
Abstract
Systems-oriented genetic approaches that incorporate gene expression and genotype data are valuable in the quest for genetic regulatory loci underlying complex traits. Gene coexpression network analysis lends itself to identification of entire groups of differentially regulated genes—a highly relevant endeavor in finding the underpinnings of complex traits that are, by definition, polygenic in nature. Here we describe one such approach based on liver gene expression and genotype data from an F2 mouse intercross utilizing weighted gene coexpression network analysis (WGCNA) of gene expression data to identify physiologically relevant modules. We describe two strategies: single-network analysis and differential network analysis. Single-network analysis reveals the presence of a physiologically interesting module that can be found in two distinct mouse crosses. Module quantitative trait loci (mQTLs) that perturb this module were discovered. In addition, we report a list of genetic drivers for this module. Differential network analysis reveals differences in connectivity and module structure between two networks based on the liver expression data of lean and obese mice. Functional annotation of these genes suggests a biological pathway involving epidermal growth factor (EGF). Our results demonstrate the utility of WGCNA in identifying genetic drivers and in finding genetic pathways represented by gene modules. These examples provide evidence that integration of network properties may well help chart the path across the gene–trait chasm.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
While traditional meiotic mapping methods such as linkage analysis and allelic association studies have been fruitful in identifying genetic targets responsible for Mendelian traits, these methods have been less successful in the identification of pathways and genes underlying complex traits. Integration of gene expression, genetic marker, and phenotype data via genetical genomics strategies is increasingly used in complex disease research (Bystrykh et al. 2005; Chen et al. 2004; Chesler et al. 2005; Hubner et al. 2005; Mahr et al. 2006; Nishimura et al. 2005; Schadt et al. 2003).
Closely related to “genetical genomics” are “systems genetics” approaches that emphasize network methods to describe the relationship between the transcriptome, physiologic traits, and genetic markers (Drake et al. 2006; Kadarmideen et al. 2006; Schadt and Lum 2006). Here we describe a particular incarnation of a systems genetics approach: integrated weighted gene coexpression network analysis (WGCNA) (Zhang and Horvath 2005; Horvath et al. 2006). By focusing on modules rather than on individual gene expressions, WGCNA greatly alleviates the multiple-testing problem inherent in microarray data analysis. Instead of relating thousands of genes to the physiologic trait, it focuses on the relationship between a few (here 12) modules and the trait. Because modules may correspond to biological pathways, focusing the analysis on module eigengenes (and equivalently intramodular hub genes) amounts to a biologically motivated data reduction scheme. WGCNA starts from the level of thousands of genes, identifies clinically interesting gene modules, and finally screens for suitable targets by requiring module membership (high intramodular connectivity) and other application-dependent criteria such as gene ontology or associations with clinical trait-related quantitative trait loci. Genetic marker data allow one to identify the chromosomal locations (referred to as module quantitative trait loci, mQTLs) that influence the module expression profiles. Genetic marker data also allow one to prioritize genes inside trait-related modules. In particular, if a genetic marker is known to be associated with the module expressions, using it to screen for gene expressions that correlate with the SNP allows one to identify upstream drivers of the module expressions. The underlying assumption in such an analysis is that functionally related genes and/or genetic pathways are regulated by common genetic drivers. We have applied this approach to identify mQTLs that control the expression profiles of a body weight–related module in an F2 population of mice (Ghazalpour et al. 2006). Here we extend these findings to another mouse cross. We also demonstrate the utility of WGCNA in relating distinct subgroups of a population via differential network analysis.
Materials and methods
The weighted gene coexpression network terminology is reviewed in Table 1 and in the Supplementary Material, Appendix A.
