites and tested for significance in genodive with 10,000 permutations (Table S3, Supporting information). We looked for patterns by testing for isolation-by-distance (IBD) and created a matrix of pairwise F’st values and a matrix of pairwise geographic distances. The matrix of geographic distances was populated by using the shortest distance between each pair of sites following the contour of the barrier reef using esri arcgis v.10.2.2. for sites around the reef of Palau (sites from S1 to S25), Yap (sites S27, S29 and S30) and Ngulu (site S28, Table S4, Supporting information). We tested for isolation-by-distance with a Mantel test (Mantel 1967) in genodive comparing a matrix of transformed pairwise F’ST values (\(F^{{\prime }} ST / F^{{\prime }} ST - 1\)) to a matrix of log transformed geographic distances with 100,000 permutations at two scales, (1) Palau only (medium scale) and (2) Palau, Ngulu and Yap (large scale). We compared results with stratified Mantel tests controlling for clusters as per Meirmans (2012) using islands as the strata factor for the large scale in genodive v.2.27 with 100,000 permutations. We plotted in R (Fig. 3) (package ggplot2 v.2.1.0) the mean and standard deviation of pairwise F’st values against scale and islands We used the mean pairwise F’st values of all three islands Palau-Yap-Ngulu as a reference point. Palau and Yap represented the medium scale and Palau-Yap, Palau-Ngulu and Yap-Ngulu represented the large scale. We used an approximative multivariate Kruskal–Wallis test (Coin v. 1.3-1 package in R) to test the difference in paired F’st values with scale for all islands. We used a Monte Carlo permutation to test for differences among distributions (10,000 permutations) (Table S5, Supporting Information).

Comparison of mean pairwise kinship coefficient by islands at different scales. Bars represent 95% confidence intervals. Scales are represented by colors. Pink: fine scale; olive: small scale; green: medium scale; blue: large scale; purple represents a reference point of the mean kinship of all the pairwise comparisons between all the colonies on the three islands
Large, medium, small and fine scales: pairwise kinship coefficients
Kinship coefficients were calculated in genodive relative to colonies of A. hyacinthus by island: Palau sites S1 through S25 (excluding S19 with too many missing alleles) and all six transects (S130, S200, S210, S220, S240, S250), Yap sites S27, S29, S30 and Ngulu site S28. We repeated this for colonies between islands: Yap-Ngulu, Palau-Ngulu, Palau-Yap, and for all colonies on all three islands: Palau-Yap-Ngulu as a reference point. Overall, we estimated pairwise kinship among a total of 1959 colonies at four scales, (1) large scale: pairwise kinship between colonies on different pairs of islands, (2) medium scale: pairwise kinship between colonies on the same island, (3) small scale: pairwise kinship between colonies within the same site and (4) fine scale: pairwise kinship between colonies within a belt transect of 2 × 100 m.
We performed a Kruskal–Wallis permutation test (Coin v. 1.3-1 package in R) with 10,000 permutations to see whether there were significant differences in the distribution of the kinship coefficients between the four scales (Table S6, Supporting Information). We then used the Dunn Test (package dunn.test v.1.3.2) with a Holm-Bonferroni correction as a conservative post hoc test for pairwise differences in average kinship values against scale (Table S7, Supporting information). To compare the distribution of kinship coefficient at the four scales, we used R (package ggplot2 v.2.1.0) to plot the mean and confidence interval of pairwise kinship coefficients at each of the four scales (Fig. 3).
To look more closely at the relationship between scale and kinship, we repeated this approach splitting scale by island (referred to a sub-scale: within transects, within Ngulu, within Yap, within Palau, between Yap, between Palau, between Yap and Ngulu, between Palau and Ngulu, between Palau and Yap and a reference point between all islands). We tested the difference in mean pairwise kinship with a Kruskal–Wallis permutation test (Coin v. 1.3-1 package in R) (Table S8, Supporting information) and a post hoc Dunn Test with a Holm-Bonferroni correction (package dunn.test v.1.3.2, Table S9, Supporting information). We used R (package ggplot2 v.2.1.0) to plot the mean and confidence interval of pairwise kinship coefficients at each of the 10 sub-scales (Fig. 4).

