
Abstract
Complete and accurate annotation of the mouse genome is critical to the advancement of research conducted on this important model organism. The National Center for Biotechnology Information (NCBI) develops and maintains many useful resources to assist the mouse research community. In particular, the reference sequence (RefSeq) database provides high-quality annotation of multiple mouse genome assemblies using a combinatorial approach that leverages computation, manual curation, and collaboration. Implementation of this conservative and rigorous approach, which focuses on representation of only full-length and non-redundant data, produces high-quality annotation products. RefSeq records explicitly link sequences to current knowledge in a timely manner, updating public records regularly and rapidly in response to nomenclature updates, addition of new relevant publications, collaborator discussion, and user feedback. Whole genome re-annotation is also conducted at least every 12–18 months, and often more frequently in response to assembly updates or availability of informative data. This article highlights key features and advantages of RefSeq genome annotation products and presents an overview of NCBI processes to generate these data. Further discussion of NCBI’s resources highlights useful features and the best methods for accessing our data.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
RefSeq provides an annotated mouse genome dataset
Mouse is an essential model organism for biomedical research. Decades of research analyzing and manipulating the mouse genome have translated into a better understanding of human physiology and diseases. Accurate and complete annotation of the mouse genome is crucial for this translational research. In recent years, the realm of genome annotation has expanded from identifying only protein-coding genes to include additional gene types such as pseudogenes, non-coding loci, and regulatory regions (Yandell and Ence 2012). The reference sequence (RefSeq) project was initiated by the National Center for Biotechnology Information (NCBI) in order to provide a curated and logically organized public sequence resource that is updated to reflect current knowledge as it accumulates (Pruitt et al. 2000). Since the initial release of 3446 human transcript and protein records, the RefSeq project has grown exponentially to include eukaryote, microbe, virus, and organelle sequences (Pruitt et al. 2014). Due to its importance as a model organism and critical research tool, curation of mouse genes, transcripts, and proteins is a major focus area for RefSeq.
The strength of NCBI’s annotation of the mouse genome lies not only in the quantity of genes, transcripts, and proteins annotated, but also on the quality of information provided. The RefSeq project provides comprehensive, non-redundant data, including genomic, transcript, and protein sequences (http://www.ncbi.nlm.nih.gov/refseq/). Mouse genomic RefSeq records include representation of nuclear and mitochondrial genomes, non-transcribed pseudogenes, and haplotype-specific regions. Transcript records may be protein-coding, non-coding, or structural RNAs. ‘Known’ RefSeq records are generated by manual curation, are mostly derived from GenBank transcripts, and use NM_, NR_, NP_, and NG_ accession prefixes, while ‘model’ RefSeq records are created by NCBI’s evidence-based eukaryotic genome annotation pipeline and use XM_, XR_, and XP_ accession prefixes. Each record is constructed from sequences submitted to the International Nucleotide Sequence Database Collaboration (INSDC) and includes information about the sequence, gene type, genomic location, current nomenclature, and conserved domain feature annotation. The subset of records that are supported by curation may contain additional information about functional regions within a sequence, summaries of protein function and interesting biology, transcript variant descriptions, and include relevant publications. The data source and supporting evidence for these sequence records are also clearly reported. RefSeq records serve as a stable reference for a variety of research purposes including genome annotation, gene identification, and comparative analyses.
RefSeq currently provides whole genome annotation of both the reference strain C57BL/6J genome assembly, which is maintained by the Genome Reference Consortium (GRC), as well as the mixed strain Celera assembly (Mural et al. 2002; Waterston et al. 2002). NCBI’s pipeline also calculates annotation on alternate loci scaffolds provided by the GRC, which represent strain-specific variation (http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/mouse/). Annotation updates are provided approximately yearly but could occur more frequently following a major assembly update, a significant increase or improvement in primary support data, or a significant update to the NCBI genome annotation pipeline. As additional strain-specific assemblies become publicly available, we will assess whether any should be added to the set of assemblies that go through the NCBI annotation pipeline. Annotation of additional mouse strains may be particularly beneficial in regions where the C57BL/6J genome has assembly issues or represents a haplotype that prevents the best representation of a locus (Keane et al. 2014). For example, BALB/c mice produce a functional protein from the Cxcl11 locus (NCBI GeneID: 56066), but a single nucleotide deletion in the C57BL/6J genome introduces a frameshift resulting in early translation termination and renders the allele null (Meyer et al. 2001; Sierro et al. 2007). To ensure appropriate annotation, NM_019494.1 was created to represent a protein-coding allele based on BALB/c transcripts, while NR_038116.1 represents a non-coding pseudogene transcript based on the C57BL/6J genome sequence.
This article outlines NCBI’s eukaryotic genome annotation, curation, and collaboration processes and highlights useful NCBI resources for the mouse research community.