Data description
We illustrate our methods using data from previously studied F2 mouse crosses. The first F2 data set (B × H cross) was obtained from liver tissue of 135 female mice derived from the F2 intercross between inbred strains C3H/HeJ and C57BL/6J (Ghazalpour et al. 2006; Wang et al. 2006). The second F2 (B × D) intercross data included liver tissue of 113 F2 mice derived from a cross of two standard inbred strains, C57BL/6J and DBA/2J (Ghazalpour et al. 2006; Schadt et al. 2003). Body weight and related physiologic (“clinical”) traits were measured in both sets of mice. We note that B × H and B × D mice differ in some respects. B × H mice are ApoE null (ApoE −/−) and thus hyperlipidemic, whereas B × D mice are wild type (ApoE +/+). B × H mice were fed a high-fat diet and B × D mice were fed a high-fat, high-cholesterol atherogenic diet. Also, B × H mice were sacrificed at an earlier age (24 weeks) than were the B × D mice (16 months).
Coexpression network analysis strategies
In the following, we present two distinct network analysis approaches: single-network analysis and differential network analysis. The two approaches answer different questions. The single-network analysis defines modules that can then be tested for validity with other data sets. Single-network analysis aims at identifying (a) pathways (modules) and (b) their key drivers (e.g., hub genes) that are present in a given data set. For example, we use all mice of a given F2 intercross to identify trait-related modules and mQTLs.
The second strategy, differential network analysis, aims to uncover differences in the modules and connectivity between different data sets (e.g., males versus females). Here we use body weight to arrive at two distinct data sets: lean and obese mice. Each data set is then used to construct a network. Next, the networks are contrasted to find (1) nonpreserved modules, (2) differentially expressed genes, and (3) differentially connected genes. Traditionally, a main goal of studying gene expression data is to relate differences in gene expression profiles to phenotypic differences across different conditions (e.g., different groups of mice). Viewing individual genes in isolation and analysis of differential expression is a well-established technique that has already yielded many important insights. On the other hand, differential analysis of network quantities (i.e., quantities describing the relationships between the genes such as intramodular connectivity) is neither as developed nor as widely used, although it has already led to some interesting results. For example, differential analysis of intramodular connectivity was used to identify key differences in expression networks of human and chimpanzee brains (Oldham et al. 2006).
Single weighted gene coexpression network analysis
In the case of single-network analysis, one uses a single network for modeling the relationship between transcriptome, clinical traits, and genetic marker data. In the following, we describe a typical single-network analysis for finding body weight–related modules and genes. While a single network is the focus, it does not imply that only a single data set is used. Instead, appropriately similar multiple data sets can be used to validate the robustness of module definition and connectivity.
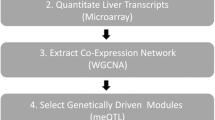
In the following, we provide an overview of single-network analysis strategy, which is depicted in Fig. 1: (1) A weighted gene coexpression network is constructed from genome-wide transcription data. (2) Modules are identified and module centrality measures (intramodular connectivity) are calculated. (3) Network modules are analyzed for biological significance. (4) Genetic loci driving functionally relevant modules within the network are identified. (5) Trait-related mQTLs are used to prioritize genes within physiologically significant modules.

Overview of weighted gene coexpression network analysis (single-network analysis)
Differential weighted gene coexpression network analysis
We describe another application of WGCNA, differential network analysis, which may be useful in identifying gene pathways distinguishing phenotypically distinct groups of samples. In our example, we identified the 30 mice at both extremes of the weight spectrum in the B × H data and constructed the first network using the 30 leanest mice and the second network using the 30 heaviest mice. For the ith gene, we denote by k 1(i) and k 2(i) the whole-network connectivity in networks 1 and 2, respectively. To facilitate the comparison between the connectivity measures of each network, we divide each gene connectivity by the maximum network connectivity, i.e.,
Next we define a measure of differential connectivity as Diff K(i) = K 1(i) – K 2(i), but other measures of differential connectivity could also be considered.
Software availability
R software tutorials and the data for WGCNA can be found at http://www.genetics.ucla.edu/labs/horvath/CoexpressionNetwork/DifferentialNetworkAnalysis/.
