
Abstract
Despite the growing number of sequenced bovine genomes, the knowledge of the population-wide variation of sequences remains limited. In many studies, statistical methodology was not applied in order to relate findings in the sequenced samples to a population-wide level. Our goal was to assess the population-wide variation in DNA sequence based on whole-genome sequences of 32 Holstein–Friesian cows. The number of SNPs significantly varied across individuals. The number of identified SNPs increased with coverage, following a logarithmic curve. A total of 15,272,427 SNPs were identified, 99.16 % of them being bi-allelic. Missense SNPs were classified into three categories based on their genomic location: housekeeping genes, genes undergoing strong selection, and genes neutral to selection. The number of missense SNPs was significantly higher within genes neutral to selection than in the other two categories. The number of variants located within 3′UTR and 5′UTR regions was also significantly different across gene families. Moreover, the number of insertions and deletions differed significantly among cows varying between 261,712 and 330,103 insertions and from 271,398 to 343,649 deletions. Results not only demonstrate inter-individual variation in the number of SNPs and indels but also show that the number of missense SNPs differs across genes representing different functional backgrounds.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Despite the fact that a continuously growing number of whole-genome sequences are available for cattle, the knowledge of the bovine genome is still limited (Kõks et al. 2014). Up to now, most of the published results were based on the analysis of genomes of single (Eck et al. 2009; Kõks et al. 2013, 2014) or only a few individuals (Larkin et al. 2012; Lee et al. 2013; Stothard et al. 2011). Recently, a few studies in which a larger number of animals were sequenced (Baes et al. 2014; Brøndum et al. 2014; Daetwyler et al. 2014; Höglund et al. 2015; Jansen et al. 2013) were published. However, they did not apply statistical methodology in order to relate findings on single-nucleotide variation in the sequenced samples to a population-wide level. Such results are largely of a descriptive nature, which mainly describe individual characteristics of the sequenced animals. Moreover, without formal hypotheses testing, conclusions from analyses based on single or a few animals are highly prone to sampling and technological bias and, thus, may not be suitable for population-based conclusions. Daetwyler et al. (2014) and Höglund et al. (2015) used sequence information from a larger number of animals to perform imputation of whole-genome polymorphic variants in order to relate the variation in DNA sequence to the variation of (pseudo)phenotypes. Studies of Brøndum et al. (2014) and Daetwyler et al. (2014) were concentrated on the accuracy of imputation techniques, while Baes et al. (2014) compared different variant calling approaches. Since the focus of the aforementioned analyses was not on DNA variation itself, our study aims to extend the scope of a whole-genome sequence analysis of dairy cattle by considering the variation of single-nucleotide polymorphisms. The availability of whole-genome sequences for 32 individuals allows the incorporation of statistical testing procedures and consequently enables population-wide inferences.
Materials and methods
Animals
Whole-genome DNA sequences were available for 32 Polish Holstein–Friesian cows. These individuals were selected from a group of 991 case–control cows with clinical mastitis cases diagnosed by a veterinarian and their healthy herdmates. The experimental design comprised 16 paternal half-sib pairs matched according to the number of parities, production level, and birth year but differing in terms of their mastitis resistance expressed by the frequency of clinical mastitis diagnosed throughout their production life. More specifically, in each pair, one of the half-sibs represents an animal without clinical mastitis incidence throughout the whole production period and the other with multiple clinical mastitis cases.
Whole-genome DNA sequencing
DNA was isolated from blood samples using a DNA Isolation System. The quality of the DNA was verified using a 2200 TapeStation DNA Screen Tape device and its concentration ascertained using fluorescence methods (Picogreen, LifeTechnologies). Libraries were generated from 1 ug of genomic DNA using the Illumina TruseqDNA PCR free sample prep kit following the manufacturer’s protocol and their evaluation was made with the Agilent Tape Station 2200. Fragments were quantified by Picogreen and then normalized to 10 nM as recommended by Illumina for clusters generation on the Hiseq2000. Libraries were denaturated and samples were run in a total of 32 lanes of Hiseq Flowcell. The Illumina Truseq PE cluster kit v3 was used to generate clusters on the grafted Illumina Flowcell and the hybridized libraries were sequenced on the Hiseq2000 with a 100 cycles of paired-end sequencing module using the Truseq SBS kit v3. All of the samples were sequenced on an IlluminaHiSeq2000 next-generation sequencing platform.