RefSeq genome annotation leverages a combination of computation, curation, and collaborative approaches
Three major processes are integrated during RefSeq genome annotation to provide comprehensive, accurate, and reliable data. A robust evidence-based computational approach is supplemented with and informed by expert manual curation and collaboration with other working groups, resulting in high-quality mouse genome annotation.
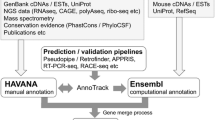
The NCBI eukaryotic genome annotation pipeline is an automated system for producing annotation of genes, transcripts, and proteins on public genome assemblies (Thibaud-Nissen et al. 2013). The pipeline incorporates publicly available transcript, RNA-seq and protein data, as well as known RefSeq records, in the genome annotation process (Fig. 1). Gnomon, NCBI’s eukaryotic gene prediction tool, evaluates the set of transcript and protein alignments to build supported gene, transcript, and protein models; for some genomes, especially those lacking significant transcriptomic data, ab initio modeling may also be used (http://www.ncbi.nlm.nih.gov/genome/guide/gnomon.shtml). Final annotation products include individual coding and non-coding transcripts and proteins, transcribed and non-transcribed pseudogenes, as well as the annotated scaffolds and chromosomes of assembled genomes with NCBI Genes (www.ncbi.nlm.nih.gov/gene/) and RefSeq RNAs and proteins included as features. Model RefSeq records (with a distinguishing prefix of XM_, XR_, or XP_) are predicted sequences with varying levels of support from cDNAs, ESTs, RNA-seq, and protein homology and allow for increased representation of potential transcript variation such as alternate UTRs, novel exons, and alternate splice donor and acceptor sites. All eukaryotic genomes currently annotated by NCBI are summarized at http://www.ncbi.nlm.nih.gov/genome/annotation_euk/all/. This page includes helpful links to additional NCBI resources including FTP, organism-specific BLAST databases, and a detailed annotation report that presents information about the input reagents used and the resulting annotation products. The annotation report for NCBI’s most recent Mus musculus Annotation Release 105 (a February 2015 update for the GRCm38.p3 and Celera assemblies) is available at http://www.ncbi.nlm.nih.gov/genome/annotation_euk/Mus_musculus/105/. An overview of the NCBI eukaryotic genome annotation pipeline is available at http://www.ncbi.nlm.nih.gov/genome/annotation_euk/process/, while a detailed description, including algorithms, is documented at http://www.ncbi.nlm.nih.gov/books/NBK169439/.

Overview of NCBI’s eukaryotic annotation pipeline from http://www.ncbi.nlm.nih.gov/genome/annotation_euk/process/#process. Briefly, genomic sequences are repeat masked (gray), and transcripts (blue), proteins (green), RNA-seq reads (orange), and curated RefSeq sequences (pink) are aligned to the genome. Based on these alignments, gene model predictions are calculated (brown), best models are selected, named and accessioned (purple), and finally annotation products are released publicly (yellow). During re-annotations, models and genes are given special attention and are tracked from one annotation release to the next
Manual curation is the second component in mouse genome annotation. Although RefSeq focuses on representing protein-coding transcripts and proteins, long non-coding RNAs (lncRNAs), and structural RNAs, we also represent both transcribed and non-transcribed pseudogene records. We employ a conservative approach with an emphasis on long-range exon support in an effort to avoid intentional annotation of partial coding sequences. Transcript, protein, and pseudogene records are primarily generated through sequence analysis, literature review, and comparative analysis; we also take into consideration other useful information, including conservation, epigenomic data, polyA-Seq data, protein domain hits, and user feedback. Genomic records representing non-transcribed pseudogenes are defined by imports from Pseudogene.org (Karro et al. 2007), manual curation (based on alignments of a functional gene to additional locations of the genome), or collaboration with nomenclature groups and the Consensus Coding Sequence (CCDS) project (Farrell et al. 2014). Expansion of pseudogene representation is not a primary focus of RefSeq curators, which is evident in the relatively small increase in the number of pseudogenes annotated during our last annotation.