Results
Single network analysis results
A single weighted gene coexpression network was constructed using expression data from livers of 135 female mice of the B × H cross, utilizing the 3421 most connected and varying transcripts from the approximately 23,000 transcripts present on the arrays (Ghazalpour et al. 2006). Using hierarchical clustering, we obtained 12 modules (each designated by a color). Gray denotes genes outside of modules. In this network, the Blue module had the highest module significance score for the physiologic trait of mouse weight (g) (module significance = 0.395, p = 7.7 × 10−5), and was also highly significant for abdominal fat pad mass (g) (module significance = 0.323, p = 0.009). These p values remain significant after Bonferroni correction adjusting for 12 modules. We mention that total mass (g) of other fat depots is also significant (module significance = 0.309, p = 0.02), but does not remain significant after Bonferroni correction.
To study the preservation of modules across different F2 intercrosses, we used the B × H module color assignment to cluster the corresponding network in the B × D mouse cross data set (Fig. 2a). A weighted gene coexpression network analysis was constructed using 1953 genes in the B × D data set that have corresponding probes in the B × H data set. We observe that several modules (Red, Blue, Green-yellow, Turquoise, and Green modules being notable examples) are roughly preserved between these two data sets. Figure 2b shows a multidimensional scaling (MDS) plot of the B × D data colored by B × H modules. This plot visualizes the pairwise gene dissimilarities by projecting them into a 3-dimensional Euclidean space.

a (Top) Average linkage hierarchical clustering dendrogram of the B × D cross. (Middle) Visualization of the modules in the B × D network; module colors correspond to branches of the dendrogram shown above. (Bottom) Visualization of rough module preservation. Here we color the genes by the colors of the original B × H (not B × D) cross. The fact that colors stay together suggests module preservation. b Multidimensional scaling (MDS) plot of B × D mouse cross data, with coloring by B × H module definitions
If, in fact, intramodular connectivity (centrality and membership to the Blue module) reflects physiologic significance, one would expect to see a high correlation between kME and GSweight for the Blue module genes. As in Ghazalpour et al. (2006), we find a high correlation between kME and GSweight in the B × H cross (r = 0.47, p ≤ 10−20, Fig. 3c). Here we validate this relationship in the B × D cross (r = 0.57, p ≤ 10−20, Fig. 3d).

a Scatterplot of kME in both crosses. kME describes each eigengene’s connectivity to the Blue module. The value for the B × D cross (y axis) is plotted against the value in the B × H data set (x axis). b Scatterplot between GSweight for all genes in the B × D cross (y axis) and in the B × H cross (x axis). Colors depict B × H module membership. c Scatterplot between GSweight (y axis) and kME (x axis) in the B × H data set in genes that overlapped with the B × D cross. d Same as (c), except in the B × D cross. Spearman correlation coefficients are reported above all plots
Figure 3a shows that intramodular connectivity (kME) with regard to the Blue module is preserved between the B × H and the B × D crosses (correlation r = 0.45, p ≤ 10−20). GSweight was conserved with a Spearman correlation of 0.19 (p = 1.0 × 10−17, see Fig. 3b). Network-based gene screening uses both GSweight and kME to find weight-related genes. Note that kME is better preserved than GSweight, which suggests that kME may be a more robust gene-screening variable (see Fig. 3).
A module QTL on chromosome 19
We had previously identified a single nucleotide polymorphism (SNP) marker on chromosome 19 (SNP19) that affected weight and module expression. Table 2 demonstrates the preservation of correlations between the Blue module eigengene MEblue, weight, and SNP19 in both the B × H and the B × D data sets. A relationship was seen between MEblue and weight in both the B × H data (r = 0.62, p = 1.3 × 10−15) and in the B × D cross (r = 0.34, p = 2.1 × 10−4). We note here that while the p values are not adjusted for multiple comparisons, using the most conservative correction—the Bonferroni correction, wherein we multiply the p significance level by the number of modules—still results in a significant correlation between MEblue and weight in the B × H data. More explicitly, in correcting the p value, multiplying p = 1.3 × 10−15 by the number of modules (12) leads to a still significant p = 1.6 × 10−14. This illustrates the value of using WGCNA to reduce the number of multiple comparisons common to microarray analysis. We note that the mQTL on chromosome 19 had a single-point LOD score of 3.36. While a relatively weaker correlation between SNP19 (d19mit71) and weight is seen in B × D data compared with B × H, homozygous animals for the B6 allele of a different marker on chromosome 19 (d19mit63) have significantly different weight from DBA homozygotes (in the B × D cross). This result is consistent with the previous finding that B6 and DBA homozygotes have significantly different subcutaneous fat pad mass (a weight-related trait) (Ghazalpour et al. 2005). It is also possible that the differences in experimental design such as diet, age of the animals, and the status of the Apoe gene could account for the weaker correlation observed in the B × D network.
