
Abstract
Body weight is a highly heritable trait across species. In humans, genetic variation plays a major role in determining the inter-individual differences in susceptibility or resistance to environmental factors which influence energy intake and expenditure. In this review, I discuss how genetic studies have contributed to our understanding of the central pathways that govern energy homeostasis. The study of individuals harboring highly penetrant genetic variants that disrupt the leptin–melanocortin pathway has informed our understanding of the physiological pathways involved in mammalian energy homeostasis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In humans, there is a strong evidence that within a population the variance in body weight and in body mass index (BMI; weight in kg/height in meters squared) is strongly influenced by genetic factors (Maes et al. 1997), with family, twin and adoption studies yielding heritability estimates that range between 40 and 70 % (Stunkard et al. 1986, 1990; Sorensen et al. 1989). In experimental overfeeding studies, Bouchard and colleagues showed that weight gain induced by overfeeding identical male twin pairs by 10 % of their energy requirements differed across sets of twins but was similar between members of a twin pair (Bouchard et al. 1990). Similarly, the response to negative energy balance was also heritable when the same individuals were calorically restricted (Bouchard et al. 1996). Given the high heritability of body weight, genetic approaches can be a useful tool with which to dissect the mechanisms involved in energy homeostasis. A number of contrasting experimental approaches have been used to identify human obesity-associated genes.
Finding common genetic variants that are associated with increased risk of obesity
Genome-wide association studies (GWASs) are based on the premise that the heritability of common diseases is made up of a large number of common variants [minor allele frequency (MAF) of more than 5 %]. To this end, the use of high throughput arrays has facilitated the genotyping of thousands of common variants covering more than 75 % of the genome in large population-based cohorts on whom body mass index data is available. The first GWAS-derived loci detected were intronic variants in the FTO (fat mass and obesity-associated) gene (Frayling et al. 2007; Dina et al. 2007) and variants approximately 200 kb downstream of MC4R (Loos et al. 2008). To date, more than 50 genetic loci relevant for body weight regulation have been identified by GWAS approaches (Loos 2012). While GWAS-associated signals are often identified by the name of the nearest gene, there is little evidence to suggest that variation in these specific genes explains the association signal. Furthermore, many of the signals identified to date map to non-coding regions of the genome so their functional significance needs to be determined. The strongest association with BMI in individuals of Caucasian origin has consistently been found with SNPs in the first intron of FTO. Similar observations have been made in other ethnic groups (Zhang et al. 2008; Li et al. 2008). Homozygotes for the risk allele of the most common SNP in FTO are 3–4 kg heavier than those without the risk allele and have a 1.67-fold increased risk of obesity (Frayling 2007). Several human studies have demonstrated an association between SNPs in FTO and energy intake rather than energy expenditure (Wardle et al. 2008). FTO is expressed in the hypothalamus, and its expression has been suggested to be nutritionally regulated by some (although not all) groups (Gerken et al. 2007; Stratigopoulos et al. 2008). Fto-null mice are small with increased energy expenditure (Fischer et al. 2009), although the effect is negligible when using the linear regression method to correct for differences in lean mass (McMurray et al. 2013). In contrast, mice overexpressing Fto are obese (Church et al. 2010). Genes within the region of interest are evaluated as potential candidates for causing the association such as RPGRIP1L (Stratigopoulos et al. 2011, 2014), but the association may also arise due to long-range genetic interactions as evidenced recently by work which suggests that the nearby gene IRX3 may be important (Smemo et al. 2014).
Cumulatively, the common variants identified in GWASs are characterized by modest effect sizes (per-allele odds ratios between 1.2 and 1.5), and the proportion of variability explained by GWAS-identified loci to date remains relatively modest (<5 %). Studies in childhood onset obesity and in severely obese children and adults have shown that there is some overlap between the common variants that contribute to early-onset and adult-onset weight gain (Wheeler et al. 2013), but also that both these approaches can identify novel variants that are not found in adult populations (Bradfield et al. 2012). Differences in genetic architecture as well as in environmental confounders may explain some of the discordance between different GWAS studies. While meta-analyses of even larger datasets are underway, there is a growing consensus in the study of common complex diseases that additional common variants are unlikely to explain the missing heritability underlying traits such as BMI or obesity. There is the possibility that the heritability of obesity-related phenotypes may have been overestimated, as the effects of the shared environment and, in the case of twins, the shared in utero environment, are difficult to separate from inherited influences.