Additional feature annotation may be provided for transcript and protein records based on literature review or computational approaches. RefSeq feature annotation adheres to the international standards defined by the INSDC (http://www.insdc.org/). Proteins that undergo post-translational processing are in scope for multiple methods of feature annotation. We import feature annotation from UniProtKB/Swiss-Prot records that have a high-quality alignment to the RefSeq protein (and which track with the same GeneID), add manual feature annotations, computationally provide protein domain and functional site annotation via NCBI’s Conserved Domain database, and compute signal peptide annotation using SignalP 4.0 (Petersen et al. 2011) if not in conflict with imported UniProtKB/Swiss-Prot feature annotation. Manual curation and evaluations are performed when conflicts in predicted signal peptides arise. Annotation of complex processing of preproproteins into proproteins and mature peptides require literature review-based manual curation. A significant advantage of transcript-based manual curation is that it allows for the correction of genome assembly errors using transcript evidence to compensate for underlying assembly issues. This facilitates the representation of transcript and protein sequence even when the genome is incomplete or incorrect. For example, the first 397 nucleotides of two Kazn transcript variants, NM_144531.3 and NM_001109684.1, are not represented in the GRCm38 assembly, but these RefSeq transcripts can still be represented based on transcript support and protein homology (Fig. 2a). The supporting transcripts are clearly included in the COMMENT section, in an “Evidence data” structured comment on known RefSeq records (Fig. 2b). Furthermore, manual curation may be strategically focused on genes with a known medical impact in human or to increase representation of historically unrepresented or under-represented loci, such as non-coding loci, loci encoding very short proteins, or weakly expressed but biologically functional loci. In these cases, curation may also incorporate specific knowledge from published studies. For example, a recent curation effort focused on representation of very short proteins that would normally be missed by standard pipeline parameters. Figure 2c highlights updated annotation of the mouse apelin receptor early endogenous ligand gene (GeneID: 100038489, Apela), which encodes a 54 amino acid protein that is well conserved in vertebrates. The functional protein has been described in human and zebrafish (Chng Serene et al. 2013; Pauli et al. 2014).

Examples of loci benefitting from manual curation. a The first 397 nucleotides of NM_144531.3 and NM_001109684.1 are missing from the GRCm38 reference genome assembly. The 5′ portion of the chromosome 4 gene Kazn (GeneID: 71529) was screen captured from NCBI's sequence viewer in the Gene resource and labels were edited (http://www.ncbi.nlm.nih.gov/gene/?term=71529#genomic-regions-transcripts-products). The partial alignment of the 5′ end of these RefSeq records is indicated by the double black arrows, and by the qualifier statement which is revealed upon hovering the mouse over the RefSeq transcript graphic. b Supporting evidence is reported on the NM_144531.3 record (http://www.ncbi.nlm.nih.gov/nuccore/NM_144531.3). The comments section shows that the full exon combination represented by NM_144531.3 is supported by the messenger RNA transcript, AK173090.1. This type of support evidence is associated with the ECO ID:0000332. The set of ECO IDs reported has been previously described (Pruitt et al. 2014). c The Apela gene on chromosome 8 (GeneID: 100038489) was defined as a non-coding locus in Mus musculus Annotation Release 104 (represented by NR_040692.1), but manual curation resulted in an update of the locus type to protein-coding in Annotation Release 105 (represented by NM_001297554.1/NP_001284483.1). The graphical display of RefSeq genome annotation that is shown in a, c was screen captured from NCBI's sequence viewer in the Gene resource and labels were edited (http://www.ncbi.nlm.nih.gov/gene/?term=100038489#genomic-regions-transcripts-products)
Finally, collaboration with other groups is a critical element in NCBI’s support of mouse genome annotation. Consultation with the Mouse Genome Informatics (MGI) database collaborators and integration of mouse official nomenclature data are essential for making consistent and unified locus type decisions and nomenclature updates. NCBI automatically imports new and updated official nomenclature information, using MGI-supplied transcript and protein accessions for quality assurance checks of consistent data associations in both MGI and NCBI data. In addition, RefSeq participates in the collaborative CCDS effort, along with Ensembl, UCSC, MGI, the HUGO Gene Nomenclature Committee (HGNC), and the Wellcome Trust Sanger Institute Manual Curation Group (Havana) (Farrell et al. 2014). This collaboration facilitates the goal of converging on stable, consistent protein-coding sequence annotation of the mouse and human reference genomes in NCBI, Ensembl, and UCSC Genome Browsers. The CCDS collaboration includes thorough manual curation and consultation between these large groups (http://www.ncbi.nlm.nih.gov/CCDS/). Indeed, when RefSeq curators identify an annotation issue that has wider impact than just the RefSeq dataset, we regularly initiate a discussion that includes, as relevant, curation staff at MGI, HGNC, Havana, and the Rat Genome Database, thus having a much wider impact on improved consistency in representing the gene type and nomenclature across orthologs. Another facet of collaboration allows automated data integration from other working groups, such as miRBase, allowing our annotation to reflect their focused annotation efforts. Furthermore, an established communication framework with the GRC (Church et al. 2011) allows the RefSeq group to report assembly issues encountered during curation in order to focus improvements for future assembly updates. For example, RefSeq curators reported an indel that prevented correct representation of the coding sequence of Col6a3 (GeneID: 12835) on the previous mouse assembly (MGSCv37); this issue was fixed in the current reference assembly (GRCm38) and the reported issue status is available publicly from the GRC web site (http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/mouse/issues/?id=MG-4012). To date, RefSeq has reported 120 mouse assembly issues which are resolved in GRCm38p.3, and additional 144 issues still await resolution. A list of mouse assembly issues is available through the GRC’s website (http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/mouse/issues/) and users may additionally submit issues that are not currently reported (http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/ReportAnIssue.shtml).
