
Abstract
Noise-induced hearing loss (NIHL) is one of the more common sources of environmentally induced hearing loss in adults. In a mouse model, Castaneous (CAST/Ei) is an inbred strain that is resistant to NIHL, while the C57BL/6J strain is susceptible. We have used the genome-tagged mice (GTM) library of congenic strains, carrying defined segments of the CAST/Ei genome introgressed onto the C57BL/6J background, to search for loci modifying the noise-induced damage seen in the C57BL/6J strain. NIHL was induced by exposing 6-8-week old mice to 108 dB SPL intensity noise. We tested the hearing of each mouse strain up to 23 days after noise exposure using auditory brainstem response (ABR). This study identifies a number of genetic loci that modify the initial response to damaging noise, as well as long-term recovery. The data suggest that multiple alleles within the CAST/Ei genome modify the pathogenesis of NIHL and that screening congenic libraries for loci that underlie traits of interest can be easily carried out in a high-throughput fashion.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Noise-induced hearing loss (NIHL) is a common disability among people exposed to either prolonged loud noise or excessively loud impulse noise. This susceptibility to hearing loss appears to be due to both genetic and environmental factors. While the environmental variable can be well defined, the genetic components are still largely unknown. Due to the complexity of carrying out well-controlled experiments in humans, we utilized a mouse model of hearing loss to address the genetics underlying NIHL.
While NIHL and age-related hearing loss (ARHL) are most likely two separate disorders, NIHL has been linked to ARHL in mice by studies that demonstrate an exacerbation of ARHL by noise exposure (Kujawa and Liberman 2006). Speculation on the importance of the Ahl1 gene in NIHL has been explored in work demonstrating that mice susceptible to ARHL also displayed a permanent threshold shift (PTS) after noise exposure. This is in contrast to the response seen in the CBA/CaJ strain, which has little or no PTS and represents a resistant model to NIHL (Erway et al. 1996). Although it has been speculated that the advanced onset of ARHL seen in mice after noise damage is the sole result of the Ahl1 allele on chromosome 10, additional studies suggest that several other loci may contribute to NIHL susceptibility (Harding et al. 2005; Vazquez et al. 2004). Furthermore, the C57BL/6J strain exhibits ARHL but, of the many strains carrying the Ahl1 allele, it does not show the highest susceptibility to NIHL. These results suggest the interaction of multiple genes in the pathogenesis of NIHL (Jimenez et al. 2001).
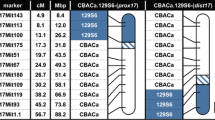
To better define the genetic components of this disorder, we undertook a screen of different strains of mice for their susceptibility to NIHL. The classical approach to mapping loci underlying traits of interest in the mouse has been QTL analysis. With the advent of high-throughput technologies and the systematic development of congenic and recombinant inbred (RI) mouse strains providing genome-wide coverage, it is now feasible to rapidly screen strains for loci that modify given traits, bypassing the lengthy breeding process required for QTL analysis. Among these resources is a library of congenic mice that contain defined segments of the CAST/Ei genome introgressed onto the C57BL/6J background, commonly referred to as Genome-Tagged Mice (GTM) (Davis et al. 2007; Iakoubova et al. 2001). The library of CAST/Ei strains contains 39 strains that provide coverage over approximately 80% of the autosomal chromosomes.
In this study we describe a method whereby GTM mice are screened in a high-throughput fashion to identify genomic loci involved with resistance or susceptibility to NIHL. We tested 16 strains from the B6.CAST congenic library and were able to identify several strains that modify the susceptibility to NIHL seen in the C57BL/6J strain. One interesting outcome of this experimental protocol, and the resulting data, is the apparent dissection of NIHL into two distinct pathologic mechanisms, one being susceptibility to the initial noise damage and the second being recovery from damage. These findings improve our understanding of the genetics of NIHL and facilitate experiments designed to discover the connection between NIHL and ARHL in the mouse.