Candidate gene studies
Candidate gene studies based on the molecules known to cause severe obesity in experimental animals have shown that these genes also contribute to childhood onset human obesity. The foundation for this work was laid by elegant physiological studies showing that the regulation of body weight is a homeostatic process (Friedman 2000), which has been demonstrated by lesioning studies to be regulated at the level of the hypothalamus. Parabiosis experiments in inbred strains of mice with severe obesity, such as ob/ob and db/db, suggested the existence of a circulating factor that regulates weight which was missing in ob/ob mice and to which db/db were resistant (Coleman and Hummel 1969). The identification of this hormone, leptin, through positional cloning of the ob gene, and the finding that this was mutated in severely obese ob/ob mice (Zhang et al. 1994) paved the way for the molecular and physiological circuits controlling energy homeostasis to be dissected. Leptin is a 16-kDa hormone whose circulating levels correlate closely with fat mass. Many of the physiological effects of leptin are mediated through the central nervous system, particularly the hypothalamus, which is the site of the highest mRNA expression of the long signaling isoform of the leptin receptor. Leptin stimulates pro-opiomelanocortin (POMC) expressing primary neurons in the arcuate nucleus of the hypothalamus. POMC is post-translationally processed to yield the melanocortin peptides, which are agonists at melanocortin 3 and 4 receptors. In addition, leptin inhibits neurons expressing the melanocortin antagonist Agouti-related protein and neuropeptide Y (NPY); NPY can suppress the expression of POMC. These primary leptin-responsive neurons project to second-order neurons expressing the melanocortin 4 receptor (MC4R). Targeted genetic disruption of MC4R in mice leads to increased food intake and increased lean mass and linear growth (Huszar et al. 1997). These hypothalamic pathways interact with other brain centers to coordinate energy intake and energy expenditure (Cummings and Schwartz 2003).
Insights from genetics—leptin signaling
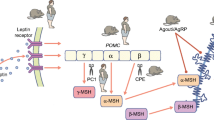
We and others have demonstrated that human obesity can result from a multiplicity of defects in the leptin–melanocortin pathway (Fig. 1). Generally these disorders are rare, being found in 1–5 % of patients with severe obesity (own observations). Individuals with homozygous loss-of-function mutations in the genes encoding leptin and the leptin receptor have a normal birthweight but exhibit rapid weight gain in the first few months of life resulting in severe obesity (Montague et al. 1997; Farooqi et al. 2002). Characteristic features are an intense drive to eat (hyperphagia) as well as impaired satiety with food-seeking behavior soon after the end of a meal. Recombinant human leptin injections are effective in these individuals leading to normalization of hyperphagia, enhanced satiety, and weight loss (Farooqi et al. 1999). Leptin is also involved in mediating food reward. In rodents, leptin receptors are expressed in the mesolimbic brain regions which mediate the rewarding properties of food and drugs (Barge-Schaapveld et al. 2011). Leptin administration decreases the firing threshold of dopaminergic neurons within this system, while long-term RNAi-mediated knockdown of Lepr in the ventral tegmental area has been shown to increase food intake and sensitivity to highly palatable food (Hommel et al. 2006). In leptin-deficient humans, images of food (compared to non-food images) are associated with a marked increase in neuronal activation in the ventral striatum imaged using functional MRI. This response was normalized by 7 days of leptin treatment (Farooqi et al. 2007a) before significant weight loss consistent with the view that activation in the ventral striatum does not directly encode the “liking” but rather the motivational salience, or “wanting,” of food.

Schematic of the hypothalamic leptin−melanocortin pathway. *Indicate genetic obesity syndromes
In leptin-deficient children and adults, basal metabolic rate, total energy expenditure, and free-living energy expenditure are appropriate for body composition (Farooqi et al. 2007c). Ravussin and colleagues showed that before weight loss, leptin-deficient adults and matched controls had similar energy expenditures. However, after weight loss, controls had energy expenditures lower than expected for their new weight and body composition (Galgani et al. 2010). This is a well-known phenomenon, often called the metabolic adaptation to weight loss, i.e., a decrease in metabolic rate beyond that expected on the basis of the decrease in fat-free mass and fat mass. Therefore, the absence of this change in energy expenditure in some leptin-deficient adults is consistent with an effect of leptin on energy expenditure.