The combination of these strategies reflects a robust approach to providing high-quality, supported, genome annotation for the mouse. Our pipeline is adept at completing whole genome annotations with a very short turnaround time and has the capability of integrating a significant number of RNA-seq experiments. We periodically re-annotate the mouse genome as the C57BL/6J assembly is updated, the annotation pipeline software is further improved on, or as additional data become available. These updates offer several advantages to the research community in that the genome annotation is actively maintained and refreshed to reflect new RefSeq curation results, to reflect additional supported loci and transcript/protein variation, and to report more support evidence on the annotated genome and as a series of RNA-seq tracks available in the Gene resource. RefSeq data for the mouse are available in a number of NCBI databases and tools as summarized in the following sections.
Current status of the mouse RefSeq project
At the time of the February 2015 annotation release date (NCBI Annotation Release 105), NCBI’s annotation of the mouse GRCm38 genome represents 46,432 genes, 107,631 transcripts, and 76,131 protein-coding (mRNA) records (Table 1). Protein-coding transcripts represent 71 % of the total transcripts annotated, and 28,881 of these transcripts (38 %; Table 1) are known RefSeq (NM_ and NR_) accessions, many of which are tracked with a CCDS ID (23,880 proteins) and have undergone manual curation. At the present time, there are 22,196 known mouse protein-coding RefSeq accessions (NM_) and 2911 known mouse non-coding (NR_) accessions that have been manually curated and have a ‘validated’ or ‘reviewed’ status indicated on the sequence record (in the COMMENT section of the record).
Recent changes to the genome annotation pipeline have resulted in a more complete representation of non-coding RNAs on both the human and mouse genomes. There are currently 31,500 non-protein coding RNA records on the mouse GRCm38 assembly, representing a 26 % increase relative to the previous annotation release in December 2013 (Table 1). The significant increase in non-coding transcripts is due mainly to increased annotation of lncRNAs, primarily resulting from RNA-seq alignment analysis carried out by the genome annotation pipeline. This has been further increased, although by a much smaller amount, by focused curation of published non-coding RNA loci (for example, Sheik Mohamed et al. 2010; Hu et al. 2011; Sauvageau et al. 2013; Mueller et al. 2015). The subset of model lncRNAs that are supported by long cDNAs or publications are promoted to the known RefSeq accession type by a mix of automatic processes and curatorial review.
NCBI’s Gene resource
NCBI provides a plethora of tools and resources to easily view and analyze mouse RefSeq genome, transcript, and protein data. NCBI’s Gene database provides a comprehensive and centralized view of gene-specific information, providing both graphical and text-based displays (Brown et al. 2015). NCBI’s Gene resource can be accessed at www.ncbi.nlm.nih.gov/gene/. Gene pages can be retrieved using NCBI’s robust query interface using a GeneID, gene symbol, protein name or Gene Ontology term, or a RefSeq or INSDC accession number. Searches using either names or symbols yield results for the mouse gene and its orthologs, facilitating comparative analyses with taxonomically diverse species. Gene report pages are organized into sections that report functional information, sequence data, related database, or sequence identifiers, and include a graphical overview of the annotated genome (Fig. 3a, b). A table of contents located on the sidebar in the right column facilitates navigation to each section of the page.

Graphical display of mouse RefSeq transcripts using NCBI’s Gene Resource. a Genomic context. Coordinates on multiple mouse genome assemblies and a graphical display of the location and orientation of genes neighboring the Fst (GeneID: 14313) locus are shown here. b Genomic regions, transcripts, and products. Tracks displayed with the default settings are indicated with red arrows. The configure button (red circle) may be used to customize tracks. c Zoom and pan features allow easy identification of differences between transcript variants. Quantitative RNA-seq intron features data are displayed in this view. d Shown here is a subset of the links and related information displayed in the sidebar of each Gene record
All RefSeq transcripts that were annotated at the time of the last genome annotation update are graphically displayed in the ‘Genomic regions, transcripts, and products’ section of the Gene page, showing exon and intron structures. The default settings also display any CCDS proteins and Ensembl transcripts for the locus, variation from the dbSNP database, RNA-seq exon and intron coverage maps, and RNA-seq intron features (Fig. 3b). All known RefSeqs and pseudogenes that align to the genome including newly added or updated records can be viewed by adding the ‘interim gene annotation’ track using the ‘Configure’ button (Fig. 3b). Other customized tracks including a tiling path, scaffolds, CpG islands, six-frame translation, and multiple RNA-seq tracks can also be selected using the ‘Configure’ window. The RNA-seq intron features track reflects the introns that NCBI detected from spliced reads in the aggregated analyzed RNA-seq data (Fig. 3b, c). The number of spliced reads detected are now shown above each intron. This can provide valuable information about the relative frequencies of molecules found among sequenced reads in the database. For instance, the CCDS protein, CCDS36787.1, displayed in Fig. 3b is represented by the RefSeq transcript, NM_008046.3, encoding a 343 amino acid protein. Two other alternatively spliced transcript variants are also represented by RefSeq, NM_001301373.1 and NM_001301375.1, encoding a 344 amino acid and 317 amino acid isoform, respectively. Use of the zoom and pan features on the Gene graphical display allows determination that NM_008046.3 and NM_001301373.1 differ only at a single splice acceptor site of exon 5, and that the splice acceptor site used in NM_001301373.1 is much more prevalent in the analyzed RNA-seq data than that observed for NM_008046.3 (1276 reads vs. 346 reads as of May 2015; Fig. 3b, c).