Using a body weight–related mQTL to prioritize genes inside the Blue module
A SNP marker allows one to define a gene significance measure, GS.SNP, which can be used to prioritize genes within a module.
For the ith gene, GS.SNP(i) is defined as the absolute value of the correlation between the ith gene’s expressions and a given SNP’s additive marker coding value:
Additive marker coding reflects the dosage of a given allele; alternatively, one could use dominant or recessive marker coding (see Supplementary Material, Supplementary Table 2).
Observed GS.SNP values are reported in Supplementary Fig. 2a for our simulated module example. We explore the relationship between the GS.SNP values obtained by different marker coding methods in Supplementary Material, Appendix B, and depict the strong relationship between GS.SNP and the traditional LOD score in Supplementary Fig. 3. In short, this figure demonstrates that regardless of whether additive, dominant, or recessive marker coding is used, GS.SNP is highly related to the LOD score values.
Systems genetics gene-screening criteria
As described above, we found a SNP marker on chromosome 19 that is highly related to body weight and to the Blue module expressions. To determine which gene expressions mediate between this mQTL and body weight, it is natural to rank gene expressions based on their correlations with SNP19 and the clinical trait. This suggests to screen for genes with high GS.SNP19 and high GSweight. Furthermore, since the Blue module was found to be related to body weight, it is natural to rank genes by membership to the Blue module, i.e., by intramodular connectivity. Our gene-screening criteria for finding the genetic drivers of body weight are as follows: (1) high association with the body weight, i.e., high values of GSweight; (2) membership and hub status in a trait-related module, i.e., a high value of kME; and (3) high association with a body weight–related mQTL, i.e., high values of GS.SNP. Specifically, we used the 85th percentile of each screening variable, which resulted in nine genes inside the Blue module (Table 3). The gene list is quite robust with respect to the percentile as the reader may explore using our online R software tutorial. An examination of their potential relationship to body weight using the Mouse Genomics Informatics gene ontology database (http://www.informatics.jax.org/) (Eppig et al. 2005) and existing literature yields the following: Fsp27 encodes a pro-apoptotic protein. Nordstrom et al. (2005) found that Fsp27-null mice are resistant to obesity and diabetes. In addition, Fsp27 expression is halved in obese humans after weight loss, and other recent research suggests that Fsp27 regulates lipolysis in white human adipocytes (Nordstrom et al. 2005). A number of the other genes are related to basic biological processes that may be altered in the obese state, which is associated clinically with both the metabolic syndrome and vascular disease, among other conditions. Gpld1 (glycosylphosphatidylinositol-specific phospholipase D1) expression in liver is increased with a high-fat diet in mice, and overexpression is associated with an increase in fasting and postprandial plasma triglycerides and a reduction in triglyceride-rich lipoprotein catabolism (Raikwar et al. 2006). Gene products of F7 and Kng2 are elements of the hemostatic system and may play roles in thrombosis and vascular disease (Kaschina et al. 2004; Reiner et al. 2007; Viles-Gonzalez et al. 2006). Our network-based gene screening method appears to identify biologically relevant genes, considering the evidence from primary literature supporting involvement of these genes in obesity (Fsp27) and/or known obesity-related disorders (diabetes, metabolic syndrome, and vascular disease). Other genes identified by this method may be novel candidates. As such, these results should be considered a starting point for subsequent experimentation to explore involvement of these genes in obesity.