Body composition measurements show that leptin deficiency is characterized by the preferential deposition of fat mass (compared with lean mass), and weight loss leads to a preferential loss of fat mass. In rodents, leptin stimulates fatty acid oxidation in skeletal muscle via the stimulation of AMP kinase activity (Minokoshi et al. 2002). In leptin-deficient humans, impaired fat oxidation has been demonstrated by chamber calorimetry (Galgani et al. 2010). Ozata et al. (1999) have reported abnormalities of sympathetic nerve function in leptin-deficient adults consistent with defects in the efferent sympathetic limb of thermogenesis. Evidence from rodents and humans suggests that leptin is necessary for the normal biosynthesis and secretion of thyrotropin-releasing hormone and that complete leptin deficiency is associated with a moderate degree of hypothalamic hypothyroidism characterized by low free thyroxine and high serum thyroid-stimulating hormone, which is bio-inactive (Gibson et al. 2004).
Insights from genetics—melanocortinergic circuits
Leptin signaling modulates energy balance through a combination of melanocortin-dependent and -independent pathways. In humans, null mutations in POMC lead to hyperphagia, early-onset obesity, isolated adrenocorticotropin (ACTH) deficiency, and hypopigmentation of skin and hair (Coll et al. 2007). Heterozygous null mutations in POMC and loss-of-function mutations in α- and β-melanocyte-stimulating hormone (α- and β-MSH) significantly increase obesity risk but are not invariably associated with obesity (Coll et al. 2004).
Prohormone convertase 1 (PCSK1) is an enzyme involved in the cleavage of POMC into ACTH, which is then further cleaved to make α-MSH by carboxypeptidase E. Humans lacking PCSK1 are severely obese and have glucocorticoid deficiency, hypogonadotropic hypogonadism, and postprandial hypoglycemia, which occurs as a result of impaired processing of proinsulin to insulin by PCSK1 (Farooqi et al. 2007b). Elevated plasma levels of proinsulin and 32–33 split proinsulin in the context of low levels of mature insulin provide the basis for a diagnostic test for this disorder (Jackson et al. 1997).
We and others have reported that mutations in MC4R are found in 5–6 % of patients with severe early-onset obesity (Farooqi et al. 2003; Stutzmann et al. 2008) and at a frequency of approximately 1/1,000 in the general UK population, making this one of the most common human genetic diseases. Functionally significant MC4R mutations are inherited in a co-dominant manner, with variable penetrance and expression in heterozygous carriers. Most naturally occurring disease-causing MC4R mutations disrupt normal expression and trafficking of the receptor to the cell surface (Vaisse et al. 2000). The mechanism of GPCR dysfunction has potential interest, as we have shown that pharmacological chaperones can increase the cell surface expression and signaling of mutant GPCRs. We have previously characterized human MC4R deficiency and reported hyperphagia, increased lean mass, and increased linear growth and demonstrated a genotype−phenotype correlation with the degree of receptor dysfunction in vitro predicting all aspects of the phenotype, including ad libitum energy intake (Farooqi et al. 2007c).
We have shown that both increase and decrease in central melanocortin signaling influence blood pressure in humans and that the effects are not explained by changes in circulating insulin levels or insulin sensitivity (Greenfield et al., 2009). MC4R-deficient patients with long-standing decreases in melanocortinergic tone have a lower prevalence of hypertension and lower systolic and diastolic blood pressures. These changes are associated with reduced sympathetic nervous system activity. Also, administration of a melanocortin receptor agonist in obese volunteers increases blood pressure. Thus, central melanocortin signaling appears to play an important role in the regulation of blood pressure and its coupling to changes in weight. With weight gain, leptin levels increase, thereby increasing signaling through MC4R. As MC4R neurons synapse with preganglionic sympathetic neurons which regulate vascular tone, increased signaling through MC4R can lead to an increase in blood pressure. This mechanism explains why reduced melanocortin signaling in people with loss-of-function MC4R mutations, may be associated with reduced blood pressure.
Insights from genetics—molecules downstream of MC4R and other mechanisms
Several lines of evidence suggest that Brain-derived neurotrophic factor (BDNF), a nerve growth factor which activates signaling by the tyrosine kinase receptor tropomycin-related kinase B (TrkB), and SIM1 may lie downstream of MC4R signaling in the paraventricular nucleus. Haploinsufficient mice and mice in which BDNF has been deleted postnatally are obese with hyperphagia and hyperactivity; this unusual combination of phenotypes is also seen in individuals with genetic disruption of BDNF or its receptor TrkB (Gray et al. 2006; Yeo et al. 2004).