Comparison of mean pairwise kinship coefficient by islands at different sites. Bars represent 95% confidence intervals. Scale categories are represented by colors and letters. Pink (F): fine scale (w/in transects). Olive (S): small scale (w/in Palau, w/in Yap and w/in Ngulu). Green (M): medium scale (between sites within Palau, between sites within Yap). Blue (L): large scale (PN: Palau to Ngulu, PY: Palau to Yap, YN: Yap to Ngulu). Purple (R) represents a reference point of the mean kinship of all the pairwise comparisons between all the colonies on the three islands: PYN (Palau-Yap-Ngulu)
Violin plots of the pairwise kinship coefficients (package plotrix v. 3.6-2) were plotted at different sub-scale to look more closely at the effect of the tails of the distributions on population structure (Fig. 5). We plotted a line that indicated the “related” and “unrelated” colonies based on Loiselle et al. (1995) coancestry coefficients, using “related” for kinship > = 0.09375 and “unrelated” for kinship < 0.09375. This cutoff is somewhat arbitrary and does not represent a true division between kin groups (D’Aloia et al. 2018), but rather gives an idea of large and small genetic differences. We calculated the percentage of “related” colonies per island and per sub-scale (Table S10a and b, Supporting Information).

Violin plot of the distribution of pairwise kinship coefficients. Pink (F): fine scale represented by the distance between colonies within transects. Olive (S): small scale represented by the distance between colonies within sampling locations. Green (M): medium scale represented by the distance between colonies in different sites on the same island. Blue (L): large scale represented by the distance between colonies on different islands. Purple (R): a reference point of kinship coefficient between all colonies among all three islands. The black dot and line represent the median and confidence interval for each distribution. The horizontal black line indicates the division between “related” and “unrelated” (see text) individuals in each comparison. The total number of pairwise kinship coefficients in the comparison is given above the violin plot
Results
Descriptive analyses
We did not find any clones in our sampling of A. hyacinthus, so all samples were used in these analyses. We genotyped between 37 and 178 colonies at each site for each of our 11 microsatellite loci (Table 2). At each site, the effective number of alleles per locus (AE) varied between 1 and 9.6. Observed heterozygosity (HO) ranged from 0.007 to 0.69 and expected heterozygosity (HE) from 0.007 to 0.91, with significant inbreeding coefficients across most loci and all locations.
Large and medium scales: Population structure and isolation-by-distance
Pairwise F’st values (Table S3, Supporting information) show significant structure among sites around Palau and among sites on each of the three islands (see Cros et al. 2017 for a more detailed discussion of this population structure).
We tested to see whether there was isolation-by-distance at the large (among island) and medium (among sites around an island) scales. Isolation-by-distance (IBD) was significant at only the largest scale (between Palau, Ngulu and Yap; r2 = 0.047, p < 0.01), but when we repeat the Mantel test in genodive stratifying by island, the value is no longer significant (p = 0.23) indicating that there is hierarchical clustering at the island level (Meirmans 2012). We repeated the test at the medium scale for sites around Palau and likewise found no evidence of IBD. The Kruskal–Wallis permutation test did not show any difference in the magnitude of F’st between scales (Table S5, Supporting Information).
Large, medium, small and fine scales: Pairwise kinship coefficients
We compared pairwise kinship coefficient between the four scales using a Kruskal–Wallis permutation test which showed significant difference the four scales with a Chi square value of 2578 and p < 0.001 (Table S6, Supporting Information). All scales have significantly different mean pairwise kinship coefficients, except the large scale and the reference point (Table S7, Supporting Information). Pairwise kinship coefficients are significantly higher at the small than other scales (Table S7, Supporting Information and Fig. 3).
The Kruskal–Wallis permutation test showed a significant difference between sub-scales with a Chi square value of 2762.4 and a p value < 0.001 (Table S8, Supporting Information). The Dunn test confirms that most pairwise comparisons between sub-scales are significant with two exceptions. Kinship coefficients within Ngulu are only significant with the kinship coefficients between colonies from Yap and Ngulu and within Palau (Fig. 4, Table S9, Supporting Information). Likewise, kinship coefficients within sites in Palau are significantly greater than kinship coefficients within Ngulu and within Yap and significantly higher overall (Fig. 4 and Table S9, Supporting Information), whereas kinship coefficients between colonies in Yap and Ngulu are significantly smaller than kinship coefficients between sites on Palau and Yap and Palau and Ngulu (Fig. 4 and Table S9, Supporting Information).
There is a trend between scale and kinship coefficient where higher kinship coefficients are observed at the smaller geographic scales (Fig. 3). When scale is broken down by island (Fig. 4), the trend still exists on two of the three islands. For Yap and Palau, the kinship coefficients between colonies within sites are significantly higher than among colonies between sites or between islands. On Palau, the fine scale is the exception where mean pairwise kinship within transects is smaller than within sites on Palau.