Sector plots for identifying differentially expressed and differentially connected genes
Differential network analysis is concerned with identifying both differentially connected and differentially expressed genes. To measure differential gene expression between the lean and the obese mice, we use the absolute value of the Student t-test statistic. Plotting DiffK, the difference in connectivity between lean and obese mice, versus the t-test statistic value for each gene gives a visual demonstration of how difference in connectivity relates to a more traditional t-statistic describing difference in expression between the two networks.
Figure 4a shows a scatterplot of DiffK vs. the t statistic. Eight sectors of the plot with high absolute values of DiffK (> 0.4) and/or t-statistic values (> 1.96) are shown. Horizontal lines depict sector boundaries based on t-statistic values, and vertical lines depict boundaries based on DiffK. These eight sectors are marked by numbers in Fig. 4a. To assign a significance level (p value) to a gene’s DiffK value or to its membership in a particular sector defined by DiffK and t statistic, we use a permutation test approach that randomly permutes the microarray sample labels. The permutation test contrasts networks built by randomly partitioning the 60 mice into two groups. We consider the number of genes inside a given sector (which is defined by thresholding the t statistic and DiffK as described above) in determining significance level. Figure 4b demonstrates the same information except network membership is permuted. Based on 1000 random permutations, sector membership was found to be significant for sectors 2, 3, and 6 with p ≤ 1.0 × 10−3. Membership in sector 5 was significant with p ≤ 1.0 × 10−2.

Sector plots of differential network analysis. In (a) and (b), difference in connectivity (DiffK) is plotted on the x axis, and t-test statistic values are plotted on the y axis. Horizontal lines indicate a difference in connectivity of −0.4 and 0.4, whereas vertical lines depict a t-statistic value of −1.96 or 1.96. a Observed DiffK and t-statistic values: Genes are colored based on network 1 module definitions. Numbers indicate sectors 1–8. b Corresponding sector plot for a permuted network where array samples in data sets 1 and 2 were randomly permuted
Functional enrichment analysis of sector 3 genes
We analyzed 61 sector 3 genes that were both highly connected in network 1 and lowly connected in network 2 for functional enrichment using the DAVID database (Dennis et al. 2003). This software, which is free and available for download at http://www.d.abcc.ncifcrf.gov/home.jsp, calculates the p value for the extent of enrichment of a given biological pathway/set by performing Fisher’s exact test. We focused on sector 3 for two reasons. First, sector 3 members had extreme values of DiffK as well as high t-statistic values. Also, as one can readily see from Fig. 4a, a high proportion of Yellow module genes were found in this module, based on network 1 module definitions. These Yellow module genes were lowly connected in network 2, and therefore were annotated as Gray module (background) members in a module assignment scheme based on network 2. This result suggests that in a pathophysiologic state (mouse obesity), the Yellow module can no longer be found.
Results for this analysis that were significant at p < 0.05 level are shown in Table 4. These genes were markedly enriched for the extracellular region (37.7% of genes p = 1.8 × 10−4), extracellular space (34.4% of genes p = 5.7 × 10−4), signaling (36.1% of genes p = 5.4 × 10−4), cell adhesion (16.4% of genes p = 7.7 × 10−4), and glycoproteins (34.4% of genes p = 1.6 × 10−3). Furthermore, 12 terms for epidermal growth factor or its related proteins were recovered in the functional analysis. A few of the notable results are EGF-like 1 (8.2% of genes p = 8.7 × 10−4), EGF-like 3 (6.6% of genes p = 1.6 × 10−3), EGF-like 2 (6.6% of genes p = 6.0 × 10−3), EGF (8.2% of genes p = 0.013), and EGF_CA (6.6% of genes p = 0.015).
In summary, we find a group of rewired genes identified by differential connectivity in lean and obese mice. These genes are highly enriched for extracellular and cell–cell interactions and notably 12 epidermal growth factor (EGF) or EGF-related factors. An indirect validation of the differential network results is provided by a published article that reports that EGF plays a causal role in inducing obesity in ovariectomized mice (Kurachi et al. 1993).