Single minded 1 (SIM1) is a transcription factor involved in the development of the paraventricular and supraoptic nuclei of the hypothalamus. A de novo balanced translocation between chromosomes 1p22.1 and 6q16.2, which disrupts SIM1 (Holder et al. 2000), and missense mutations in SIM1 cause severe obesity and a variable phenotype of developmental delay (Ramachandrappa et al. 2013). The transcriptional targets of SIM1 are unknown, but one potential target is the neuropeptide oxytocin. Oxytocin mRNA levels are reduced in line with Sim1 gene dosage in mouse models of Sim1 deficiency. The hyperphagia of Sim1-haploinsufficient animals is ameliorated by oxytocin administration and accentuated by the administration of oxytocin receptor antagonists (Kublaoui et al. 2008).
Recent studies have suggested a connection between ciliary function and leptin signaling (Seo et al. 2009). Conditional postnatal knockout of proteins involved in intraflagellar transport in mice results in hyperphagia and obesity. This phenotype is recapitulated when the loss of cilia is limited to neurons and when it is specifically targeted to POMC neurons. As several human obesity disorders disrupt genes involved in ciliary function (e.g., Alström syndrome and Bardet−Beidl syndrome) (Tobin and Beales 2007), the role of cilia in key neuronal populations involved in energy homeostasis is likely to be the subject of much future research.
Finding the missing heritability in human obesity
Next-generation sequencing technologies are currently being used to undertake CNV analysis (Walters et al. 2010) and whole exome sequencing in obese cohorts and populations (Gill et al. 2014). The aim of these studies is to find low-frequency/rare genetic variants, but assessing the potential pathogenicity of those variants remains a major challenge. While statistical and computational methods are continually emerging to address these challenges, as with other hypothesis-free genetic approaches the technique is ultimately only as powerful as the genetic material being interrogated. Initial methodological approaches have tended toward sequencing related individuals, or sequencing individuals at opposing extremes of the phenotypic spectrum. The contribution of epigenetic modulation and of non-coding genetic variation to the missing heritability of obesity remains to be explored.
References
Barge-Schaapveld DQ, Maas SM, Polstra A, Knegt LC, Hennekam RC (2011) The atypical 16p11.2 deletion: a not so atypical microdeletion syndrome? Am J Med Genet A 155A:1066–1072
Bouchard C, Tremblay A, Despres JP, Nadeau A, Lupien PJ, Theriault G, Dussault J, Moorjani S, Pinault S, Fournier G (1990) The response to long-term overfeeding in identical twins [see comments]. N Engl J Med 322:1477–1482
Bouchard C, Tremblay A, Despres JP, Nadeau A, Lupien PJ, Moorjani S, Theriault G, Kim SY (1996) Overfeeding in identical twins: 5-year postoverfeeding results. Metabolism 45:1042–1050
Bradfield JP, Taal HR, Timpson NJ, Scherag A, Lecoeur C, Warrington NM, Hypponen E, Holst C, Valcarcel B, Thiering E, Salem RM, Schumacher FR, Cousminer DL, Sleiman PM, Zhao J, Berkowitz RI, Vimaleswaran KS, Jarick I, Pennell CE, Evans DM, St pourcain B, Berry DJ, Mook-Kanamori DO, Hofman A, Rivadeneira F, Uitterlinden AG, Van Duijn CM, Van Der Valk RJ, De Jongste JC, Postma DS, Boomsma DI, Gauderman WJ, Hassanein MT, Lindgren CM, Magi R, Boreham CA, Neville CE, Moreno LA, Elliott P, Pouta A, Hartikainen AL, Li M, Raitakari O, Lehtimaki T, Eriksson JG, Palotie A, Dallongeville J, Das S, Deloukas P, McMahon G, Ring SM, Kemp JP, Buxton JL, Blakemore AI, Bustamante M, Guxens M, Hirschhorn JN, Gillman MW, Kreiner-Moller E, Bisgaard H, Gilliland FD, Heinrich J, Wheeler E, Barroso I, O’Rahilly S, Meirhaeghe A, Sorensen TI, Power C, Palmer LJ, Hinney A, Widen E, Farooqi IS, McCarthy MI, Froguel P, Meyre D, Hebebrand J, Jarvelin MR, Jaddoe VW, Smith GD, Hakonarson H, Grant SF (2012) A genome-wide association meta-analysis identifies new childhood obesity loci. Nat Genet 44:526–531