Methods
Animals
Two strains of mice, C57BL/6J and CAST/Ei, differ significantly in their susceptibility to NIHL and therefore served as the parental strains for this experiment. The GTM library of congenic strains provides an important resource for the identification of genes underlying traits observed in the mouse and has allowed us to scan a portion of the CAST/Ei genome to determine if specific alleles affecting the pathogenesis of NIHL are present. These congenic lines have been fully characterized by high-density single nucleotide polymorphism (SNP) mapping to detect remnants of the CAST/Ei genome outside of the introgressed elements and to define the borders of the introgressed segments (Iakoubova et al. 2001).
Male and female animals were housed separately under similar conditions with ambient noise not exceeding that of normal air conditioning. All techniques performed on mice were approved by the Division of Laboratory Animal Medicine at UCLA. For inclusion in our study, complete data from at least three members of each congenic strain was required, and unhealthy animals were excluded. Pre-exposure threshold levels were obtained from mice averaging 6.5 weeks of age. To gather sufficient numbers of animals for each strain, both genders were included in the experiment. In addition, animals with elevated pre-exposure thresholds were removed from the experiment. Lastly, to prevent confounding data due to age-related hearing loss, the entire protocol was carried out on animals between 5 and 12 weeks of age.
Noise exposure
To assess noise-induced hearing loss, mice were exposed for 1 h to an octave band noise (OBN) with a center frequency of 10 kHz at a sound pressure level (SPL) of 108 ± 2 dB. It has been shown that hypothermia (30°C) is protective for NIHL while hyperthermia (40°C) exacerbates NIHL (Henry 2003). To avoid these confounding artifacts due to fluctuating body temperature, the mice were left awake during the exposure. The noise was calibrated at the beginning and end of each exposure with a B&K Sound Level Meter. OBN was created by Kenneke Communications Media Transfer Services.
During exposure, the mice were housed in a pie-shaped wire-mesh exposure cage with eight compartments. Each mouse was confined to one compartment with a food pellet and allowed to move about within the compartment. The mice were separated to minimize huddling that might reduce sound transmission. One-half-inch wire mesh was used for the cage to allow mouse waste to drop away from the animals and to allow sound transmission. A circular design that measured 12 in. in diameter and 5 in. in height was used to ensure equivalent SPLs between mice. The cage was placed in the MAC-1, a sound-proof chamber designed by Industrial Acoustics (IAC). The sound chamber was lined with 1-in.-thick acoustical sound-proofing foam wedges to minimize sound reflections. Noise recordings were played with a Morel MDT40 Tweeter Speaker built into the top of the sound chamber. This brand was chosen due to the low variation (± 3 dB) in the frequency response curve.
Anesthesia
Mice were anesthetized with an intraperitoneal injection of a mixture of ketamine (80 mg/kg body weight) and xylazine (16 mg/kg body weight). Mouse body temperature was maintained through the use of a TCAT-2DF temperature controller and the HP-4 M heating plate (Physitemp Instruments Inc., Clifton, NJ). Artificial tear ointment was applied to the eyes during anesthesia. Lastly, mice recovered from anesthesia on a heating pad.
Equipment and tone pips
Auditory signals were presented as 10-kHz tone pips in the form of a hamming wave with a 0.3-ms rise and fall time (total time of 1 ms). These signals were presented at a rate of 40 per second. They were then sent to an amplifier and then to the sound transducer from Intelligent Hearing Systems. Physiologic responses were recorded with a 20,000 analog-to-digital rate and sent to an 8 channel 150-gain AC/DC headbox and then onto a secondary Synamps signal amplifier of 2500 gain before analysis. Filter settings were set at a low-pass of 3000 Hz and a high-pass of 100 Hz with an artifact rejection of signals with amplitudes exceeding ± 50 μV. Three thousand waveforms were averaged at each test intensity. Elevated thresholds due to noise damage correspond to the frequency of the initial noise exposure (Harding et al. 2005). In an effort to streamline our phenotypic exam, only the 10-kHz frequency thresholds, identical to the exposure frequency, were recorded (Nemoto et al. 2004).
Hearing assessment using auditory brainstem response
The ability of individual mice to hear was assessed using an auditory brainstem response test (ABR). Mice were tested 4 days before noise exposure and again at 0, 4, and 23 days after noise exposure. These time points were chosen to optimize the difference between a temporary threshold shift (TTS) that subsides after 1-4 days and a permanent threshold shift (PTS) that persists beyond 20 days (Harding et al. 2005).