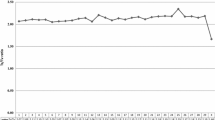
The total number of raw reads generated for a single animal varied between 164,984,147 and 472,265,620. Raw reads generated were filtered and trimmed using trimmomatic (Bolger et al. 2014) to remove low-quality base calls and sequencing adapters. Filtered reads were aligned to the UMD3.1 reference genome using BWA–MEM (Li and Durbin 2010). The resulting number of aligned reads varied from 155,202,885 to 454,412,859, which covered between 94.07 and 99.57 % of the genome, respectively. The corresponding coverage, averaged along the genome, was 14.03, ranging from 5 to 17 (Fig. 1). The coverage also varied considerably along an individual genome (Fig. 2), with some regions not covered at all and other regions with very high coverage. BAM files were processed to mark and remove duplicated reads using Picard (http://broadinstitute.github.io/picard/). Variant calling was done using the latest version of FreeBayes with filters requiring a minimum alignment quality of 30 and a minimum base quality of 20 for allele calling.

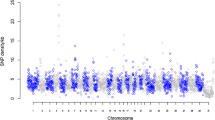
The genome averaged sequencing coverage for each individual

Coverage in percent observed for a particular individual on BTA1 (black), BTA10 (red), BTA20 (green), and BTX (blue) (Color figure online)
Selected genes
Table 1 lists genes that were selected for inter-individual comparison of mononucleotide variability. The primary selection criterion was gene function for which three categories were considered: (1) housekeeping—representing genes of primary importance for the organism metabolism, and in our study genes from the commercial bovine housekeeping gene array from QIAGEN (RT2 Profiler™ PCR array cow housekeeping genes) were considered; (2) neutral to selection—representing genes which are supposed to remain relatively neutral to selection for modern dairy cattle according to ISAG–FAO recommendations; and (3) strongly selected—representing genes with documented strong effect on production traits in dairy cattle which are therefore supposed to be under strong unidirectional selection pressure over many generations. For calculating SNP number and density, exon regions (including UTR) within genes were considered, while introns were skipped, so that SNP density was expressed as the number of SNPs divided by the total exon length of a gene.
Hypothesis testing
In order to test hypotheses regarding the inter-individual variability in SNP/indel numbers (N i) defined as \(H_{0} (\forall i,i^{\prime } :N_{i} = N_{{i{\prime }}} )\) and \(H_{1} (\exists i,i:N_{i} \ne N_{i\prime } )\), the Pearson’s χ 2 statistics were used: \(\chi^{2} = \sum\nolimits_{i = 1}^{32} {\frac{{(N_{i} - \overline{N} )}}{{\overline{N} }}} \sim \chi_{31}^{2}\), with \(\overline{N}\) denoting the number of variants averaged over all individuals. Furthermore, to test differences in SNP density between chromosomes, as well as between selected genes and gene categories, a one-way and a nested two-way analyses of variance were applied with the subsequent F tests for overall variability and post hoc t tests for assessing the differences between particular groups.
Results and discussion
Single-nucleotide polymorphisms
The total number of SNPs identified per individual ranged between 2,063,811 and 6,117,976, with a standard deviation of 663,223, which accounts for between 0.08 and 0.23 % of the total genome length. A cow with a notably low number of polymorphisms was excluded from further comparisons involving individual variability of variants. Still, for the remaining animals, the differences in the number of detected SNPs were highly significant (P < 10−20). Similar numbers of SNPs were reported by Jansen et al. (2013) ranging between 5,885,050 and 6,366,501 SNPs, by Baes et al. (2014) ranging between 5,854,886 and 6,404,094, by Kõks et al. (2013)—5,932,230 SNPs for a single sequenced cow, and by Kõks et al. (2014)—6,362,988 SNPs for a single sequenced bull, whereas a lower number of 3,755,633 SNPs was reported for a single bull by Stothard et al. (2011). Short sequences from all of the above individuals were aligned to the same (UMD3.1) reference genome as was the case in our study, but depending on the study, represented various breeds (Fleckvieh, Jersey, Swiss dairy cattle, or Holstein–Friesian).
By looking at the numbers of SNPs identified per individual in our sample and the corresponding average genome coverage presented in Fig. 3, it is evident that the two quantities are related. In order to quantify this relationship, various regression models including a linear regression, a second-grade polynomial, and a logarithmic model were fitted to the data available in our study complemented by the data from Kõks et al. (2013, 2014) and Stothard et al. (2011), which provided information on the number of SNPs obtained by high coverage. The logarithmic model fitted the best (Fig. 3) indicating a non-linear relationship between the number of detected SNPs and the genome coverage. In particular, a higher increase in SNP number was observed for the low coverage situation, rather than in higher coverage genomes. For instance, a rise in coverage from 5× to 10× is expected to increase the number of detected SNPs by 741,046, whereas a rise in coverage from 60× to 65× results in an increase in the number of identified SNPs by only 85,574. However, it should be noted that due to (1) the fact that data compiled from different studies were analyzed together, (2) a relatively small number of observations was available for high coverage data, and (3) a series of potentially important variables, such as sequencing technology, variation in read depth along the genome, alignment and variant calling methodology, individual level of genomic variation which is a function of inbreeding coefficient, as well as the level of genetic relatedness to the donor of the reference genome, and SNP filtering criteria were neglected, and these comparisons only provide a rough description of the relationship between average genome coverage and the number for polymorphisms detected.