Church C, Moir L, McMurray F, Girard C, Banks GT, Teboul L, Wells S, Bruning JC, Nolan PM, Ashcroft FM, Cox RD (2010) Overexpression of Fto leads to increased food intake and results in obesity. Nat Genet 42:1086–1092
Coleman DL, Hummel KP (1969) Effects of parabiosis of normal with genetically diabetic mice. Am J Physiol 217:1298–1304
Coll AP, Farooqi IS, Challis BG, Yeo GS, O’Rahilly S (2004) Proopiomelanocortin and energy balance: insights from human and murine genetics. J Clin Endocrinol Metab 89:2557–2562
Coll AP, Farooqi IS, O’Rahilly S (2007) The hormonal control of food intake. Cell 129:251–262
Cummings DE, Schwartz MW (2003) Genetics and pathophysiology of human obesity. Annu Rev Med 54:453–471
Dina C, Meyre D, Gallina S, Durand E, Korner A, Jacobson P, Carlsson LM, Kiess W, Vatin V, Lecoeur C, Delplanque J, Vaillant E, Pattou F, Ruiz J, Weill J, Levy-Marchal C, Horber F, Potoczna N, Hercberg S, le Stunff C, Bougneres P, Kovacs P, Marre M, Balkau B, Cauchi S, Chevre JC, Froguel P (2007) Variation in FTO contributes to childhood obesity and severe adult obesity. Nat Genet 39:724–726
Farooqi IS, Jebb SA, Langmack G, Lawrence E, Cheetham CH, Prentice AM, Hughes IA, McCamish MA, O’Rahilly S (1999) Effects of recombinant leptin therapy in a child with congenital leptin deficiency. N Engl J Med 341:879–884
Farooqi IS, Matarese G, Lord GM, Keogh JM, Lawrence E, Agwu C, Sanna V, Jebb SA, Perna F, Fontana S, Lechler RI, Depaoli AM, O’Rahilly S (2002) Beneficial effects of leptin on obesity, T cell hyporesponsiveness, and neuroendocrine/metabolic dysfunction of human congenital leptin deficiency. J Clin Invest 110:1093–1103
Farooqi IS, Keogh JM, Yeo GS, Lank EJ, Cheetham T, O’Rahilly S (2003) Clinical spectrum of obesity and mutations in the melanocortin 4 receptor gene. N Engl J Med 348:1085–1095
Farooqi IS, Bullmore E, Keogh J, Gillard J, O’Rahilly S, Fletcher PC (2007a) Leptin regulates striatal regions and human eating behavior. Science 317:1355
Farooqi IS, Volders K, Stanhope R, Heuschkel R, White A, Lank E, Keogh J, O’Rahilly S, Creemers JW (2007b) Hyperphagia and early-onset obesity due to a novel homozygous missense mutation in prohormone convertase 1/3. J Clin Endocrinol Metab 92:3369–3373
Farooqi IS, Wangensteen T, Collins S, Kimber W, Matarese G, Keogh JM, Lank E, Bottomley B, Lopez-Fernandez J, Ferraz-Amaro I, Dattani MT, Ercan O, Myhre AG, Retterstol L, Stanhope R, Edge JA, McKenzie S, Lessan N, Ghodsi M, de Rosa V, Perna F, Fontana S, Barroso I, Undlien DE, O’Rahilly S (2007c) Clinical and molecular genetic spectrum of congenital deficiency of the leptin receptor. N Engl J Med 356:237–247
Fischer J, Koch L, Emmerling C, Vierkotten J, Peters T, Bruning JC, Ruther U (2009) Inactivation of the Fto gene protects from obesity. Nature 458:894–898
Frayling TM (2007) Genome-wide association studies provide new insights into type 2 diabetes aetiology. Nat Rev Genet 8:657–662
Frayling TM, Timpson NJ, Weedon MN, Zeggini E, Freathy RM, Lindgren CM, Perry JR, Elliott KS, Lango H, Rayner NW, Shields B, Harries LW, Barrett JC, Ellard S, Groves CJ, Knight B, Patch AM, Ness AR, Ebrahim S, Lawlor DA, Ring SM, Ben-Shlomo Y, Jarvelin MR, Sovio U, Bennett AJ, Melzer D, Ferrucci L, Loos RJ, Barroso I, Wareham NJ, Karpe F, Owen KR, Cardon LR, Walker M, Hitman GA, Palmer CN, Doney AS, Morris AD, Smith GD, Hattersley AT, McCarthy MI (2007) A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science 316:889–894