Stainless-steel electrodes were placed subcutaneously at the vertex of the head and the left mastoid. A ground electrode was placed at the base of the tail. Test sounds were presented using an Intelligent Hearing Systems speaker attached to an 8–in. long tube that was inserted into the ear canal. Due to time and equipment constraints, only the left ear was assessed.
Tone pips were first presented at a high intensity to elicit a waveform. Next, the intensity was decreased by 20 dB until nearing threshold. Intensity was then decreased in smaller steps of 10, 5, and 2 dB as threshold was approached. Hearing threshold was determined by visual inspection of ABR waveforms and was defined as the intensity at which two peaks could be distinguished. Experiments were duplicated at low intensities when the peaks were not apparent.
Data analysis and statistics
Fisher’s protected least-significant-difference (Fisher’s PLSD) post hoc test was used to determine significant differences between the parental strains and the congenic strains. Significance levels were set at 5%.
Results
Pre-exposure hearing levels
Sixteen GTM strains were included in this high-throughput screen for resistance to NIHL. Pre-exposure threshold levels were consistent between the parental strains and the congenic strains (Fig. 1), suggesting that the genetic basis for baseline hearing thresholds is common between these mice strains. In addition, these data provide a baseline value for measuring noise-induced threshold shifts as well as demonstrating overall consistency of hearing health among the strains, with no animals suffering from ARHL.

Pre-exposed hearing thresholds were evaluated 4 days before noise exposure. Threshold data from the parental and congenic strains are presented. Parental strains C57BL/6J and CAST/Ei are shown on the left
Immediate effects of noise exposure
Following exposure to noise, mice were functionally tested to determine the threshold shift associated with initial noise exposure. Measurements of hearing were done using dB hearing levels (dBHL). dBHL is the net hearing loss for a given time point. To obtain dBHL, pre-exposure threshold levels were subtracted from any subsequent threshold measurements. Figure 2 shows the threshold shifts for each strain tested. For C57BL/6J, as well as for all of the congenic strains, threshold shifts were in the range of 35-70 dBHL. For the CAST/Ei parental strain, a much smaller threshold shift of 12 dBHL was observed. Consistent with the majority of the genome having an origin from C57Bl/6J, it is not unexpected that initial threshold values for the congenic lines are much closer to the C57Bl/6J parental strain than the CAST/Ei parental strain. Interestingly, some strains showed susceptibility to immediate noise damage greater than seen in the C57BL/6J strain, suggesting the presence of alleles in the CAST/Ei genome that contribute to both susceptibility and resistance (Fig. 2).

Identification of temporary threshold shifts in the parental and congenic strains. Data are presented for hearing levels after 1 h of noise exposure. The two parental strains are presented on the left
Detection of NIHL
NIHL is defined as permanent hearing loss, measured after a recovery period. Mice exposed to damaging noise levels show exponential threshold recovery with increasing post-exposure time up to about 2 weeks (Miller et al. 1963). For this reason, we measured NIHL at 23 days post noise exposure. At 4 days post-exposure, only the CAST/Ei strain demonstrated full recovery from noise exposure, with all congenic strains and C57BL/6J showing elevated thresholds (data not shown). At 23 days post-exposure, consistent with previous observations (Harding et al. 2005), C57Bl/6J demonstrated a significant permanent threshold shift (Fig. 3). An in-depth examination of the data revealed at least two congenic strains that display a modification of the C57BL/6J response to noise exposure. Strain B6.CAST.4PM demonstrated a permanent threshold shift that was approximately one-third that of the C57BL/6J parental strain. The initial threshold shift for this strain was equal to that seen in C57BL/6J, indicating that the CAST/Ei allele facilitates recovery from the initial damage. In contrast, data from B6.CAST.2P indicate the presence of an allele from the CAST/Ei strain that decreases recovery from noise damage (Fig. 3).