A total of 15,272,618 SNPs were identified across all the 32 individuals, and among them, 575,215 SNPs were common for all sequenced animals (excluding the cow with exceptionally low coverage). The vast majority of SNPs were bi-allelic (99.159 %). However, there were also monomorphic (i.e. only alternative homozygous individuals) SNPs (0.551 %), tri-allelic SNPs (0.289 %), and even a small fraction of four-allelic SNPs (0.001 %) (Fig. 4). Although rare, such SNPs need special attention in further processing of data since they may arise from sequencing, alignment errors, or strong selection (monomorphic SNPs) but may also represent genomic sites of high mutation rate in the bovine genome (tri- and four-allelic SNPs).

The percentage of SNPs with 1 and 3 alleles as well as the total number of SNPs identified across 32 individuals
Genomic distribution of missense SNPs
We considered the distribution of SNPs identified among the 32 cows within coding regions, comparing genes belonging to three different categories: housekeeping genes (HK), genes undergoing strong selection in dairy cattle (SS), and genes neutral to selection (NS). Significant differences in SNP density for missense and stop codon removing SNPs were observed between gene categories and between individual genes, with the density for NS being the highest with 1.235 SNPs per Mbp, density of 0.261 SNP per Mbp being the intermediate for HK, and density of 0.059 SNP per Mbp for SS being the lowest. Also the raw number of missense SNPs was significantly higher within NS genes (2.33 SNPs per gene on average) than within HK (0.36 SNP) and SS (0.14 SNP) categories. The low number and density of SNPs identified within HK and SS was expected from the evolutionary perspective, both regarding natural and artificial selection, since potentially protein changing mutations have metabolic consequences ranging from a benign effect (58 % of nonsynonymous SNPs) to a damaging effect (24 % of nonsynonymous SNPs)—as predicted by Jansen et al. (2013).
Additionally, we considered polymorphisms located within the 3′-UTR and 5′-UTR regions marking the parts of genes that possibly affect the expression of exons. It was found that both their number and density varied significantly between gene categories as well as between genes. In particular, a significant difference in density was observed between the HK group with 0.621 SNPs per Mbp and the other two categories—NS with 1.491 SNPs and SS with 1.572 SNPs per Mbp. The highest average number of UTR SNPs per gene was equal to one for SS genes, followed by 0.34 for NS and 0.23 for HK, which is in contrast to the number of SNPs in coding regions of these genes. The discrepancy might at first glance seem surprising, but Larizza et al. (2002) found that the evolutionary dynamics of the UTRs is rather different from that of coding regions. This results from differences in functional constraints: 5′ and 3′-UTRs that bracket coding sequences are fundamental structural and regulatory regions of eukaryotic genes (Larizza et al. 2002; Mignone et al. 2002; Ptashne and Gann 2001; Wilkie et al. 2003). As demonstrated by Larizza et al. (2002), UTRs are much more divergent in terms of length and sequence across species of mammals (Homo sapiens, Bos taurus, Mus sp) than the corresponding coding regions. This is, among other reasons, due to the presence of repetitive elements in the UTRs, which are not found in coding regions.
Technical error in SNP detection
Using BTA10 as an example chromosome we evaluated differences in the number of SNPs identified by bioinformatic pipelines differing only in the applied variant calling software—FreeBayes (http://arxiv.org/abs/1207.3907), GATK (McKenna et al. 2010), or Samtools (Li et al. 2009). Considering the output of FreeBayes as a baseline only 93.5 % of SNPs were common between pipelines. A clear pattern in the number of “private” SNPs, i.e., SNPs which that identified only by one of the applied software packages, was observed across animals (Fig. 5). For all cows, Samtools detected the highest number of private polymorphisms, whereas for 29 out of 31 cows, it was FreeBayes, which resulted in the lowest number of private SNPs. A t test comparison of the Phred scaled probabilities associated with all common and private SNPs identified on BTA10 across all cows revealed that for FreeBayes and Samtools, the SNP likelihood was significantly lower for privately called SNPs. The opposite was observed for GATK. Our results do not correspond to the study of Baes et al. (2014) who identified a higher total number of SNPs using GATK than Samtools.