To facilitate comparisons between current and previous mouse genome assemblies, the annotation and gene products from both previous and alternate assemblies can be displayed by selecting the desired “Genomic Sequence” from the dropdown menu (Fig. 3b). Furthermore, the graphical display tool supports uploading BLAST results or local datasets. Please refer to online documentation for more information (http://www.ncbi.nlm.nih.gov/tools/sviewer/).
Importantly, NCBI's Gene resource provides a multitude of links to both NCBI and external resources. The sidebar on the right-hand column provides links to many NCBI resources, including Map Viewer to view transcript alignments to the gene of interest, dbVar to view structural variation, and links to the nucleotide and protein records for the gene of interest (Fig. 3d). The sidebar also provides links to useful protein analysis tools such as the conserved domains database and HomoloGene (Fig. 3d). The latter provides a user-friendly graphical output of orthologous RefSeq proteins across taxa and is built entirely from RefSeq genome annotation data. Links to other mouse resources including MGI, Ensembl, UCSC, and KEGG are also provided.
More detailed information on NCBI RefSeq transcript and protein data for each gene is also available in the ‘NCBI Reference Sequences (RefSeq)’ section of the Gene page. RefSeq transcript and protein records that have been added or updated since the last genome annotation run will be reported here but not in the default graphical display of the genome annotation. RefSeq transcripts and pseudogenes that do not align to an assembly, due to a larger assembly gap, are also reported in this section. This section provides source sequence information for each RefSeq, text describing each transcript variant, conserved domain information, and links to similar UniProtKB/Swiss-Prot sequences, when available. Additional information about the Gene resource and functionality is available at http://www.ncbi.nlm.nih.gov/books/NBK3841/.
Mouse NCBI RefSeq data is best viewed using NCBI resources
While some RefSeq transcripts can be either viewed, or are referenced, by external resources, the full array of mouse RefSeq transcripts may not be included in these external resources. For example, the UCSC Genome Browser currently displays alignments of only the known RefSeq (NM_ and NR_ accessions) records, but does not display NCBI’s calculated placement of these transcripts or curated non-transcribed pseudogenes or the model RefSeq (XM_ and XR_ accessions) records that are generated by NCBI’s eukaryotic genome annotation pipeline. Other external resources do display both model and known RefSeq records, but may not contain the most current versions of known and model RefSeq records, or, may not have mouse genome annotation that is consistent with NCBI. For example, 1700022N22Rik (GeneID: 69431, MGI: 1916681) is annotated on the opposite strand of neighboring Capn15 (GeneID: 50817, MGI: 1355075) on chromosome 17, and the representation of both loci include several model RefSeq transcripts (Fig. 4a). However, the RefSeq Genes track in the UCSC Genome Browser does not include any model RefSeqs for either locus (Fig. 4b). Since 1700022N22Rik is solely represented by model RefSeqs, this locus is not represented at all in UCSC’s RefSeq Genes track. For this reason, we recommend viewing mouse RefSeq data directly from NCBI resources, and also downloading mouse RefSeq data directly from NCBI (see “Bulk data access” section).

The UCSC Genome Browser does not accurately represent RefSeq data. a NCBI Sequence Viewer. Coordinates on mouse chromosome 17 (NC_000083.6 from 25,957,500 to 25,988,800) and a graphical display the neighboring loci, 1700022N22Rik (GeneID: 69431) and Capn15 (GeneID: 50817), were screen captured from NCBI sequence viewer in the Gene resource and labels were edited. b UCSC Genome Browser. Coordinates on mouse chromosome 17 (NC_000083.6) and the RefSeq Genes track were screen captured from the UCSC Genome Browser and labels were edited. No RefSeq models are displayed in the RefSeq Genes track
RefSeq nucleotide and protein records
Links to known RefSeq nucleotide or protein records (NM_, NR_, NP_, and NG_ accessions) are readily available from the ‘NCBI Reference Sequences (RefSeq)’ section of Gene records. Alternatively, NCBI’s Nucleotide and Protein resource can be queried directly to retrieve RefSeq records where left-column facets facilitate restricting the returned results by different data categories (e.g., species, molecule type, or source database). RefSeq records are clearly identified with a keyword (RefSeq). Each record contains information pertaining to taxonomy, publications, a summary, and, when relevant, text describing the transcript variant, similar to what is found on the Gene record. RefSeq tries to be both transparent and complete in its reporting of evidence and source information on RefSeq records. Support evidence is reported in a structured comment labeled ‘Evidence Data’ that is located in the COMMENT section of the record. Up to two INSDC records that provide support for the transcript are listed under ‘Transcript exon combination’ (Fig. 2b). The level of support for each record is reported using Evidence Code Ontology (ECO) terms (Chibucos et al. 2014) as previously described (Pruitt et al. 2014). ECO provides a standardized vocabulary to describe the level of experimental evidence to support annotation assertions. For example, the ECO:0000332 term indicates that a RefSeq transcript has full-length transcript cDNA or EST transcript support for all exons (Fig. 2b). In contrast, support evidence for model RefSeq records is denoted on the Gene feature annotation as a ‘/note’ (not shown; for example, XM_006544953.2).