Functional enrichment analysis of sector 5 genes
Sector 5 is analagous to sector 3 in that it contains genes with both extreme differences in connectivity and extreme t-statistic values. After Bonferroni correction, these genes are enriched for enzyme inhibitor activity (p = 2.93 × 10−3), protease inhibitor activity (p = 6.00 × 10−3), endopeptidase activity (p = 6.00 × 10−3), dephosphorylation (p = 0.0122), protein amino acid dephosphorylation (p = 0.0122), and serine-type endopeptidase inhibitor activity (p = 0.0417) (Supplementary Table 6). Two genes were enriched for all significant categories: Itih1 and Itih3. These two genes are located near a QTL marker for hyperinsulinemia (D14Mit52) identified in C57Bl/6, 129S6/SvEvTac, and (B6 × 129) F2 intercross mice (Almind and Kahn 2004). Itih3 was independently determined to be a gene candidate for obesity-related traits based on differential expression in murine hypothalamus (Bischof and Wevrick 2005). Two serine protease inhibitors, Serpina3n and Serpina10, were enriched for the categories of enzyme inhibitor, protease inhibitor, and endopeptidase inhibitor. In humans, Serpina10 is also known as Protein Z-dependent protease inhibitor (ZPI). This serpin inhibits activated coagulation factors X and XI; ZPI deficiencies have been found to be associated with venous thrombosis (Water et al. 2004). We note that obesity is a strong independent risk factor for venous thrombosis (Abdollahi et al. 2003; Goldhaber et al. 1997) and that accordingly PZI may be a link between obesity and increased risk of venous thrombotic events.
Results from functional enrichment analysis for all other sectors are described in Supplementary Material, Appendix C and Supplementary Tables 3, 4, 5, 7, and 8 (Supplementary Table 3: enrichment of biological pathways/sets for Blue module genes intersecting B × H and B × D data sets; Supplementary Table 4: enrichment of biological pathways/sets for sector 2 genes; Supplementary Table 5: enrichment of biological pathways/sets for all sector 3 genes; Supplementary Table 7: enrichment of biological pathways/sets for sector 6 genes; Supplementary Table 8: enrichment of biological pathways/sets for sector 8 genes).
Discussion
Integrating weighted gene coexpression network analysis with genotype data holds great promise for elucidating the molecular and genetic basis of complex diseases. Since WGCNA focuses on coexpression modules (as opposed to individual gene expressions), it will be useful only if trait-related modules can be detected in the gene expression data. In our mouse genetics application, we provide evidence for a body weight-related module that can be found in two F2 mouse crosses.
We show that several modules identified in the F2 B × H mouse intercross are roughly preserved in an independent B × D mouse cross. In particular, the weight-related module found in the F2 mouse intercross is recovered in the second mouse cross. Highly connected hub genes within this module are found to have high correlation with weight (GSweight). We also find that module-based measures tend to be stable and robust across independent data sets. This is even more striking given the difference between the B × H and B × D mouse populations. Hub gene status is also roughly preserved, validating the importance and robustness of intramodular connectivity. These validation successes provide evidence for the utility and robustness of network-based methods.
Central to WGCNA is the concept of intramodular connectivity, which can be considered a measure of module membership. In coexpression networks, intramodular hub genes can be considered the most central genes inside the module. Because the expression profiles of intramodular hub genes inside an interesting module are highly correlated, they are statistically equivalent. This does not imply that such genes have the same functional significance. Gene ontology may reveal that they differ in terms of biological plausibility or clinical utility. In many applications, the list of module hub genes may be further prioritized based on (1) biological plausibility based on external gene (ontology) information, (2) availability of protein biomarkers for further validation, (3) availability of suitable mouse models for further validation, and/or (4) druggability, i.e., the opportunity for therapeutic intervention.