Violin plots (Fig. 5) show that the distribution of kinship values overlap at all four scales: fine (10 s of cm), small (10–100 s m within sites), medium (10 s km between sites) and large (100 s of km between islands) and for all three islands. The line indicates the division between “related” and “unrelated” (k = 0.0937) individuals and helps to identify the sub-scales that display the highest frequency of colonies with high kinship values. The within Palau scale shows the highest mean pairwise kinship coefficients, and has the highest percentage of apparently related colonies (Table S9, Supporting Information). Palau has the longest positive tail because it has the highest individual pairwise kinship coefficients. Overall, as distance between colonies increases, the distribution of pairwise kinship coefficients decreases and suggests more limited dispersal than has previously been thought for these corals.
The percentage of related colonies is highest for colonies within the same sites on Palau (30.7%, Table S10a, Supporting Information); however, all three islands have almost the same ratio of related/unrelated colonies (Table S10b, Supporting Information).
Discussion
The overall patterns of genetic structure based on F-statistics among populations of A. hyacinthus in Yap, Ngulu and Palau show little support for a correlation between geographic and genetic distances (isolation-by-distance, IBD). As with previous studies (Cros et al. 2016, 2017), sites that are geographically separated often show smaller F’st values than sites that are in close proximity. These data confirm that sites around both Palau and Yap at the medium scale show patchy genetic structure with no clear signal of IBD. This pattern, in which significant genetic differentiation is observed among populations separated by distances much smaller than the presumed range of dispersal, has often been dubbed chaotic genetic patchiness (Johnson and Black 1982; Toonen and Grosberg 2011; reviewed by Eldon et al. 2016). Here, analyses of hierarchically sampled individuals reveal that local kin groups may explain the apparent chaotic patchiness when samples are collected without prior knowledge of the spatial extent of relatedness.
Similar patterns of high genetic differences at small scale, coupled with neighborhoods of high kinship, were found to drive chaotic genetic patchiness in previous studies by Iacchei et al. (2013) and Selwyn et al. (2016). Iacchei et al. (2013) attribute kinship of long-lived spiny lobsters in part to localized recruitment driven by environmental factors such as upwelling that lead to high pairwise differentiation among groups irrespective of geography. Selwyn et al. (2016) conclude neighborhoods of close kin for a marine goby developed through the processes of self-recruitment and collective dispersal of larvae. For the coral A. hyacinthus, wave exposure appeared to explain some of the observed genetic structuring among sites (Cros et al. 2017), but wave exposure alone cannot explain the high kinship observed within genetic neighborhoods at scales of less than 400 m.
One hypothesis to explain high kinship between individuals is that larvae simply do not disperse as expected and are recruiting at distances under 400 m from the site of spawning. Although this short dispersal distance is unexpected for a coral with a long pelagic duration, an increasing literature is reporting localized dispersal (e.g., Jones et al. 1999; Swearer et al. 2002; Hellberg 2007; Cowen and Sponaugle 2009; Berumen et al. 2012; Gorospe and Karl 2013; Selkoe et al. 2016; D’Aloia et al. 2018). Other possibilities include high kinship resulting from spiky dispersal kernels (sensu Siegel et al. 2008, i.e., an irregular frequency distribution of larval dispersal pathways, Selkoe and Toonen 2011) and patchy recruitment (Selkoe et al. 2006; Broquet et al. 2013; Riginos and Liggins 2013) or collective dispersal (Yearsley et al. 2013; Eldon et al. 2016; Selwyn et al. 2016), in which larvae spawned at the same location are transported and recruit together as a cohort. In the case of collective dispersal, recruitment can take place far from the natal patch but give the impression of self-recruitment. Evidence for collective dispersal remains limited (D’Aloia et al. 2018). If larvae of A. hyacinthus were dispersed cohesively among these three islands, we would expect: (1) less differentiation among the islands, and (2) higher kinship values at small scales, but at greater distances than we were able to find in this study.