Noise-induced hearing loss for congenic strains. Permanent hearing threshold shifts for the parental strains and the congenic strain are presented. Vertical values are given as dBHL (dB hearing level), which represents the net threshold shifts between pre-exposure levels and day-23 levels
Genetic determinants of NIHL
Figure 4 demonstrates the statistically significant differences observed for threshold measurements between three of the GTM congenic strains and the parental strains. B6.CAST.4PM shows a reduced permanent threshold shift that is significantly different from C57BL/6J (p = 0.0017). A similar but less protective effect was seen in strain B6.CAST.19C (p = 0.0418). In contrast, B6.CAST.2P shows a permanent threshold shift that is significantly worse than that of C57BL/6J (p < 0.0001). These data indicate that naturally occurring allelic variation within the CAST/Ei genome contributes loci that confer susceptibility and resistance to NIHL.

Significance values for select strains. This figure represents the difference between final and pre-exposure threshold levels quantifying noise-induced hearing loss. Parental strains are shown on the left, with data from three congenic strains also presented
Discussion
Two of the primary conditions of the ear that affect hearing in human adults are NIHL and ARHL. Both have a genetic component but are greatly influenced by environmental exposures. To address the underlying genetic basis for both of these conditions, a number of mouse models have been exploited (Ohlemiller 2006). A genetic interaction between NIHL and ARHL has been clearly demonstrated with several spontaneous and targeted mutants, including Cdh 23ahl, Cdh 23v, Pmca2, Sod1, GPx1, Trpv4, and Cp, demonstrating dual susceptibilities (Ohlemiller 2006). These results indicate that this locus is involved with both pathologies. Interestingly, the experimental protocol for our study revealed two novel endophenotypes related to NIHL, primarily, genetic loci that contribute to the initial damage and loci involved with recovery.
Experiments to address NIHL in these mouse models have shown that the allele of Ahl1 present in the genome predisposes these strains to NIHL (Davis et al. 1999). Similar experiments that used a congenic mouse, differing only at the Ahl1 locus, demonstrated that the CAST/Ei allele alone is not enough to protect against noise damage (Harding et al. 2005; Vazquez et al. 2004), suggesting the genetic complexity of NIHL.
To identify additional loci involved in the pathogenesis of NIHL, we have exploited the GTM library of congenic strains, specifically the B6.CAST panel, to test for differences in sensitivity to NIHL (Davis et al. 2007; Iakoubova et al. 2001). ABR data from the C57BL/6J parental strain revealed that mice suffer from an initial threshold shift of almost 50 dBHL (mean of 48 dBHL), a portion of which is temporary and subsides before 23 days. The CAST/Ei parental strain has a much smaller threshold shift in response to damaging noise, and this shift is entirely temporary (mean of 12 dBHL), lasting 4 days or less. Comparison between C57BL/6J and CAST/Ei at 23 days reveals significant differences between NIHL.
Preliminary analysis of our TTS and PTS data for the congenic strains revealed many congenic lines within the CAST/Ei library that displayed a modified NIHL compared to C57BL/6J. A more in-depth analysis appears to reveal two components of NIHL pathogenesis. The first component appears to influence susceptibility to the initial damage (Fig. 2), while the second component seems to act through the recovery mechanism (Fig. 3). For each of these components we see strains that appear to represent both susceptibility and resistance. A corollary to this hypothesis would be that strains of GTM mice should exist that exhibit characteristics for the four subtypes of NIHL pathogenesis. The first of these subclasses would be represented by strains that appear susceptible to the initial damage and have a poor recovery. The second subclass would have susceptibility to the initial damage but a good recovery. The third subclass would be represented by strains that exhibit resistance to the initial damage and a strong recovery, while the fourth subclass shows resistance to the initial damage but a poor recovery from this damage.
The first and second subclasses, which show susceptibility to the initial damage but have variable recoveries, appear to be represented by strains B6.CAST.2P and B6.CAST.4PM. Data presented in Figs. 2 and 3 show that initial damage for both B6.CAST.2P and B6.CAST.4PM is similar to or worse than that for C57BL/6J (p = 0.0665 and 0.1093, respectively). However, when compared to the recovery of C57BL/6J, strain B6.CAST.2P shows almost no recovery from noise-induced damage, resulting in a highly significant PTS (p < 0.0001), while strain B6.CAST.4PM shows almost complete recovery from noise-induced damage (p = 0.0111).