Friedman JM (2000) Obesity in the new millennium. Nature 404:632–634
Galgani JE, Greenway FL, Caglayan S, Wong ML, Licinio J, Ravussin E (2010) Leptin replacement prevents weight loss-induced metabolic adaptation in congenital leptin-deficient patients. J Clin Endocrinol Metab 95:851–855
Gerken T, Girard CA, Tung YC, Webby CJ, Saudek V, Hewitson KS, Yeo GS, McDonough MA, Cunliffe S, McNeill LA, Galvanovskis J, Rorsman P, Robins P, Prieur X, Coll AP, Ma M, Jovanovic Z, Farooqi IS, Sedgwick B, Barroso I, Lindahl T, Ponting CP, Ashcroft FM, O’Rahilly S, Schofield CJ (2007) The obesity-associated FTO gene encodes a 2-oxoglutarate-dependent nucleic acid demethylase. Science 318:1469–1472
Gibson WT, Farooqi IS, Moreau M, Depaoli AM, Lawrence E, O’Rahilly S, Trussell RA (2004) Congenital leptin deficiency due to homozygosity for the Delta133G mutation: report of another case and evaluation of response to four years of leptin therapy. J Clin Endocrinol Metab 89:4821–4826
Gill R, Cheung YH, Shen Y, Lanzano P, Mirza NM, Ten S, Maclaren NK, Motaghedi R, Han JC, Yanovski JA, Leibel RL, Chung WK (2014) Whole-exome sequencing identifies novel LEPR mutations in individuals with severe early onset obesity. Obesity (Silver Spring) 22:576–584
Gray J, Yeo GS, Cox JJ, Morton J, Adlam AL, Keogh JM, Yanovski JA, el Gharbawy A, Han JC, Tung YC, Hodges JR, Raymond FL, O’Rahilly S, Farooqi IS (2006) Hyperphagia, severe obesity, impaired cognitive function, and hyperactivity associated with functional loss of one copy of the brain-derived neurotrophic factor (BDNF) gene. Diabetes 55:3366–3371
Greenfield JR, Miller JW, Keogh JM, Henning E, Satterwhite JH, Cameron GS, Astruc B, Mayer JP, Brage S, See TC, Lomas DJ, O’Rahilly S, Farooqi IS (2009) Modulation of blood pressure by central melanocortinergic pathways. N Engl J Med 360:44–52
Holder JL, Butte NF Jr, Zinn AR (2000) Profound obesity associated with a balanced translocation that disrupts the SIM1 gene. Hum Mol Genet 9:101–108
Hommel JD, Trinko R, Sears RM, Georgescu D, Liu ZW, Gao XB, Thurmon JJ, Marinelli M, Dileone RJ (2006) Leptin receptor signaling in midbrain dopamine neurons regulates feeding. Neuron 51:801–810
Huszar D, Lynch CA, Fairchild-Huntress V, Dunmore JH, Fang Q, Berkemeier LR, Gu W, Kesterson RA, Boston BA, Cone RD, Smith FJ, Campfield LA, Burn P, Lee F (1997) Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell 88:131–141
Jackson RS, Creemers JW, Ohagi S, Raffin-Sanson ML, Sanders L, Montague CT, Hutton JC, O’Rahilly S (1997) Obesity and impaired prohormone processing associated with mutations in the human prohormone convertase 1 gene [see comments]. Nat Genet 16:303–306
Kublaoui BM, Gemelli T, Tolson KP, Wang Y, Zinn AR (2008) Oxytocin deficiency mediates hyperphagic obesity of Sim1 haploinsufficient mice. Mol Endocrinol 22:1723–1734
Li H, Wu Y, Loos RJ, Hu FB, Liu Y, Wang J, Yu Z, Lin X (2008) Variants in the fat mass- and obesity-associated (FTO) gene are not associated with obesity in a Chinese Han population. Diabetes 57:264–268
Loos RJ (2012) Genetic determinants of common obesity and their value in prediction. Best Pract Res Clin Endocrinol Metab 26:211–226