Whatever the mechanism, it is clear that kinship values are skewed higher at small scales (10 s of m) and that these high kinship values are driving the genetic patterns observed for A. hyacinthus. This relatedness among individuals at small scales results in apparent chaotic genetic patchiness when sites are sampled haphazardly and genetic differentiation is analyzed at a larger scale. Although recent reviews have shown that there is uncertainty in assigning relationships among individuals when using small numbers of microsatellite markers (Baetscher et al. 2018; D’Aloia et al. 2018; Morales-González et al. 2019), power issues associated with kin detection should be less important if only comparing the relative proportion of putative kin among spatial scales here. Further, we are using kinship as a finer-scale metric of genetic structure rather than trying to estimate the magnitude of kin relatedness among individuals and acknowledge that our interpretations are bound to this limitation. Focusing on the relative magnitude of pairwise kinship values, we observe clearer patterns of genetic differences among scales and sites, particularly for the islands unimpacted by the 1998 mass mortality event. Plotting mean pairwise kinship against distance classes, we find colonies in proximity displaying higher kinship than colonies between sites (Fig. 4). The highest proportions of individuals classified as putative siblings are recovered at the small scale, between colonies located in the same site at distances of 10 s on meters rather than among sites on any of the three islands (Table S10, Supporting Information).
We propose that the process of recovery may explain the differing patterns observed between disturbed and undisturbed islands. In the case of Palau, we find similar F’st values as Yap (Table S2, Supporting information) yet Palau exhibits, at the small scale, a kinship coefficient that is significantly higher than that of Yap (Fig. 4, p < 0.001, Table S9, Supporting information). The population of A. hyacinthus around Palau was decimated, with estimates of mortality reported to be 100% (Bruno et al. 2001). We hypothesize that there was a handful of widespread surviving colonies around the island that became local breeding populations. These surviving colonies are now recolonizing the disturbed areas left in the wake of the 1998 mass mortality, resulting in each site being genetically differentiated from all others because of skewed allele frequencies from an over-representation of related individuals. Our data are consistent with limited dispersal between genetic neighborhoods creating a mosaic pattern of the populations of A. hyacinthus which have not had time to grow and overlap each other spatially, nor had enough exchange over time to create a pattern of isolation-by-distance. Without mixing of these genetic neighborhoods, each sampling site is genetically distinct from every other site (as shown previously in Cros et al. 2016, 2017). Now, the few surviving colonies in a given area have reproduced to repopulate the reef, but as they expand the population is out of migration-drift genetic equilibrium. Allele frequencies differ among sites not because of the processes of genetic drift and migration, but because family groups from the handful of surviving colonies scattered across the island began with different allele frequencies and have not had time to intermingle. Following such a massive disturbance, F-statistics take many generations to return to equilibrium values (Whitlock and McCauley 1999; Neigel 2002) and this non-equilibrium recovery creates an impression of chaotic patchiness. In a large population that has not undergone mass mortality, such as Yap, colonies within sites have mixed through time and are therefore closer to migration-drift equilibrium. The overall ratio of related/unrelated colonies is similar for all three islands (Fig. 5); however, the distribution of pairwise kinship coefficients differs at different scales (Fig. 4).
If our hypothesis is correct, this natural experiment provides insight into the future for A. hyacinthus around Palau: the population mean thermal tolerance should have shifted from this selection event because surviving kin groups should on average have greater bleaching resistance than those who died, or those unimpacted corals living on Yap and Ngulu. This process would predict that genomic screens including loci linked to thermal tolerance should reveal evidence of positive selection in the Palau population. Likewise, over time genetic structure of this recovering population on Palau should start to resemble the patterns seen on Yap and Ngulu, with less genetic differentiation among sites, more evenly distributed kinship coefficients, and eventually isolation-by-distance among geographic locations.