The INSDC records used to generate each NCBI RefSeq are listed in the ‘PRIMARY’ section of the record, with coordinates used for the RefSeq and the primary record, listed under the ‘REFSEQ_SPAN’ and ‘PRIMARY_SPAN’, columns, respectively. Some records include information on RefSeq Attributes that are added during the curation process. The ‘FEATURES’ section of the record provides additional information about the transcript and/or protein. Many protein features are automatically propagated to the transcript record to provide additional context to the sequence and indeed this direct linking of transcript and protein sequence data is a major advantage of the RefSeq product. Annotated features can include regulatory upstream open reading frames, upstream in-frame stop codons, alternative start codons, signal peptides, mature peptides, modified residues, binding sites, conserved domains, regulatory polyA signal sequences, and polyA sites. Some protein features are propagated from UniProtKB/Swiss-Prot records, others are computationally identified by NCBI’s Conserved Domain group, and yet others are manually annotated by NCBI curators. When imported from external databases, the data source is recorded in a “note” (e.g., ‘propagated from UniprotKB/Swiss-Prot’, followed by the Swiss-Prot accession). Computational predictions of signal peptides are displayed with the qualifier ‘/inference=”COORDINATES: ab initio prediction:SignalP:4.0’.
While the default Display Setting for nucleotide records is the ‘GenBank’ format, FASTA and graphical displays are available by changing the ‘Display Settings’ at the top of the page, or by clicking on the FASTA or Graphics quick link. Alternate displays are also available, including a ‘Revision History’ display that reveals both the date of updates to the records, the first date that the record became available, the removal date (as relevant), and details of the changes made to the record between updates.
Retrieval of mouse RefSeq data using NCBI resources provides easy access to NCBI analysis tools. DNA sequence analysis links such as BLAST and Primer-BLAST can be accessed easily using links provided on the sidebar on the right side of nucleotide and protein records. BLAST queries can be limited to RefSeq records by searching the appropriate RefSeq RNA or genomic database from the pull-down menu on the BLAST page. Analysis can be performed either on the full record, or on a specified sequence region that is selected using the ‘change region shown’ option. Quick access to external resources such as Ensembl, the UCSC Genome Browser, and reagents are also available using the links provided on the sidebar. Access to NCBI tools and resources is also provided on NCBI Protein Records. Links to a multitude of resources, such as BLink, which provides a graphical display of pre-computed BLAST results for proteins, are also provided on the right sidebar.
New analysis and data retrieval resources at NCBI
Identical Proteins report
The ‘Identical Proteins’ report, a relatively new resource offered by NCBI, details the accessions of proteins that are identical, both in length and sequence. This resource may prove valuable in identifying and studying highly conserved proteins. The Identical Proteins report for the conserved mouse histone H2A.Z protein, NP_058030.1, can be found at http://www.ncbi.nlm.nih.gov/protein/7949045?report=ipg, and provides links to nucleotide and protein records from identical proteins in other organisms. Information provided on the report page also includes the source of the sequence (e.g., RefSeq, INSDC, or Swiss-Prot) and mapping coordinates at both the transcript and genome levels. The Identical Proteins report can be accessed using the link at the top of the NCBI Protein record or through the Display Settings menu, and a more detailed description of this resource can be found at http://www.ncbi.nlm.nih.gov/refseq/about/nonredundantproteins/#identicalprotein.