We demonstrate that both single-network and differential network analyses may be useful for finding body weight-related genes. Single-network analysis describes the module structure and topological properties of a single data set. In single-network analysis, all samples, irrespective of their clinical trait, are used for network and module construction. In contrast, differential network analysis compares two different networks. Differential network analysis aims to identify genes that are both differentially expressed and differentially connected. Since module genes tend to be highly connected in coexpression networks, screening for differentially connected genes is related to studying the preservation of modules between the two networks. We have shown that genes that are differentially connected may or may not be differentially expressed. Changes in connectivity may correspond to large-scale “rewiring” in response to environmental changes and physiologic perturbations (Luscombe et al. 2004).
The availability of genetic markers greatly enhances the kind of questions that can be addressed by WGCNA. Genetic marker data provide valuable information for prioritizing gene expressions inside a module. The resulting systems genetics gene-screening strategy goes beyond drafting lists of differentially expressed genes or finding chromosomal locations that seem to cosegregate with a trait.
References
Abdollahi M, Cushman M, Rosendaal FR (2003) Obesity: risk of venous thrombosis and the interaction with coagulation factor levels and oral contraceptive use. Thromb Haemost 89:493–498
Almind K, Kahn CR (2004) Genetic determinants of energy expenditure and insulin resistance in diet-induced obesity in mice. Diabetes 53:3274–3285
Bischof JM, Wevrick R (2005) Genome-wide analysis of gene transcription in the hypothalamus. Physiol Genomics 22:191–196
Bystrykh L, Weersing E, Dontje B, Sutton S, Pletcher MT, et al. (2005) Uncovering regulatory pathways that affect hematopoietic stem cell function using ‘genetical genomics’. Nat Genet 37:225–232
Chen J, Lipska BK, Halim N, Ma QD, Matsumoto M, et al. (2004) Functional analysis of genetic variation in catechol-O-methyltransferase (COMT): effects on mRNA, protein, and enzyme activity in postmortem human brain. Am J Hum Genet 75:807–821
Chesler EJ, Lu L, Shou S, Qu Y, Gu J, et al. (2005) Complex trait analysis of gene expression uncovers polygenic and pleiotropic networks that modulate nervous system function. Nat Genet 37:233–242
Dennis G Jr, Sherman BT, Hosack DA, Yang J, Gao W, et al. (2003) DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol 4:P3
Drake TA, Schadt EE, Lusis AJ (2006) Integrating genetic and gene expression data: application to cardiovascular and metabolic traits in mice. Mamm Genome 17:466–479
Eppig JT, Bult CJ, Kadin JA, Richardson JE, Blake JA, et al. (2005) The Mouse Genome Database (MGD): from genes to mice—a community resource for mouse biology. Nucleic Acids Res 33:D471–D475
Ghazalpour A, Doss S, Sheth SS, Ingram-Drake LA, Schadt EE, et al. (2005) Genomic analysis of metabolic pathway gene expression in mice. Genome Biol 6:R59
Ghazalpour A, Doss S, Zhang B, Wang S, Plaisier C, et al. (2006) Integrating genetic and network analysis to characterize genes related to mouse weight. PLoS Genet 2:e130
Goldhaber SZ, Grodstein F, Stampfer MJ, Manson JE, Colditz GA, et al. (1997) A prospective study of risk factors for pulmonary embolism in women. JAMA 277:642–645
Horvath S, Zhang B, Carlson M, Lu KV, Zhu S, et al. (2006) Analysis of oncogenic signalling networks in glioblastoma identifies ASPM as a novel molecular target. Proc Natl Acid Sci 103(46):17402–17407
Hubner N, Wallace CA, Zimdahl H, Petretto E, Schulz H, et al. (2005) Integrated transcriptional profiling and linkage analysis for identification of genes underlying disease. Nat Genet 37:243–253
Kadarmideen HN, von Rohr P, Janss LL (2006) From genetical genomics to systems genetics: potential applications in quantitative genomics and animal breeding. Mamm Genome 17:548–564