Conservation
Understanding the scale at which larval dispersal will shape population structure is critical to infer the processes by which population recovery may occur following such a disturbance, and the conservation and management implications. In the case of Palau, the mosaic structure observed in the genetic data indicates that a handful of colonies widely dispersed around the island survived the 1998 mass mortality. Rather than a mass colonization from adjacent, unimpacted islands, these few hardy survivors are responsible for repopulation of A. hyacinthus on reefs around Palau within a decade. This scenario predicts that Palau should be repopulated by more thermally tolerant individuals (those that survived the 1998 mass mortality) than Yap and Ngulu, and lead to the opposite conservation priorities as if the population were recolonized entirely by dispersal from the unimpacted populations on these unimpacted adjacent islands. Thus, understanding the mechanism of persistence and subsequent recovery can lead to very different management and conservation priorities which could ultimately be beneficial or detrimental to long-term persistence of coral reefs if misunderstood and the wrong management strategy was applied. Because it is currently impossible to predict either the identity or location of which colonies will survive the next mass mortality, the limited dispersal of kin groups from rare but widespread surviving colonies reported here highlights the need for a management plan that will prioritize the overall protection of surviving colonies on the reef wherever they exist instead of creating a handful of marine protected areas designed to maximize presumed connectivity with other islands. The genetic data indicate that such connectivity, while undoubtedly important for evolutionary processes and long-term colonization, is less important for the timescales of recovery on Palau since the 1998 event. The process of recovery playing out on the reefs of Palau also supports a more holistic approach to conservation: these genetically distinct patches around Palau will eventually grow to be more connected to one another and intermix, but this process takes time. To allow sufficient time for this recovery to play out, and to prepare for the recovery from the next natural disaster, the more effective strategy appears to be protecting as many colonies as possible to maximize the chances that the ones who might survive the event have not been removed by anthropogenic stressors and are still present on the reef. In the absence of a diagnostic tool to predict exactly which colonies will survive mass mortality, only by protecting as many surviving colonies as is possible can we ensure those handful of colonies that make it through a disaster will be present to grow, reproduce and enable local reefs to recover.
Data availability
Data are publicly available from the following repositories:
Microsatellite genotypes: Dryad https://doi.org/10.5061/dryad.m4q9f
DNA sequences: Genebank accession SAMN05177222, ID: 5177222
References
Ayre DJ, Hughes TP (2000) Genotypic diversity and gene flow in brooding and spawning corals along the Great Barrier Reef, Australia. Evolut Int J Organ Evolut 54:1590–1605. https://doi.org/10.1111/j.0014-3820.2000.tb00704.x
Baetscher DS, Clemento AJ, Ng TC, Anderson EC, Garza JC (2018) Microhaplotypes provide increased power from short-read DNA sequences for relationship inference. Mol Ecol Resour 18(2):296–305. https://doi.org/10.1111/1755-0998.12737
Baria MVB, Dela Cruz DW, Villanueva RD, Guest JR (2012) Spawning of three-year-old Acropora millepora corals reared from larvae in northwestern philippines. Bull Mar Sci 88:61–62. https://doi.org/10.5343/bms.2011.1075
Berumen ML, Almany GR, Planes S, Jones GP, Saenz-Agudelo P, Thorrold SR (2012) Persistence of self-recruitment and patterns of larval connectivity in a marine protected area network. Ecol Evolut 2:444–452. https://doi.org/10.1002/ece3.208
Broquet T, Viard F, Yearsley JM (2013) Genetic drift and collective dispersal can result in chaotic genetic patchiness. Evolution 67:1660–1675. https://doi.org/10.1111/j.1558-5646.2012.01826.x
Buston PM, D’Aloia CC (2013) Marine ecology: reaping the benefits of local dispersal. Curr Biol 23(9):R351–R353. https://doi.org/10.1016/j.cub.2013.03.056