The third and fourth subclasses are represented by strains that show resistance to the initial noise damage but display variable recovery. The third subclass shows resistance to initial noise damage and good noise recovery. The strain closest to that model is B6.CAST.19C. This strain appears to show resistance to initial noise damage compared to C57BL/6J (p = 0.0293) as well as improved recovery (p = 0.0418). The fourth subclass is represented by strains B6.CAST.14PM and B6.CAST.2D. These strains appear to demonstrate resistance to initial damage compared to C57BL/6J (p = 0.0546 and 0.0017, respectively), but recover similar to C57BL/6J (p = 0.4722 and 0.8634). Figure 4 presents the first three subclasses showing variable levels of permanent NIHL.
The above data strongly support the hypothesis that NIHL can be divided into two subphenotypes, the first being susceptibility to the initial damage from noise, the second being the recovery from the initial damage. Our data are consistent with observations made previously by investigators who examined the genetics of NIHL that suggested that initial damage and recovery from that damage are separate events (Li 1992). Clearly, our data provide an ideal starting point for studies designed to better understand the cellular response in the cochlea to damaging noise. We predict that morphologic analysis of inner ears from the congenic strains that represent each of the disease subclasses will present unique morphologies that reflect the mechanisms of damage and damage recovery. As we refine these loci and focus on candidate genes we are preparing to analyze the attendant morphologic correlates.
A better understanding of the full genetic basis of NIHL in humans poses numerous challenges. This study involved mice and was limited to a physiologic phenotype and the identification of naturally occurring genetic variants. An important question raised by these results is the role of hair cell loss in NIHL. Severe noise exposure has been shown to result in hair cell loss (Harding et al. 2005); however, the exact relationship between NIHL and hair cell loss has not been determined. There has been considerable discussion concerning the time of onset of hair cell loss in correlation with NIHL. In early NIHL experiments using cats, threshold shifts on the order of 40 dBHL occurred in the absence of hair cell loss but did appear to involve damage to the stereocilia (Liberman and Beil 1979). A more recent study that involved careful observations of inner-ear morphology following acoustic trauma found that the most obvious disruption to cellular organelles visible after noise exposure was a “buckling” of the pillar bodies, which resulted in the stereocilia of the outer hair cells separating from the tectorial membrane. The authors noted that the nerve fibers appeared normal, with minimal swelling of nerve fibers as has been proposed for a TTS mechanism (Nordmann et al. 2000). Another recent study demonstrated that hair cells and supporting cells in CBA/CaJ mice can appear normal 4-8 weeks post-exposure, even when permanent hearing loss is present (Kujawa and Liberman 2006). The authors noted that the only histologically visible damage 2 weeks post-exposure was the loss of type IV fibrocytes. Other cochlear structures, including the stria vascularis, spial limbus, tectorial membrane, and hair cells, appeared normal. The presence of CAST/Ei genetic elements that confer susceptibility or resistance to NIHL may provide a better experimental system to address hair cell loss.
Further studies will be necessary to identify specific candidate genes within this region. The introgressed segment of the castaneous genome is very large, measuring over 125 Mbp, making candidate gene identification difficult. One way to reduce the number of candidate genes would be to focus on genes expressed in the cochlea. Several cochlea-expressed genes have been identified and mapped to this region of chromosome 4 (Howard et al. 2003; Nelson et al. 2007). Some of these genes are good candidates for our observed strain differences. For example, mice with deletions of Fbxo2 are deficient in Fbx2, an ubiquitin ligase F-box protein, and develop cochlear degeneration and age-related hearing loss beginning at 2 months of age (Nelson et al. 2007). The Fbxo2 gene belongs to a gene family that shares homology in a region of the protein that binds mannose residues. Glycoproteins that fail to fold properly are retained in the endoplasmic reticulum through the interaction of these proteins and mannose residues of the glycosylation complex and are then targeted to the proteosomal degradation pathway. Because mice with this knockout lose their hearing as adults and there is a known link between NIHL and ARHL, this gene is a compelling candidate.