Kaschina E, Stoll M, Sommerfeld M, Steckelings UM, Kreutz R, et al. (2004) Genetic kininogen deficiency contributes to aortic aneurysm formation but not to atherosclerosis. Physiol Genomics 19:41–49
Kurachi H, Adachi H, Ohtsuka S, Morishige K, Amemiya K, et al. (1993) Involvement of epidermal growth factor in inducing obesity in ovariectomized mice. Am J Physiol 265:E323–E331
Luscombe NM, Babu MM, Yu H, Snyder M, Teichmann SA, et al. (2004) Genomic analysis of regulatory network dynamics reveals large topological changes. Nature 431:308–312
Mahr S, Burmester GR, Hilke D, Gobel U, Grutzkau A, et al. (2006) Cis- and trans-acting gene regulation is associated with osteoarthritis. Am J Hum Genet 78:793–803
Nishimura DY, Swiderski RE, Searby CC, Berg EM, Ferguson AL, et al. (2005) Comparative genomics and gene expression analysis identifies BBS9, a new Bardet-Biedl syndrome gene. Am J Hum Genet 77:1021–1033
Nordstrom EA, Ryden M, Backlund EC, Dahlman I, Kaaman M, et al. (2005) A human-specific role of cell death-inducing DFFA (DNA fragmentation factor-alpha)-like effector A (CIDEA) in adipocyte lipolysis and obesity. Diabetes 54:1726–1734
Oldham MC, Horvath S, Geschwind DH (2006) Conservation and evolution of gene coexpression networks in human and chimpanzee brains. Proc Natl Acad Sci U S A 103:17973–17978
Raikwar NS, Cho WK, Bowen RF, Deeg MA (2006) Glycosylphosphatidylinositol-specific phospholipase D influences triglyceride-rich lipoprotein metabolism. Am J Physiol Endocrinol Metab 290:E463–E470
Reiner AP, Carlson CS, Rieder MJ, Siscovick DS, Liu K, et al. (2007) Coagulation factor VII gene haplotypes, obesity-related traits, and cardiovascular risk in young women. J Thromb Haemost 5:42–49
Schadt EE, Lum PY (2006) Thematic review series: systems biology approaches to metabolic and cardiovascular disorders. Reverse engineering gene networks to identify key drivers of complex disease phenotypes. J Lipid Res 47:2601–2613
Schadt EE, Monks SA, Drake TA, Lusis AJ, Che N, et al. (2003) Genetics of gene expression surveyed in maize, mouse and man. Nature 422:297–302
Viles-Gonzalez JF, Fuster V, Badimon JJ (2006) Links between inflammation and thrombogenicity in atherosclerosis. Curr Mol Med 6:489–499
Wang S, Yehya N, Schadt EE, Wang H, Drake TA, et al. (2006) Genetic and genomic analysis of a fat mass trait with complex inheritance reveals marked sex specificity. PLoS Genet 2:e15
Water N, Tan T, Ashton F, O’Grady A, Day T, et al. (2004) Mutations within the protein Z-dependent protease inhibitor gene are associated with venous thromboembolic disease: a new form of thrombophilia. Br J Haemotol 127:190–194
Zhang B, Horvath S (2005) A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol 4(1) Article 17
Acknowledgments
The authors acknowledge the support from Program Project Grant 1U19AI063603−01 and from NIH/NIDDKD-DK072206. They are grateful for discussions with collaborators Jun Dong, Dan Geschwind, Peter Langfelder, Ai Li, Wen Lin, Paul Mischel, Stan Nelson, Roel Ophoff, Anja Presson, Christiaan Saris, Lin Wang, and Wei Zhao.
Author information
Authors and Affiliations
Corresponding author
Additional information
Tova F. Fuller, Anatole Ghazalpour contributed equally to this work.
Electronic supplementary material
Electronic supplementary material
Electronic supplementary material
Electronic supplementary material
Electronic supplementary material
Electronic supplementary material
Electronic supplementary material
Electronic supplementary material
Electronic supplementary material
Electronic supplementary material
Electronic supplementary material
Electronic supplementary material
Electronic supplementary material
Electronic supplementary material
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Fuller, T.F., Ghazalpour, A., Aten, J.E. et al. Weighted gene coexpression network analysis strategies applied to mouse weight. Mamm Genome 18, 463–472 (2007). https://doi.org/10.1007/s00335-007-9043-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00335-007-9043-3