Loos RJ, Lindgren CM, Li S, Wheeler E, Zhao JH, Prokopenko I, Inouye M, Freathy RM, Attwood AP, Beckmann JS, Berndt SI, Jacobs KB, Chanock SJ, Hayes RB, Bergmann S, Bennett AJ, Bingham SA, Bochud M, Brown M, Cauchi S, Connell JM, Cooper C, Smith GD, Day I, Dina C, De S, Dermitzakis ET, Doney AS, Elliott KS, Elliott P, Evans DM, Sadaf Farooqi I, Froguel P, Ghori J, Groves CJ, Gwilliam R, Hadley D, Hall AS, Hattersley AT, Hebebrand J, Heid IM, Lamina C, Gieger C, Illig T, Meitinger T, Wichmann HE, Herrera B, Hinney A, Hunt SE, Jarvelin MR, Johnson T, Jolley JD, Karpe F, Keniry A, Khaw KT, Luben RN, Mangino M, Marchini J, McArdle WL, McGinnis R, Meyre D, Munroe PB, Morris AD, Ness AR, Neville MJ, Nica AC, Ong KK, O’Rahilly S, Owen KR, Palmer CN, Papadakis K, Potter S, Pouta A, Qi L, Randall JC, Rayner NW, Ring SM, Sandhu MS, Scherag A, Sims MA, Song K, Soranzo N, Speliotes EK, Syddall HE, Teichmann SA, Timpson NJ, Tobias JH, Uda M, Vogel CI, Wallace C, Waterworth DM, Weedon MN, Willer CJ, Wraight, Yuan X, Zeggini E, Hirschhorn JN, Strachan DP, Ouwehand WH, Caulfield MJ et al (2008) Common variants near MC4R are associated with fat mass, weight and risk of obesity. Nat Genet 40:768–775
Maes HH, Neale MC, Eaves LJ (1997) Genetic and environmental factors in relative body weight and human adiposity. Behav Genet 27:325–351
McMurray F, Church CD, Larder R, Nicholson G, Wells S, Teboul L, Tung YC, Rimmington D, Bosch F, Jimenez V, Yeo GS, O’Rahilly S, Ashcroft FM, Coll AP, Cox RD (2013) Adult onset global loss of the fto gene alters body composition and metabolism in the mouse. PLoS Genet 9:e1003166
Minokoshi Y, Kim YB, Peroni OD, Fryer LG, Muller C, Carling D, Kahn BB (2002) Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature 415:339–343
Montague CT, Farooqi IS, Whitehead JP, Soos MA, Rau H, Wareham NJ, Sewter CP, Digby JE, Mohammed SN, Hurst JA, Cheetham CH, Earley AR, Barnett AH, Prins JB, O’Rahilly S (1997) Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature 387:903–908
Ozata M, Ozdemir IC, Licinio J (1999) Human leptin deficiency caused by a missense mutation: multiple endocrine defects, decreased sympathetic tone, and immune system dysfunction indicate new targets for leptin action, greater central than peripheral resistance to the effects of leptin, and spontaneous correction of leptin-mediated defects. J Clin Endocrinol Metab 84:3686–3695
Ramachandrappa S, Raimondo A, Cali AM, Keogh JM, Henning E, Saeed S, Thompson A, Garg S, Bochukova EG, Brage S, Trowse V, Wheeler E, Sullivan AE, Dattani M, Clayton PE, Datta V, Bruning JB, Wareham NJ, O’Rahilly S, Peet DJ, Barroso I, Whitelaw ML, Farooqi IS (2013) Rare variants in single-minded 1 (SIM1) are associated with severe obesity. J Clin Invest 123:3042–3050
Seo S, Guo DF, Bugge K, Morgan DA, Rahmouni K, Sheffield VC (2009) Requirement of Bardet-Biedl syndrome proteins for leptin receptor signaling. Hum Mol Genet 18:1323–1331
Smemo S, Tena JJ, Kim KH, Gamazon ER, Sakabe NJ, Gomez-Marin C, Aneas I, Credidio FL, Sobreira DR, Wasserman NF, Lee JH, Puviindran V, Tam D, Shen M, Son JE, Vakili NA, Sung HK, Naranjo S, Acemel RD, Manzanares M, Nagy A, Cox NJ, Hui CC, Gomez-Skarmeta JL, Nobrega MA (2014) Obesity-associated variants within FTO form long-range functional connections with IRX3. Nature 507:371–375
Sorensen TI, Price RA, Stunkard AJ, Schulsinger F (1989) Genetics of obesity in adult adoptees and their biological siblings. BMJ 298:87–90
Stratigopoulos G, Padilla S, Leduc CA, Watson E, Hattersley AT, Mccarthy MI, Zeltser LM, Chung WK, Leibel RL (2008) Regulation of Fto/Ftm gene expression in mice and humans. Am J Physiol Regul Integr Comp Physiol. doi:10.1152/ajpregu.00839.2007
Stratigopoulos G, Leduc CA, Cremona ML, Chung WK, Leibel RL (2011) Cut-like homeobox 1 (CUX1) regulates expression of the fat mass and obesity-associated and retinitis pigmentosa GTPase regulator-interacting protein-1-like (RPGRIP1L) genes and coordinates leptin receptor signaling. J Biol Chem 286:2155–2170