Bulk data access
Relatively small datasets can be downloaded directly from NCBI’s Nucleotide or Protein resource after doing a query using the available ‘Send to’ functions; however, this approach does not work to download genome-scale data or to retrieve a combination of formats. Therefore we provide mouse, and other rodent, RefSeq data for bulk FTP access in more than one directory area in order to support different uses ranging from downloading a single annotated genome assembly to downloading the entire RefSeq dataset. NCBI RefSeq genome annotation releases for mouse are available from the genomes FTP site (ftp://ftp.ncbi.nlm.nih.gov/genomes/refseq/vertebrate_mammalian/Mus_musculus/), where sequence, annotation, assembly, and analysis data are provided in a variety of formats including GenBank, GenPept, FASTA, and GFF (version 3). Sequence and annotation information is provided at the level of the genome and also as transcript and protein products. These data are updated when NCBI calculates updated annotation of the GRC and Celera genome assemblies. Re-annotation occurs following an assembly update, a significant improvement to NCBI’s eukaryotic genome annotation pipeline software, or approximately yearly for the mouse. In addition, the complete mouse transcript and protein dataset is provided as a comprehensive weekly update from the RefSeq FTP site (ftp://ftp.ncbi.nlm.nih.gov/refseq/M_musculus/mRNA_Prot). This update occurs each Monday morning and reports all current transcript and protein records, including any new and updated data that were generated by the curation process. These updated data are not currently reported in the /genomes/refseq/ area. The RefSeq FTP site also reports mouse data in the /daily/ update directory which reports all new RefSeq records that are added in between the comprehensive bi-monthly RefSeq releases. For each comprehensive RefSeq release, mouse data are included in the /refseq/release/vertebrate_mammalian/ directory in addition to the /complete/ directory that reports the entire RefSeq dataset for all organisms.
NCBI also provides programming utilities which can be used to query for, and retrieve, specific subsets of data for mouse or other rodents. For example, this approach can be used to retrieve a specific list of accessions, or the results of a query, or to link across to a different type of data entirely via a large number of NCBI provided database-to-database links, for instance, query for Gene entries that include a name of interest and use NCBI links to retrieve all of the RefSeq transcripts or proteins for the set of Gene records. The NCBI programming utilities API is well documented online (http://www.ncbi.nlm.nih.gov/books/NBK25500/).
Future directions
As new advances are made in research techniques, and as the knowledge and scope of the functional transcriptome expands, RefSeq will adapt our annotation of the mouse genome accordingly. We will evaluate mouse strain-specific assemblies to determine if we should annotate them in our pipeline, with special consideration given to strains that are particularly suited as research models for human disease. One area of continued focus will be to expand our representation of non-coding loci. Increasing evidence suggests that many transcribed regions, even including those that are not protein coding, may have significant functional effects (Mercer et al. 2009; Djebali et al. 2012; Iyer et al. 2015). Further, as large proteomics datasets are now publicly available, we are currently assessing how this type of data may be integrated into our curation decisions (Kim et al. 2014; Wilhelm et al. 2014). All of RefSeq’s future goals will benefit from our continued collaborative efforts.
In conclusion, the combined resources of the known RefSeq dataset, the CCDS subset, and the model RefSeq dataset enable investigators to quickly access gene-specific information of both known and model genes, transcripts, and proteins. Valuable aspects of RefSeq annotation include the ability to represent well-supported transcripts despite underlying genome issues, as well as regular re-annotation of multiple genome assemblies. Notably, representation of current knowledge is kept up to date by providing rapid public reporting of updates to RefSeq records and the Gene database without requiring a whole genome annotation update. We welcome feedback that will help us improve our representation of mouse genome data and encourage users to communicate their suggestions and questions through the RefSeq help desk interface (http://www.ncbi.nlm.nih.gov/projects/RefSeq/update.cgi).
References
Brown GR, Hem V, Katz KS, Ovetsky M, Wallin C, Ermolaeva O, Tolstoy I, Tatusova T et al (2015) Gene: a gene-centered information resource at NCBI. Nucleic Acids Res 43:D36–D42
Chibucos MC, Mungall CJ, Balakrishnan R, Christie KR, Huntley RP, White O, Blake JA, Lewis SE, Giglio M (2014) Standardized description of scientific evidence using the Evidence Ontology (ECO). Database J Biol Databases Curation 2014:bau075
Chng Serene C, Ho L, Tian J, Reversade B (2013) ELABELA: a hormone essential for heart development signals via the apelin receptor. Dev Cell 27:672–680
Church DM, Schneider VA, Graves T, Auger K, Cunningham F, Bouk N, Chen H-C, Agarwala R et al (2011) Modernizing reference genome assemblies. PLoS Biol 9:e1001091
Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J et al (2012) Landscape of transcription in human cells. Nature 489:101–108
Farrell CM, O’Leary NA, Harte RA, Loveland JE, Wilming LG, Wallin C, Diekhans M, Barrell D et al (2014) Current status and new features of the Consensus Coding Sequence database. Nucleic Acids Res 42:D865–D872
Hu W, Yuan B, Flygare J, Lodish HF (2011) Long noncoding RNA-mediated anti-apoptotic activity in murine erythroid terminal differentiation. Genes Dev 25:2573–2578
Iyer MK, Niknafs YS, Malik R, Singhal U, Sahu A, Hosono Y, Barrette TR, Prensner JR et al (2015) The landscape of long noncoding RNAs in the human transcriptome. Nat Genet 47:199–208
Karro JE, Yan Y, Zheng D, Zhang Z, Carriero N, Cayting P, Harrrison P, Gerstein M (2007) Pseudogene.org: a comprehensive database and comparison platform for pseudogene annotation. Nucleic Acids Res 35:D55–D60
Keane TM, Wong K, Adams DJ, Flint J, Reymond A, Yalcin B (2014) Structural variation in mouse genomes. Front Genet 5:192
Kim M-S, Pinto SM, Getnet D, Nirujogi RS, Manda SS, Chaerkady R, Madugundu AK, Kelkar DS et al (2014) A draft map of the human proteome. Nature 509:575–581
Mercer TR, Dinger ME, Mattick JS (2009) Long non-coding RNAs: insights into functions. Nat Rev Genet 10:155–159
Meyer M, Hensbergen PJ, van der Raaij-Helmer EMH, Brandacher G, Margreiter R, Heufler C, Koch F, Narumi S et al (2001) Cross reactivity of three T cell attracting murine chemokines stimulating the CXC chemokine receptor CXCR3 and their induction in cultured cells and during allograft rejection. Eur J Immunol 31:2521–2527
Mueller AC, Cichewicz MA, Dey BK, Layer R, Reon BJ, Gagan JR, Dutta A (2015) MUNC, a long noncoding RNA that facilitates the function of MyoD in skeletal myogenesis. Mol Cell Biol 35:498–513
Mural RJ, Adams MD, Myers EW, Smith HO, Miklos GLG, Wides R, Halpern A, Li PW et al (2002) A comparison of whole-genome shotgun-derived mouse chromosome 16 and the human genome. Science 296:1661–1671
Pauli A, Norris ML, Valen E, Chew G-L, Gagnon JA, Zimmerman S, Mitchell A, Ma J et al (2014) Toddler: an embryonic signal that promotes cell movement via apelin receptors. Science 343:1248636
Petersen TN, Brunak S, von Heijne G, Nielsen H (2011) SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods 8:785–786
Pruitt KD, Katz KS, Sicotte H, Maglott DR (2000) Introducing RefSeq and LocusLink: curated human genome resources at the NCBI. Trends Genet 16:44–47
Pruitt KD, Brown GR, Hiatt SM, Thibaud-Nissen F, Astashyn A, Ermolaeva O, Farrell CM, Hart J et al (2014) RefSeq: an update on mammalian reference sequences. Nucleic Acids Res 42:D756–D763
Sauvageau M, Goff LA, Lodato S, Bonev B, Groff AF, Gerhardinger C, Sanchez-Gomez DB, Hacisuleyman E et al (2013) Multiple knockout mouse models reveal lincRNAs are required for life and brain development. eLife 2:e01749
Sheik Mohamed J, Gaughwin PM, Lim B, Robson P, Lipovich L (2010) Conserved long noncoding RNAs transcriptionally regulated by Oct4 and Nanog modulate pluripotency in mouse embryonic stem cells. RNA 16:324–337
Sierro F, Biben C, Martínez-Muñoz L, Mellado M, Ransohoff RM, Li M, Woehl B, Leung H et al (2007) Disrupted cardiac development but normal hematopoiesis in mice deficient in the second CXCL12/SDF-1 receptor, CXCR7. Proc Natl Acad Sci USA 104:14759–14764
Thibaud-Nissen F, Souvorov A, Murphy TD, DiCuccio M, Kitts P (2013) Eukaryotic genome annotation pipeline. National Center for Biotechnology Information, Bethesda
Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, Agarwala R, Ainscough R et al (2002) Initial sequencing and comparative analysis of the mouse genome. Nature 420:520–562
Wilhelm M, Schlegl J, Hahne H, Gholami AM, Lieberenz M, Savitski MM, Ziegler E, Butzmann L et al (2014) Mass-spectrometry-based draft of the human proteome. Nature 509:582–587
Yandell M, Ence D (2012) A beginner’s guide to eukaryotic genome annotation. Nat Rev Genet 13:329–342
Acknowledgments
We would like to recognize Francoise Thibaud-Nissen for providing the overview graphic depicting NCBI’s eukaryotic genome annotation pipeline and statistics from the annotation releases (Fig. 1; Table 1). We thank our collaborators, especially the HUGO Gene Nomenclature Committee, the Mouse Genome Database, Rat Genome Database, UniProt and CCDS curators at the University of California Santa Cruz and the Wellcome Trust Sanger Institute for many fruitful discussions regarding correct genome annotation, gene location and nomenclature. We also thank the numerous individual scientists who have contacted us over the years to suggest an improvement. We sincerely value your input to help improve the RefSeq database content.
Funding
This work and funding for open access charge was provided by the Intramural Research Program of the National Institutes of Health, National Library of Medicine.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made.
The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this licence, visit https://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
McGarvey, K.M., Goldfarb, T., Cox, E. et al. Mouse genome annotation by the RefSeq project. Mamm Genome 26, 379–390 (2015). https://doi.org/10.1007/s00335-015-9585-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00335-015-9585-8