Bruno J, Siddon C, Witman J, Colin P, Toscano M (2001) El Niño related coral bleaching in Palau, Western Caroline Islands. Coral Reefs 20:127–136. https://doi.org/10.1007/s003380100151
Cowen RK, Gawarkiewicz G, Pineda J, Thorrold SR, Werner FE (2007) Population connectivity in marine systems. Oceanography 20:14–21. https://doi.org/10.1126/science.1122039
Cowen RK, Sponaugle S (2009) Larval dispersal and marine population connectivity. Ann Rev Marine Sci 1:443–466. https://doi.org/10.1146/annurev.marine.010908.163757
Cros A, Toonen RJ, Davies SW, Karl SA (2016) Population genetic structure between Yap and Palau for the coral Acropora hyacinthus. PeerJ. https://doi.org/10.7717/peerj.2330
Cros A, Toonen RJ, Donahue MJ, Karl SA (2017) Connecting Palau’s marine protected areas: a population genetic approach to conservation. Coral Reefs 36:735–748. https://doi.org/10.1007/s00338-017-1565-x
D’Aloia CC, Xuereb A, Fortin MJ, Bogdanowicz SM, Buston PM (2018) Limited dispersal explains the spatial distribution of siblings in a reef fish population. Mar Ecol Prog Ser 607:143–154. https://doi.org/10.3354/meps12792
Davies SW, Treml EA, Kenkel CD, Matz MV (2015) Exploring the role of Micronesian islands in the maintenance of coral genetic diversity in the Pacific Ocean. Mol Ecol 24:70–82. https://doi.org/10.1111/mec.13005
Edmunds P, Bruno J (1996) The importance of sampling scale in ecology:kilometer-wide variation in coral reef communities. Mar Ecol Prog Ser 143:165–171. https://doi.org/10.3354/meps143165
Eldon B, Riquet F, Yearsley J, Jollivet D, Broquet T (2016) Current hypotheses to explain genetic chaos under the sea. Curr Zool 62:1–54. https://doi.org/10.1093/cz/zow094
Gaither MR, Szabo Z, Crepeau MW, Bird CE, Toonen RJ (2011) Preservation of corals in salt-saturated DMSO buffer is superior to ethanol for PCR experiments. Coral Reefs 30:329–333. https://doi.org/10.1007/s00338-010-0687-1
Golbuu Y, Victor S, Penland L, Idip D, Emaurois C, Okaji K, Yukihira H, Iwase A, van Woesik R (2007) Palau’s coral reefs show differential habitat recovery following the 1998-bleaching event. Coral Reefs 26:319–332. https://doi.org/10.1007/s00338-007-0200-7
Gorospe KD, Karl SA (2013) Genetic relatedness does not retain spatial pattern across multiple spatial scales: dispersal and colonization in the coral, Pocillopora damicornis. Mol Ecol 22:3721–3736. https://doi.org/10.1111/mec.12335
Guest JR, Baird AH, Goh BPL, Chou LM (2005) Reproductive seasonality in an equatorial assemblage of scleractinian corals. Coral Reefs 24:112–116. https://doi.org/10.1007/s00338-004-0433-7
Hedrick PW (2005) A standardized genetic differentiation measure. Evolut Int J Organ Evolut 59:1633–1638. https://doi.org/10.1111/j.0014-3820.2005.tb01814.x
Hellberg ME (1995) Stepping-stone gene flow in the solitary coral Balanophyllia elegans: equilibrium and nonequilibrium at different spatial scales. Mar Biol 123:573–581
Hellberg ME (2007) Footprints on water: the genetic wake of dispersal among reefs. Coral Reefs 26:463–473. https://doi.org/10.1007/s00338-007-0205-2
Hellberg ME (2009) Gene flow and isolation among populations of marine animals. Annu Rev Ecol Evol Syst 40:291–310. https://doi.org/10.1146/annurev.ecolsys.110308.120223
Hewitt JE, Thrush SF, Lundquist C (2010) Scale-dependence in Ecological Systems. In: Encyclopedia of life sciences. Wiley, Chichester. https://doi.org/10.1002/9780470015902.a0021903
Iacchei M, Ben-Horin T, Selkoe KA, Bird CE, García-Rodríguez FJ, Toonen RJ (2013) Combined analyses of kinship and F ST suggest potential drivers of chaotic genetic patchiness in high gene-flow populations. Mol Ecol 22:3476–3494. https://doi.org/10.1111/mec.12341
Jablonski D, Lutz RA (1983) Larval ecology of marine benthic invertebrates: paleobiological implications. Biol Rev 58:21–89
Johnson MS, Black R (1982) Chaotic genetic patchiness in an intertidal limpet, Siphonaria sp. Mar Biol 70:157–164. https://doi.org/10.1007/BF00397680
Jones GP, Milicich MJ, Emslie MJ, Lunow C (1999) Self-recruitment in a coral reef fish population. Nature 402:802–804
Kinlan BP, Gaines SD (2003) Propagule dispersal in marine and terrestrial environments: a community perspective. Ecology 84:2007–2020. https://doi.org/10.1890/01-0622
Kinlan BP, Gaines SD, Lester SE (2005) Propagule dispersal and the scales of marine community process. Divers Distrib 11:139–148. https://doi.org/10.1111/j.1366-9516.2005.00158.x