Stratigopoulos G, Martin Carli JF, O’day DR, Wang L, Leduc CA, Lanzano P, Chung WK, Rosenbaum M, Egli D, Doherty DA, Leibel RL (2014) Hypomorphism for RPGRIP1L, a ciliary gene vicinal to the FTO locus, causes increased adiposity in mice. Cell Metab 19:767–779
Stunkard AJ, Sorensen TI, Hanis C, Teasdale TW, Chakraborty R, Schull WJ, Schulsinger F (1986) An adoption study of human obesity. N Engl J Med 314:193–198
Stunkard AJ, Harris JR, Pedersen NL, McClearn GE (1990) The body-mass index of twins who have been reared apart [see comments]. N Engl J Med 322:1483–1487
Stutzmann F, Tan K, Vatin V, Dina C, Jouret B, Tichet J, Balkau B, Potoczna N, Horber F, O’Rahilly S, Farooqi IS, Froguel P, Meyre D (2008) Prevalence of melanocortin-4 receptor deficiency in Europeans and their age-dependent penetrance in multigenerational pedigrees. Diabetes 57:2511–2518
Tobin JL, Beales PL (2007) Bardet-Biedl syndrome: beyond the cilium. Pediatr Nephrol 22:926–936
Vaisse C, Clement K, Durand E, Hercberg S, Guy-Grand B, Froguel P (2000) Melanocortin-4 receptor mutations are a frequent and heterogeneous cause of morbid obesity. J Clin Invest 106:253–262
Walters RG, Jacquemont S, Valsesia A, de Smith AJ, Martinet D, Andersson J, Falchi M, Chen F, Andrieux J, Lobbens S, Delobel B, Stutzmann F, El-Sayed Moustafa JS, Chevre JC, Lecoeur C, Vatin V, Bouquillon S, Buxton JL, Boute O, Holder-Espinasse M, Cuisset JM, Lemaitre MP, Ambresin AE, Brioschi A, Gaillard M, Giusti V, Fellmann F, Ferrarini A, Hadjikhani N, Campion D, Guilmatre A, Goldenberg A, Calmels N, Mandel JL, le Caignec C, David A, Isidor B, Cordier MP, Dupuis-Girod S, Labalme A, Sanlaville D, Beri-Dexheimer M, Jonveaux P, Leheup B, Ounap K, Bochukova EG, Henning E, Keogh J, Ellis RJ, Macdermot KD, van Haelst MM, Vincent-Delorme C, Plessis G, Touraine R, Philippe A, Malan V, Mathieu-Dramard M, Chiesa J, Blaumeiser B, Kooy RF, Caiazzo R, Pigeyre M, Balkau B, Sladek R, Bergmann S, Mooser V, Waterworth D, Reymond A, Vollenweider P, Waeber G, Kurg A, Palta P, Esko T, Metspalu A, Nelis M, Elliott P, Hartikainen AL, McCarthy MI, Peltonen L, Carlsson L, Jacobson P, Sjostrom L, Huang N, Hurles ME, O’Rahilly S, Farooqi IS, Mannik K, Jarvelin MR, Pattou F, Meyre D, Walley AJ, Coin LJ, Blakemore AI, Froguel P, Beckmann JS (2010) A new highly penetrant form of obesity due to deletions on chromosome 16p11.2. Nature 463:671–675
Wardle J, Carnell S, Haworth CM, Farooqi IS, O’Rahilly S, Plomin R (2008) Obesity associated genetic variation in FTO is associated with diminished satiety. J Clin Endocrinol Metab 93:3640–3643
Wheeler E, Huang N, Bochukova EG, Keogh JM, Lindsay S, Garg S, Henning E, Blackburn H, Loos RJ, Wareham NJ, O’Rahilly S, Hurles ME, Barroso I, Farooqi IS (2013) Genome-wide SNP and CNV analysis identifies common and low-frequency variants associated with severe early-onset obesity. Nat Genet 45:513–517
Yeo GS, Connie Hung CC, Rochford J, Keogh J, Gray J, Sivaramakrishnan S, O’rahilly S, Farooqi IS (2004) A de novo mutation affecting human TrkB associated with severe obesity and developmental delay. Nat Neurosci 7:1187–1189
Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM (1994) Positional cloning of the mouse obese gene and its human homologue. Nature 372:425–432
Zhang F, Xu L, Jin L, Wang XF (2008) A common variant in the FTO gene is associated with obesity in the Uyghur population. J Endocrinol Invest 31:1043
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Farooqi, S. Genetic strategies to understand physiological pathways regulating body weight. Mamm Genome 25, 377–383 (2014). https://doi.org/10.1007/s00335-014-9541-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00335-014-9541-z