Finally, the identification of multiple genetic loci that moderate NIHL in different subcongenic strains will allow us to create combinatorial congenics to address interactions between genes that convey susceptibility or resistance to NIHL and to further dissect the independent pathologies of resistance to initial damage and successful recovery from damage.
References
Davis RR, Cheever ML, Krieg EF, Erway LC (1999) Quantitative measure of genetic differences in susceptibility to noise-induced hearing loss in two strains of mice. Hear Res 134:9–15
Davis RC, Jin A, Rosales M, Yu S, Xia X et al (2007) A genome-wide set of congenic mouse strains derived from CAST/Ei on a C57BL/6 background. Genomics 90:306–313
Erway LC, Shiau YW, Davis RR, Krieg EF (1996) Genetics of age-related hearing loss in mice. III. Susceptibility of inbred and F1 hybrid strains to noise-induced hearing loss. Hear Res 93:181–187
Harding GW, Bohne BA, Vos JD (2005) The effect of an age-related hearing loss gene (Ahl) on noise-induced hearing loss and cochlear damage from low-frequency noise. Hear Res 204:90–100
Henry KR (2003) Hyperthermia exacerbates and hypothermia protects from noise-induced threshold elevation of the cochlear nerve envelope response in the C57BL/6J mouse. Hear Res 179:88–96
Howard MA, Rodenas-Ruano A, Henkemeyer M, Martin GK, Lonsbury-Martin BL et al (2003) Eph receptor deficiencies lead to altered cochlear function. Hear Res 178:118–130
Iakoubova OA, Olsson CL, Dains KM, Ross DA, Andalibi A et al (2001) Genome-tagged mice (GTM): two sets of genome-wide congenic strains. Genomics 74:89–104
Jimenez AM, Stagner BB, Martin GK, Lonsbury-Martin BL (2001) Susceptibility of DPOAEs to sound overexposure in inbred mice with AHL. J Assoc Res Otolaryngol 2:233–245
Kujawa SG, Liberman MC (2006) Acceleration of age-related hearing loss by early noise exposure: evidence of a misspent youth. J Neurosci 26:2115–2123
Li HS (1992) Influence of genotype and age on acute acoustic trauma and recovery in CBA/Ca and C57BL/6J mice. Acta Otolaryngol 112:956–967
Liberman MC, Beil DG (1979) Hair cell condition and auditory nerve response in normal and noise-damaged cochleas. Acta Otolaryngol 88:161–176
Miller JD, Watson CS, Covell WP (1963) Deafening effects of noise on the cat. Acta Otolaryngol (Suppl) 176:44–52
Nelson RF, Glenn KA, Zhang Y, Wen H, Knutson T et al (2007) Selective cochlear degeneration in mice lacking the F-box protein, Fbx2, a glycoprotein-specific ubiquitin ligase subunit. J Neurosci 27:5163–5171
Nemoto M, Morita Y, Mishima Y, Takahashi S, Nomura T et al (2004) Ahl3, a third locus on mouse chromosome 17 affecting age-related hearing loss. Biochem Biophys Res Commun 324:1283–1288
Nordmann AS, Bohne BA, Harding GW (2000) Histopathological differences between temporary and permanent threshold shift. Hear Res 139:13–30
Ohlemiller KK (2006) Contributions of mouse models to understanding of age- and noise-related hearing loss. Brain Res 1091:89–102
Vazquez AE, Jimenez AM, Martin GK, Luebke AE, Lonsbury-Martin BL (2004) Evaluating cochlear function and the effects of noise exposure in the B6.CAST+Ahl mouse with distortion product otoacoustic emissions. Hear Res 194:87–96
Acknowledgments
The authors acknowledge the assistance of Dr. Yvonne S. Sininger with both ABR technique and interpretation. We also acknowledge the UCLA Department of Laboratory Animal Medicine for their assistance. The contributions of Ms. Hesper Tsai to data collection and ABR analysis were invaluable. The study was funded by the House Ear Institute Rick and Tina Caruso Fund for research in genetic hearing loss.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
White, C.H., Ohmen, J.D., Sheth, S. et al. Genome-wide screening for genetic loci associated with noise-induced hearing loss. Mamm Genome 20, 207–213 (2009). https://doi.org/10.1007/s00335-009-9178-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00335-009-9178-5