Kopelman NM, Mayzel J, Jakobsson M, Rosenberg NA, Mayrose I (2015) Clumpak: a program for identifying clustering modes and packaging population structure inferences across K. Mol Ecol Resour 15:1179–1191. https://doi.org/10.1111/1755-0998.12387
Ladner JT, Palumbi SR (2012) Extensive sympatry, cryptic diversity and introgression throughout the geographic distribution of two coral species complexes. Mol Ecol 21:2224–2238. https://doi.org/10.1111/j.1365-294X.2012.05528.x
Levin SA (1992) The problem of pattern and scale in ecology: the Robert H. MacArthur award lecture author(s): simon A. Levin Source Ecol 73:1943–1967. https://doi.org/10.2307/1941447
Liggins L, Treml EA, Riginos C (2013) Taking the plunge: an introduction to undertaking seascape genetic studies and using biophysical models. Geogr Compass 7:173–196. https://doi.org/10.1111/gec3.12031
Loiselle BA, Sork VL, Nason J, Graham C (1995) Spatial genetic structure of a tropical understory shrub, Psychotria officinalis (Rubiaceae). Am J Bot 82:1420–1425. https://doi.org/10.2307/2445869
Lowe WH, Allendorf FW (2010) What can genetics tell us about population connectivity? Mol Ecol 19:3038–3051. https://doi.org/10.1111/j.1365-294X.2010.04688.x
Márquez LM, van Oppen MJH, Willis BL, Miller DJ (2002) Sympatric populations of the highly cross-fertile coral species Acropora hyacinthus and Acropora cytherea are genetically distinct. Proc Royal Soc Biol Sci 269:1289–1294. https://doi.org/10.1098/rspb.2002.2014
Meirmans PG (2006) Using the amova framework to estimate a standardized genetic differentiation measure. Evolution 60:2399. https://doi.org/10.1554/05-631.1
Meirmans PG (2012) The trouble with isolation by distance. Mol Ecol. https://doi.org/10.1111/j.1365-294x.2012.05578.x
Meirmans PG (2014). GenoDive version 2.0 b25. Computer software distributed by the author. Available from: http://www.bentleydrummer.nl/software/software/GenoDive.html
Morales-González S, Giles EC, Quesada-Calderón S, Saenz-Agudelo P (2019) Fine-scale hierarchical genetic structure and kinship analysis of the ascidian Pyura chilensis in the southeastern Pacific. Ecol Evolut 9(17):9855–9868. https://doi.org/10.1002/ece3.5526
Neigel JE (2002) Is FST obsolete? Conserv Genet 3:167–173. https://doi.org/10.1023/A:1015213626922
Palumbi SR (1994) Genetic divergence, reproductive isolation, and marine speciation. Annu Rev Ecol Syst 25:547–572
Pineda J, Hare JA, Sponaugle SU (2007) Larval transport and dispersal in the coastal ocean and consequences for population connectivity. Oceanography 20:22–39
Pringle JM, Lutscher F, Glick E (2009) Going against the flow: effects of non-Gaussian dispersal kernels and reproduction over multiple generations. Mar Ecol Prog Ser 377:13–17. https://doi.org/10.3354/meps07836
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Rieger RM (1994) The biphasic life cycle—a central theme of metazoan evolution. Integr Comp Biol 34:484–491. https://doi.org/10.1093/icb/34.4.484
Riginos C, Liggins L (2013) Seascape genetics: populations, individuals, and genes marooned and adrift. Geogr Compass 7:197–216. https://doi.org/10.1111/gec3.12032
Rousset F (1997) Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics 145:1219–1228
Siegel DA, Mitarai S, Costello CJ, Gaines SD, Kendall BE, Warner RR, Winters KB (2008) The stochastic nature of larval connectivity among nearshore marine populations. Proc Natl Acad Sci 105(26):8974–8979. https://doi.org/10.1073/pnas.0802544105
Selkoe KA, Gaines SD, Caselle JE, Warner RR (2006) Current shifts and kin aggregation explain genetic patchiness in fish recruits. Ecology 87:3082–3094. https://doi.org/10.1890/0012-9658(2006)87%5b3082:csakae%5d2.0.co;2
Selkoe KA, Toonen RJ (2011) Marine connectivity: a new look at pelagic larval duration and genetic metrics of dispersal. Mar Ecol Prog Ser 436:291–305. https://doi.org/10.3354/meps09238
Selkoe KA, Aloia CC, Crandall ED, Iacchei M, Liggins L, Puritz JB, von der Heyden S, Toonen RJ (2016) A decade of seascape genetics: contributions to basic and applied marine connectivity. Mar Ecol Prog Ser 554:1–9. https://doi.org/10.3354/meps11792
Selwyn JD, Hogan JD, Downey-Wall AM, Gurski LM, Portnoy DS, Heath DD (2016) Kin-aggregations explain chaotic genetic patchiness, a commonly observed genetic pattern, in a marine fish. PLoS ONE 11:1–11. https://doi.org/10.1371/journal.pone.0153381