The number of private SNPs identified on BTA10 using FreeBayes, Samtools, and GATK
Oligonucleotide insertions and deletions
The number of insertions and deletions identified in the genomes of the cows under study was much lower than the number of SNPs identified. Indels covered approximately 0.02 % of the genome, with numbers ranging from 261,712 to 330,103 and from 271,398 to 343,649 for insertions and deletions, respectively. This was very similar to the number of indel polymorphisms reported by Baes et al. (2014) remaining within the range of 496,203–832,689 per animal, depending on the applied variant detection pipeline.
The number of insertions and deletions differed significantly among cows (P < 10−20 in both cases), but a very high significant correlation of 0.996 was observed between the numbers of insertions and deletions identified for each animal. Correlations between the number of identified SNPs and mononucleotide deletions/insertions were somewhat lower, but also significant, amounting to 0.792 and 0.747, respectively.
Conclusions
Nowadays, datasets allowing for the population-wide conclusions are available not only for humans but also for other species. Thanks to very detailed records of binary (e.g. disease) and quantitative phenotypes, as well as environmental factors underlying phenotype expression, very deep pedigrees, and the relative ease of obtaining data from large groups of animals of predefined familial relationship structure, domestic cattle can play a role not only as production animals but also as model organisms. Using a dataset from 32 individuals, we were able to conduct an analysis of the genome mononucleotide variation in Bos taurus based on formal hypothesis testing.
The importance of statistically supported analysis can be best visualized by comparing results for whole-genome sequence variation of a single Holstein cow (Kõks et al. 2013) and a single Holstein bull (Kõks et al. 2014) obtained with the same sequencing methodology and bioinformatics procedures. There is a low repeatability of results reported in both studies that arises due to sampling error. For instance, the lists of 58 genes with the largest and smallest numbers of SNPs presented in both abovementioned studies contains only one overlap between a male and a female, which is within the range of 5 % type I error.
Based on whole-genome sequences of 32 cows, we were able to demonstrate statistically significant inter-individual variation in the number of SNPs and indels. An important issue in variant-based analysis of next-generation sequencing (NGS) data has always been a high level of technological error underlying variant identification (Baes et al. 2014; Lee et al. 2013). Still, we believe that due to our use of the same (1) data preparation procedure involving DNA extraction and sequencing, (2) variant detection pipeline for all individuals, and (3) incorporation of statistical analysis, our results remain valid. Note that the estimated variation in the number of SNP, insertion, and deletion sites in the animals under study expresses the degree of deviations from the UMD3.1 reference sequence used in this study. Hence, the higher or lower number of identified variants in individual cows may be due to the specificity of the applied reference genome [as demonstrated for humans by Li (2014)], which originates from the Hereford breed and thus does not reflect the genetic variation representative of the Holstein–Friesian population.
Moreover, we also observed significant variation on a functional basis, by comparing the numbers and density of SNPs identified within genes representing different functional categories. The highest rate of SNPs within genes being neutral to selection in dairy cattle supports the hypothesis that natural selection tends to eliminate variation within housekeeping genes, whereas artificial selection for milk production pre-imposed on dairy cattle for many generations suppresses variation within genes responsible for the selected trait. We observed that the sequences of UTR appeared to be more variable than those of exons, which empirically proves the hypothesis that the predominant type of selection in non-coding sequences is a positive one, i.e., favoring sequence diversity, whereas in coding sequences, there is a very strong negative selection pressure that leads to elimination of new mutated alleles. Moreover, the most variable UTR sequence was observed in genes undergoing strong selection, which is in agreement with Hong et al. (2006). This phenomenon can be related to the possible function of those regions in the regulation of translation, which can be important for highly expressed genes of individuals characterized by a very high milk production level.