Sheets EA, Warner PA, Palumbi SR (2018) Accurate population genetic measurements require cryptic species identification in corals. Coral Reefs 37:549–563. https://doi.org/10.1007/s00338-018-1679-9
Slatkin M (1987) Gene flow and the geographic structure of natural populations. Science 236:787–792
Swearer SE, Shima JS, Hellberg ME, Thorrold SR, Jones GP, Robertson DR, Morgan SG, Selkoe KA, Ruiz GM, Warner RR (2002) Evidence of self-recruitment in demersal marine populations. Bull Mar Sci 70:251–271
Thorson G (1950) Thorson G. Reproductive and larval ecology of marine bottom invertebrates. Biol Rev Cambridge Phil Soc 25: 1–45. Biol Rev 25: 1–45
Toonen RJ, Grosberg RK (2011) Causes of chaos: spatial and temporal genetic heterogeneity in the intertidal anomuran crab Petrolisthes cinctipes. In: Koenemann S, Held C, Schubart C (eds) Phylogeography and Population Genetics in Crustacea. CRC Press, Boca Raton, pp 75–107
Victor S, Golbuu Y, Yukihira H, Van Woesik R (2009) Acropora size-frequency distributions reflect spatially variable conditions on coral reefs of Palau. Bull Mar Sci 85:149–157
Wallace CC (1985) Reproduction, recruitment and fragmentation in nine sympatric species of the coral genus Acropora. Mar Biol 88:217–233. https://doi.org/10.1007/BF00392585
Weersing K, Toonen RJ (2009) Population genetics, larval dispersal, and connectivity in marine systems. Mar Ecol Prog Ser 393:1–12. https://doi.org/10.3354/meps08287
White C, Selkoe KA, Watson J, Siegel DA, Zacherl DC, Toonen RJ (2010) Ocean currents help explain population genetic structure. Proc Royal Soc B Biol Sci 277(1688):1685–1694. https://doi.org/10.1098/rspb.2009.2214
Whitlock MC, McCauley DE (1999) Indirect measures of gene flow and migration: FST not equal to 1/(4Nm + 1). Heredity 82:117–125. https://doi.org/10.1038/sj.hdy.6884960
Wright S (1943) Isolation by distance. Genetics 28:114–138.
Yearsley JM, Viard F, Broquet T (2013) The effect of collective dispersal on the genetic structure of a subdivided population. Evolution 67:1649–1659. https://doi.org/10.1111/evo.12111
Zvuloni A, Mokady O, Al-Zibdah M, Bernardi G, Gaines SD, Abelson A (2008) Local scale genetic structure in coral populations: a signature of selection. Mar Pollut Bull 56:430–438. https://doi.org/10.1016/j.marpolbul.2007.11.002
Acknowledgements
We thank S Davies for sharing the DNA extracts for the corals she collected and were used in our previous studies. We are grateful to both M Donahue and P Meirmans for their advice and help with the analyses. The manuscript was improved through thoughtful and constructive feedback from R Coleman, five anonymous referees and the editor. We are grateful to M Belcaid and Y Cros for assistance with raw data processing and to the following University of Hawai'i undergraduates for help in the laboratory: I Buffenstein, G Ciszek, B Haun, K Kaneshiro, M Keliipuleole, H Lim, K Niimoto, A Sifrit and T Whitman. Special thanks to The Nature Conservancy and Palau International Coral Reef Center for enabling the fieldwork and shipping permits. All collections were done under CITES permit PW 12-091 and a Palau Marine Research Permit RE-12-27. We also thank the HIMB NSF-EPSCoR Core Genetics Lab Facility (NSF, EPS-0903833). On behalf of all authors, the corresponding author states that there is no conflict of interest. This is the Hawai‘i Institute of Marine Biology HIMB “contribution # 1809” and the School for Ocean and Earth Science and Technology contribution # 10991.
Funding
This work was supported by the Disney Wildlife Conservation Fund to A Cros and SA Karl; the Graduate Women in Science Adel Lewis Grant Fellowship; the Founder Region Fellowship; the Ecology Evolution Conservation Biology Watson T. Yoshimoto grant and the Colonel Willys E. Lord Scholarship Award to A Cros; and a National Science Foundation grant OCE 14-16889 to RJ Toonen.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Topic Editor: Kyle Morgan.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cros, A., Toonen, R. & Karl, S.A. Is post-bleaching recovery of Acropora hyacinthus on Palau via spread of local kin groups?. Coral Reefs 39, 687–699 (2020). https://doi.org/10.1007/s00338-020-01961-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00338-020-01961-3