References
Baes CF, Dolezal MA, Koltes JE, Bapst B, Fritz-Waters E, Jansen S, Flury C, Signer-Hasler H, Stricker C, Fernando R, Fries R, Moll J, Garrick DJ, Reecy JM, Gredler B (2014) Evaluation of variant identification methods for whole genome sequencing data in dairy cattle. BMC Genom 15:948
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics 30:2114–2120
Brøndum RF, Guldbrandtsen B, Sahana G, Lund MS, Su G (2014) Strategies for imputation to whole genome sequence using a single or multi-breed reference population in cattle. BMC Genom 15:728
Daetwyler HD, Capitan A, Pausch H, Stothard P, van Binsbergen R, Brøndum RF, Liao X, Djari A, Rodriguez SC, Grohs C, Esquerré D, Bouchez O, Rossignol MN, Klopp C, Rocha D, Fritz S, Eggen A, Bowman PJ, Coote D, Chamberlain AJ, Anderson C, VanTassell CP, Hulsegge I, Goddard ME, Guldbrandtsen B, Lund MS, Veerkamp RF, Boichard DA, Fries R, Hayes BJ (2014) Whole-genome sequencing of 234 bulls facilitates mapping of monogenic and complex traits in cattle. Nat Genet 46:858–865
Eck SH, Benet-Pagès A, Flisikowski K, Meitinger T, Fries R, Strom TM (2009) Whole genome sequencing of a single Bos taurus animal for single nucleotide polymorphism discovery. Genome Biol 10:R82
Höglund J, Guldbrandtsen B, Lund MS, Sahana G (2015) Identification of genomic regions associated with female fertility in Danish Jersey using whole genome sequence data. BMC Genet 16:60
Hong X, Scofield DG, Lynch M (2006) Intron size, abundance, and distribution within untranslated regions of genes. Mol Biol Evol 23:2392–2404
Jansen S, Aigner B, Pausch H, Wysocki M, Eck S, Benet-Pagès A, Graf E, Wieland T, Strom TM, Meitinger T, Fries R (2013) Assessment of the genomic variation in a cattle population by re-sequencing of key animals at low to medium coverage. BMC Genom 14:446
Kõks S, Lilleoja R, Reimann E, Salumets A, Reemann P, Jaakma Ü (2013) Sequencing and annotated analysis of the Holstein cow genome. Mamm Genome 24:309–321
Kõks S, Reimann E, Lilleoja R, Lättekivi F, Salumets A, Reemann P, Jaakma Ü (2014) Sequencing and annotated analysis of full genome of Holstein breed bull. Mamm Genome 25:363–373
Larizza A, Makalowski W, Pesole G, Saccone C (2002) Evolutionary dynamics of mammalian mRNA untranslated regions by comparative analysis of orthologous human, artiodactyl and rodent gene pairs. Comput Chem 26:479–490
Larkin DM, Daetwyler HD, Hernandez AG, Wright CL, Hetrick LA, Boucek L, Bachman SL, Band MR, Akraiko TV, Cohen-Zinder M, Thimmapuram J, Macleod IM, Harkins TT, McCague JE, Goddard ME, Hayes BJ, Lewin HA (2012) Whole-genome resequencing of two elite sires for the detection of haplotypes under selection in dairy cattle. Proc Nat Acad Sci USA 109:7693–7698
Lee KT, Chung WH, Lee SY, Choi JW, Kim J, Lim D, Lee S, Jang GW, Kim B, Choy YH, Liao X, Stothard P, Moore SS, Lee SH, Ahn S, Kim N, Kim TH (2013) Whole-genome resequencing of Hanwoo (Korean cattle) and insight into regions of homozygosity. BMC Genom 14:519
Li H (2014) Towards better understanding of artifacts in variant calling from high-coverage samples. Bioinformatics 30:2843–2851
Li H, Durbin R (2010) Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26:589–595
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup (2009) The sequence alignment/map (SAM) format and SAM tools. Bioinformatics 25:2078–2079
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA (2010) The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20:1297–1303
Mignone F, Gissi C, Liuni S, Pesole G (2002) Untranslated regions of mRNAs. Genome Biol 3:REVIEWS0004
Ptashne M, Gann A (2001) Transcription initiation: imposing specificity by localization. Essays Biochem 37:1–15
Stothard P, Choi JW, Basu U, Sumner-Thomson JM, Meng Y, Liao X, Moore SS (2011) Whole genome resequencing of black Angus and Holstein cattle for SNP and CNV discovery. BMC Genome 12:559
Wilkie GS, Dickson KS, Gray NK (2003) Regulation of mRNA translation by 5′- and 3′-UTR-binding factors. Trends Biochem Sci 28:182–188
Acknowledgments
The research was supported by the European Community’s Seventh Framework Programme within the frame of the NADIR (FP7-228394) Project and by the Polish National Science Center (NCN) Grant Number 2014/13/B/NZ9/02016. Computations were carried out at the Poznań Supercomputing and Networking Center.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Szyda, J., Frąszczak, M., Mielczarek, M. et al. The assessment of inter-individual variation of whole-genome DNA sequence in 32 cows. Mamm Genome 26, 658–665 (2015). https://doi.org/10.1007/s00335-015-9606-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00335-015-9606-7